

Introduction
Melanoma is the most aggressive form of skin cancer and ranks as the sixth most common cancer in the United States.1, 2 While early stage melanoma can frequently be cured by surgical excision, patients with metastatic melanoma generally have a poor prognosis due its aggressive tumor biology and chemoresistant nature.3 After appropriate initial therapy, as many as 10% of patients with primary extremity lesions will recur in the same extremity in the form of in-transit metastases.4, 5 For these patients, the chemoresistant nature of metastatic melanoma can be partially overcome by infusing extremely high doses of cytotoxic agents through a surgically isolated extremity in the form of the isolated limb infusion (ILI) or perfusion (ILP).6 Using these techniques, we have reported overall response rates for ILI and ILP of 64% and 79% respectively.7 Despite high overall response rates, most patients will eventually recur, supporting the role for novel research aimed at improving durable responses and minimizing toxicity.8
Combining regional chemotherapy with targeted therapies directed against pathways associated with melanoma remains a promising strategy for improving both the efficacy of the chemotherapeutic agent and the durability of the anti-tumor response. During the malignant transformation of normal melanocytes, there is a switch in cadherin expression. E-cadherin (generally expressed in normal epithelial cells) is downregulated and N-cadherin (overexpressed in several malignancies) is upregulated. This switch alters intracellular signaling pathways, resulting in increased proliferation, migration, and survival.8–10 ADH-1 is a cyclic pentapeptide that disrupts N-cadherin interactions; it has been shown to inhibit cell growth and tumor progression both in vitro and in vivo.11, 12 Based on strong preclinical evidence supporting synergism of systemic ADH-1 and regionally infused melphalan 13, phase I and phase II clinical trials have been conducted.14, 15 Overall, combining the N-cadherin antagonist ADH-1 with melphalan (LPAM) ILI increased initial response rates but did not significantly alter time to progression at 15 months follow-up.15
The objective of this study was to explore the mechanism by which ADH-1 effects the tumor microenvironment leading to alterations in tumor growth and regional drug delivery. A better understanding of these effects would, in turn, help develop strategies to improve the magnitude and durability of anti-tumor responses initially observed in the phase I and II clinical trials (14, 15) investigating the safety and efficacy of systemic ADH-1 given prior to regional cytotoxic melphalan based therapy. We report data suggesting systemic ADH-1 has a dual function to both: (1) effect vascular permeability in the tumor microenvironment and (2) modulate tumor growth through activation of the AKT pathway.
Materials and methods
Tumor cell lines
The melanoma cell line DM443 was obtained courtesy of Dr. Hilliard Seigler (Duke University, Durham, NC). The A375 cell line was purchased from American Type Culture Collection. Cells were maintained as a monolayer in Isocove’s modified Dulbecco’s medium with 10% fetal bovine serum, 2mM glutamine, 1000IU/ml penicillin, and 100mg/mL streptomycin and grown at 37°C, 98% relative humidity, and 5% CO2.
Drugs for Xenograft Therapeutic Studies
Melphalan (LPAM) was purchased from Sigma-Aldrich (St. Louis, MO). A 0.2 mg/mL stock solution of melphalan was prepared in 0.9% sodium chloride using sonification for dissolution. A 4 mg/mL stock solution of temozolomide was prepared in PBS with 10% DMSO. Stock solutions of drugs were prepared immediately before surgery. The ILI infusate was prepared by further dilution of temozolomide stock solution with a 10% DMSO solution to achieve a final infusate concentration of 2,000 mg/kg in a volume of 22.5 mL. Likewise, the melphalan stock solution was further diluted with a 0.9% sodium chloride solution to achieve a final infusate concentration of 90 mg/kg in a volume of 22.5 mL. ADH-1, a pentapeptide that disrupts N-cadherin interactions was provided by Adherex Technologies, Inc. (Research Triangle Park, NC). ADH-1 was prepared in PBS, and 10 mL/kg body weight and was given via intraperitoneal injection (final dose, 100 mg/kg).
Xenograft Studies
Xenograft Studies were performed as previous reported (Supplemental Methods).13,16, 17
Growth Kinetics
Tumor growth was quantified as fold change in tumor volume from day of ILI. Growth rate (R), was determined from the slope of tumor growth curves during the exponential growth phase. For DM443 xenografts, this was calculated as the slope between days 0 and 30, and for A375, this was calculated to be between days 12 and 36. Assuming constant R, doubling time (N), was calculated using the formula: , where t1 and t2 represent the days and their representative fold change in tumor values q1 and q2.
In vitro Endothelial Cell Permeability Assay
Human umbilical vein endothelial cells (HUVECs, ATCC) were grown in F-12K base medium supplemented with 0.1 mg/mL heparin, 0.03 mg/mL endothelial cell growth supplement (ECGS) and 10% fetal bovine syndrome. HUVECs were grown to confluence on fibronectin coated transwell inserts (20ng/mL fibronectin per 6.5mm transwell insert with 0.4μm pore) in normal growth media. Diffusion of 40kDa fluorescein isothiocyanate (FITC)-conjugated dextran (Sigma) through the HUVEC monolayer was measured following treatment with ADH-1 (1mg/mL) or VEGF (10ng/mL) for 1 hour in the presence or absence of serum and growth factors (GF). Diffusion of the 40 kDa FITC-dextran (1mg/mL) across the HUVEC monolayers coating the transwell insert into the lower compartment was measured using a microplate reader 30 minutes following addition of the FITC-dextran to the transwell insert.
Real-time quantitative RT-PCR of N-cadherin gene transcripts
Total RNA was extracted from cells using the RNeasy™ kit (Qiagen, Valencia, CA) following the manufacturer’s protocol and including a genomic DNA elimination step. Using 800 ng of RNA, first strand cDNA synthesis was achieved using the AffinityScript QPCR cDNA synthesis kit (Stratagene, La Jolla, CA) following the manufacturer’s instructions. Using a Mx3005P PCR instrument (Stratagene, La Jolla, CA, USA), 100 ng of cDNA was amplified using the Brilliant II SYBR-Green qPCR Master Mix (Stratagene, La Jolla, CA) in a 25μL reaction volume. The mixture was heated at 95 °C for 10 min and 40 cycles of 95 °C (30 s), 55 °C (60 s) and 72 °C (30s) were run followed by 1 cycle of 95 °C (60 s), 55 °C (30 s) and 72 °C (30s). The following primers were used: 5′-CCTGGAACGCAGTGTACAGA-3′ and 5′-CTAACCCGTCGTTGCTGTTT-3′, (N-cadherin), and 5′-AAGAAGGTGGTGAAGCAGG-3′ and 5′-GTGTCGCTGTTGAAGTCAGA-3′ (GAPDH). The ΔCT values were obtained by normalizing N-cadherin expression to GAPDH expression, which was coamplified in a parallel reaction, to adjust for differences in both the amount of total RNA and different polymerase reaction efficiencies. Controls lacking RNA template were used to monitor for fluorescent contaminants and nonspecific amplification. HUVEC N-cadherin expression is reported as a function of the N-cadherin expression for DM366, a high N-cadherin expressing melanoma cell line (13).
Immunofluorescence and Immunohistochemistry
Rats were treated with ADH-1 (100 mg/kg IP) or saline 1 hour prior to ILI and then euthanized 24 hours after LPAM-, TMZ, or saline-ILI. Tumors were then fixed with formalin, paraffin-embedded, and stained using immunohistochemistry (IHC) as previously described.13, 18
Evans blue dye assay
Melanoma cells were injected as described above and grown until tumor diameter was approximately 2 cm3 at which time ILI was performed 1 hour after intraperitoneal injection of 100 mg/kg ADH-1. A 15 minute infusion of Evan’s Blue dye solution (50 mg/kg dissolved in normal saline and infused at 1.5 mL/min) was followed by a 2 minute saline wash-out (3.0 mL/min). Animals were then euthanized and tumors were excised and then incubated in formamide solution for 72 hours at 37°C. Absorbance of formamide containing Evan’s blue was measured at 595 nm and normalized to tumor volume.
Tumor Interstitial Fluid Pressure
Similar to permeability studies, melanoma cells were injected as described above and grown until tumor diameter was approximately 2 cm3, at which time ILI was performed 1 hour after intraperitoneal injection of 100 mg/kg ADH-1. Following induction of general anesthesia using isoflurane, tumor IFP measurements were made with a needle probe pressure monitor (Intra-Compartmental Pressure Monitor System, Stryker), fitted with an 18-gauge side-ported needle (Stryker) and connected to a syringe filled with 0.9% saline. The needle probe was inserted into the center of the tumor on the hind limb of the rat. IFP was recorded in mm Hg when the measurement stabilized. 3–5 rats were used for each treatment group.
Reverse Phase Protein Array Analysis
Proteins were isolated and homogenized from DM443 and A375 xenografts treated with systemic saline or ADH-1 as previously described.21 Reverse phase protein array (RPPA) analysis was performed by the MD Anderson Cancer Center Functional Proteomics Core Facility (see website below1 for detailed methodology and list of antibodies used including source and catalog number) 22, 23. Differences in relative protein loading were determined and adjusted for using the median protein expression for each sample across all measured proteins using data that had been normalized to the median value of each protein. Log2 values were converted to linear values, and differences in protein expression between saline- and ADH-1 treated tumors were determined.
Statistical analysis
Overall tumor response to chemotherapy was assessed by comparing fold change in tumor volume at 40 days from ILI. ANOVA was used to perform statistical comparisons. A two-tailed student’s t test was used to compare differences in tumor growth rates between treatment arms, permeability, and interstitial fluid pressure amongst xenografts. p values of <0.05 were considered significant.
Results
Systemic ADH-1 in absence of regional chemotherapy can both inhibit and augment of melanoma tumor growth
To explore for mechanisms underlying the variable tumor responses seen in clinical trials testing the combination of N-cadherin antagonism and regional chemotherapy, we first expanded on previous xenograft growth studies 13 to include the low N-cadherin expressing DM443 and high N-cadherin expressing A375 melanoma cell lines. In vitro, the DM443 cell line is relatively resistant to LPAM and TMZ, while the A375 cell line is more sensitive to both chemotherapeutic agents as compared to DM443.21 Mean fold change in tumor volumes, growth rates, and doubling times are summarized in Table 1. For DM443, systemic ADH-1 followed by saline infusion did not significantly decrease tumor growth (0.10 ± 0.04 d−1 vs. 0.20 ± 0.04 d−1, p=0.26). In contrast, A375 xenografts treated with systemic ADH-1 followed by arterial saline infusion demonstrated a dramatic and statistically significant, nearly 2-fold increase in growth rate as compared to those receiving both systemic and regional saline (0.53 ± 0.07 d−1 vs. 0.27 ± 0.02 d−1, p=0.018).
Table 1.
Tumor volume and growth kinetics summary.
| Xenograft | DM443 | A375 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Systemic | ILI | Fold Δ Tumor Vol. * | Growth Rate (d−1) | Doubling Time (d) | p† | p╪ | Fold Δ Tumor Vol.* | Growth Rate (d−1) | Doubling Time (d) | p† | p╪ |
| Saline | Saline | 11.0 ± 3.0 | 0.20 ± 0.04 | 11.4 ± 3.1 | Ref | -- | 9.0 ± 1.7 | 0.27 ± 0.02 | 12.6 ± 2.4 | Ref | -- |
| ADH-1 | Saline | 9.2 ± 1.9 | 0.10 ± 0.04 | 12.5 ± 2.5 | 0.26 | -- | 15.1 ± 2.1 | 0.53 ± 0.07 | 10.2 ± 2.8 | 0.018 | -- |
| Saline | LPAM | 6.5 ± 1.6 | 0.12 ± 0.01 | 14.8 ± 3.6 | 0.10 | Ref | 4.0 ± 1.6 | 0.11 ± 0.05 | 19.9 ± 8.1 | <0.001 | Ref |
| ADH-1 | LPAM | 3.7 ± 0.5 | 0.03 ± 0.01 | 21.3 ± 2.7 | <0.001 | <0.001 | 2.4 ± 0.9 | 7.1 e -4 ± 0.02 | 32.5 ± 13.0 | 0.005 | 0.27 |
| Saline | TMZ | 7.6 ± 0.7 | 0.12 ± 0.02 | 13.7 ± 1.2 | 0.016 | Ref | 3.5 ± 1.0 | 0.11 ± 0.03 | 21.9 ± 6.5 | 0.023 | Ref |
| ADH-1 | TMZ | 4.4 ± 0.5 | 0.08 ± 0.01 | 18.8 ± 2.0 | 0.009 | 0.012 | 13.4 ± 3.7 | 0.45 ± 0.12 | 10.7 ± 2.9 | 0.16 | 0.03 |
Calculated 40 days after ILI.
Two-tailed T-test comparing mean growth rate of treatment group to mean growth rate of control animals treated with systemic saline followed by regional saline infusion.
Two-tailed T-test comparing mean growth rate after combination treatment of ADH-1 and ILI with LPAM or TMZ to animals receiving ILI with LPAM or TMZ alone.
ADH-1 augmentation of melanoma tumor growth is overcome through its ability to make regionally infused melphalan more effective
As compared to control tumors exposed to both systemic and regional saline, LPAM-ILI after systemic administration of saline did not significantly alter the mean tumor growth of DM443 xenografts (0.12 ± 0.01 d−1 vs. 0.20 ± 0.04 d−1, p=0.10) but was associated with a significant decrease in mean tumor growth rate for A375 (0.11 ± 0.05 d−1 vs. 0.27 ± 0.02 d−1, p<0.001). Combination treatment with systemic ADH-1 followed by regional LPAM infusion led to a further improvement in tumor response for both xenografts. Using the DM443 xenografts, mean tumor growth rates decreased by 75% (0.03 ± 0.01 d-1 vs. 0.12 ± 0.01d-1, p<0.001) after regional LPAM treatment. (Figure 1a). Although ADH-1 dramatically augmented tumor growth of A375 xenografts in the absence of LPAM, treatment using regional LPAM concurrently with ADH-1 counteracted this effect. Combination therapy using systemic ADH-1 and regional LPAM decreased the growth rate of A375 tumors by 99% as compared to animals treated with systemic saline followed by regional saline (7.1 e−4 ± 0.02 d−1 vs. 0.27 ± 0.02 d−1, p=0.005). Absolute growth rates were also decreased 99% in the combination ADH-1 and LPAM-ILI arm as compared to LPAM-ILI alone (7.1 e−4 ± 0.02 d−1 vs. 0.11 ± 0.05 d−1) but this difference did not reach statistical significance (p=0.27). (Figure 1b) Of note, 80% (4/5) of animals bearing A375 xenografts demonstrated a complete tumor response after combination therapy with ADH-1 and LPAM-ILI as compared to only 33% (2/6) treated with LPAM-ILI alone.
Figure 1. DM443 and A375 xenografts treated with systemic ADH-1 followed by isolated limb infusion with melphalan or temozolomide.

(a.) Growth curves of DM443 xenografts treated with systemic ADH-1 or saline followed by regional infusion of LPAM or saline. (b.) Growth curves of A375 xenografts treated with systemic ADH-1 or saline followed by regional infusion of LPAM or saline. (c.) Growth curves of DM443 xenografts treated with systemic ADH-1 or saline followed by regional infusion of TMZ or saline. (d.) Growth curves of A375 xenografts treated with systemic ADH-1 or saline followed by regional infusion of TMZ or saline. *ANOVA of mean fold change in tumor volume at 40 days
ADH-1 mediated augmentation of melanoma tumor growth not altered by regionally infused temozolomide
ILI with temozolomide (TMZ) is currently being tested for safety in a phase I/II clinical trial for patients who have progressed after standard ILI with LPAM. Thus, we also tested whether ADH-1 would augment regionally infused TMZ for both DM443 and A375 xenografts in the same series of experiments. For DM443 xenografts, regional infusion of temozolomide after systemic administration of saline injection significantly decreased tumor growth rate 40% (0.12 ± 0.02 d−1 vs. 0.20 ± 0.04 d−1, p=0.016) as compared to xenografts treated concurrently with systemic and regional saline controls. (Figure 1c). When used together the combination of systemic ADH-1 and regional TMZ led to a 33% reduction in DM443 tumor growth over ILI with TMZ alone (0.08 ± 0.01 d−1 vs. 0.12 ± 0.02 d−1, p=0.012) and a 40% decrease in tumor growth compared to the systemic and regionally treated saline control animals (0.08 ± 0.01 d−1 vs. 0.12 ± 0.01 d−1, p=0.009). For A375 tumors, ILI with TMZ demonstrated similar efficacy as compared to ILI with LPAM (56.4% vs. 48.6% reduction in tumor growth as compared to the control arm) (Table 1). However, in the presence of ADH-1, TMZ-ILI had little efficacy and could not overcome the ADH-1 mediated growth augmentation seen in the A375 xenografts. The combination therapy of systemic ADH-1 plus TMZ-ILI had a tumor growth enhancing effect closer to that seen with systemic ADH-1 alone as compared to the xenografts treated with saline as the systemic and regional control arm. (Figure 1d).
Tumor growth augmentation mediated by N-cadherin antagonism is associated with increased AKT phosphorylation
Multiple studies have associated increased activity of phosphatidylinositol-3 kinase (PI3K)-AKT pathway with melanoma progression and metastasis.22, 24–26 PI3K activation via receptor tyrosine kinases such as the Epidermial Growth Factor Receptor (EGFR), activates AKT, a serine-threonine kinase which regulates the activity of a number of cellular effectors, including the mammalian target of rapamycin (mTOR) protein, leading to increased tumor cell proliferation, invasion, and survival. 27–29. Previously, we have shown differential phosphorylation of AKT after systemic ADH-1 and regional LPAM-ILI.13 Moreover, N-cadherin expression has been associated with increased AKT activity in other tumor models.30 Based on these previous reports, we hypothesized that blocking N-cadherin interactions would result in differential activation of the AKT pathway in A375 and DM443 xenografts. Exponentially growing DM443 and A375 cells were treated with 0–1 mg/mL ADH-1 and analyzed for AKT-phosphorylation and PTEN expression. Treatment with ADH-1 was associated with increased AKT phosphorylation in PTEN-expressing A375 cells did not affect AKT activation in the PTEN-null DM443 cells (Figure 2a). Similarly, tumor samples from DM443 and A375 xenografts isolated after treatment with systemic ADH-1 or saline were examined using western blot analyses to investigate whether ADH-1 treatment alters AKT activation. In A375, but not DM443 xenografts, ADH-1 treatment increased phosphorylation of AKT at serine 473. N-cadherin expression seemed to be slightly diminished in both xenografts after treatment with ADH-1. (Figure 2b).
Figure 2. Systemic ADH-1 increases AKT phosphorylation in A375 but not DM443 xenografts.

(a.) Exponentially growning DM443 and A375 melanoma cells were grown in culture, serum-starved overnight and then treated with 0–1 mg/mL ADH-1 for 1 hour. ADH-1 increased activation of AKT in A375 but not DM443 cells. (b.) DM443 and A375 xenografts were grown in hind limbs of nude rats. Once tumor diameter reached ~ 1 cm, animals were treated with systemic ADH-1 followed by saline infusion as outline in Figure 1d. After receiving their final dose of ADH-1, animals were sacrificed and tumors were flash frozen. Whole tumor lysates were then homogenized and developed by SDS-Page. Consistent with the in vivo results, western blotting demonstrated A375 xenografts treated with systemic ADH-1 to have increased phosphorylation of AKT at the serine 473 position. Blots are representative of duplicate experiments.
Reverse Phase Protein Analysis of the PI3K-AKT-mTOR pathway
In order to expand our analysis of the effects of ADH-1 treatment on signaling pathways, we performed reverse phase protein analysis (RPPA) on the A375 and DM443 xenografts. For each xenograft, the effects of ADH-1 were determined by measuring the differences in protein expression levels following ADH-1 treatment for one hour as compared to those seen with saline treatment for the same duration. We first analyzed the results for additional members of the PI3K-AKT pathway (Figure 3a). ADH-1 treatment in the DM443 resulted in decreased expression of multiple phospho-proteins in the PI3K-AKT pathway, including AKT, GSK3α/β, FOXO, P70S6K, mTOR, and 4EBP1. These results are consistent with significant inhibition of activity of the PI3K-AKT pathway. In contrast, treatment with ADH-1 resulted in increased expression of several of the phosphoproteins as compared to the control treatment in the A375. Interestingly, some of the total protein levels of the pathway effectors (i.e. P70S6K) also demonstrated increased expression with ADH-1 treatment when compared to the control treatment in the A375. The A375 xenografts also showed decreased expression of the lipid phosphatase PTEN, a negative regulator of AKT activity, with ADH-1 treatment, which was not observed in the DM443.
Figure 3. Comparative effects of ADH-1 treatment on signaling pathways in the A375 and DM443.

RPPA analysis was performed on xenografts of the A375 (Black bars) and the DM443 (Gray bars) harvested one hour after treatment with saline or ADH-1 as outlined in Figure 1. The effects of ADH-1 treatment were determined by subtracting the exponential value of the relative expression for each of the indicated proteins following saline treatment from the observed expression following ADH-1 treatment. Proteins with a relative change of >0 were increased with ADH-1 treatment; <0 indicates inhibition with ADH-1 treatment. (a.), expression of total- and phospho-proteins in the PI3K-AKT pathway. (b.) expression of total and phospho-proteins in other pro-survival kinase signaling pathways.
In order to determine if the increased expression of activation-specific markers in the PI3K-AKT pathway reflected a general increase in proliferative signaling in the A375, we also examined activation-specific markers in several other pathways (Figure 3b). ADH-1 treatment inhibited the expression of activation-specific markers in multiple pro-survival pathways in the A375 xenograft to a similar or greater extent than what was observed in the DM443 xenograft, including phosphorylated EGFR, STAT3, STAT5, MEK1/2, and p65. In turn, available total protein levels for the corresponding proteins did not show significant inhibition, or notable differences when comparing the two xenografts.
Blocking N-cadherin interactions increases endothelial cell permeability
Previously, we have shown systemic ADH-1, increases LPAM-DNA adduct formation after LPAM-ILI 13, suggesting that ADH-1 was increasing drug delivery through alterations in the tumor microenvironment. Along with VE-cadherin, N-cadherin is coexpressed on endothelial cells and has been shown to be critical for normal embryonic angiogenesis and for controlling vascular permeability. 31–34 Based on this data, we measured permeability of the N-cadherin expressing HUVEC endothelial cells after treatment with either saline, ADH-1 or VEGF (positive control). Pretreatment of HUVEC cells with ADH-1 resulted in an approximate two-fold increase in dextran permeability as compared to saline controls (Figure 4).
Figure 4. Disrupting N-cadherin interactions increases Endothelial Cell Permeability.
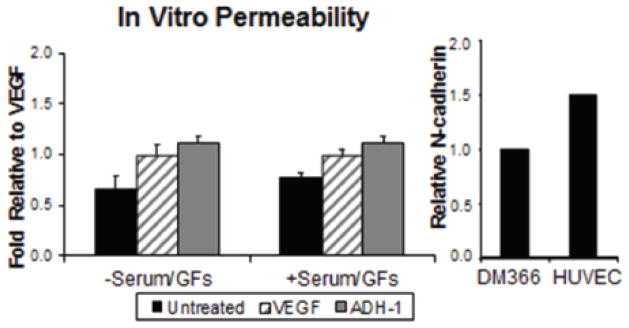
N-cadherin expressing HUVEC endothelial cells were grown to confluence in transwell plates and tested for changes in permeability in response to ADH-1 or VEGF. In the presence and absence of serum and GF, pretreatment of endothelial cells (HUVEC) with ADH-1 nearly doubled their permeability to a 40kDa FITC-conjugated dextran compared to saline controls. This increase in permeability in response to ADH-1 even exceeded the effects of VEGF, used as a positive control for changes in endothelial cell permeability. N-cadherin expression, normalized to GAPDH, in HUVEC cells is shown with respect to N-cadherin expression in the melanoma cell line DM366, a high N-cadherin expressing melanoma cell line.
Targeting N-cadherin in Vivo transiently increases vascular permeability to protein bound molecules without effecting interstitial fluid pressure
We investigated whether increased delivery of melphalan seen after systemic ADH-1 treatment resulted from alterations in vascular permeability to protein bound molecules. Using immunofluorescent techniques, we first confirmed N-cadherin to be expressed in the endothelium of DM443 and A375 xenografts (Supplemental Figure 2a–b). Next, we performed ILI on rats bearing DM443 and A375 xenografts using Evan’s blue dye as the infusate 1 hour after treatment with systemic saline or ADH-1. Spectrophotometric analysis of extracted Evan’s blue dye demonstrated a 28.4% and 32.8% increase in extravasation into DM443 and A375 tumors respectively after treatment with ADH-1 (Figure 5a, b).
Figure 5. ADH-1 increases Evan’s blue dye extravasation but not interstitial fluid pressure in melanoma xenografts.

(a.) Immunofluorescent analysis was used to determine the extent N-cadherin expression on A375 and DM443 xenografts. For both xenografts, N-cadherin (Green Staining) was noted to be readily present on tumor cells as well as tumor endothelium (white arrow).
(b.) One hour after IP injection 100 mg/kg of ADH-1, isolated limb infusion was performed on tumor bearing rats using Evan’s blue dye as the infusate (3–5 rats per treatment group). After infusion, animals were sacrificed and tumors were excised and incubated in formamide for 72 hours at 37°C to extract the Evan’s blue dye into solution. The absorbance was of Evan’s blue dye was then measured in the formamide at 595 and normalized to tumor volume. Error bars represent the SEM of 4–6 rats. Dye extravasation increased 28.4% from saline control in DM443 (p=0.0001, t-test) and 32.4% in A375 xenografts (p=0.0001, t-test) after treatment with ADH-1. (c.) One hour after IP injection 100 mg/kg of ADH-1 or saline control, tumor interstitial fluid pressure was measured in DM443 and A375 rat xenografts (3–5 rats per treatment group). Treatment with ADH-1 did not significantly alter tumor interstitial fluid pressure for either xenograft model.
Alterations in tumor vascular permeability have been associated with changes in tumor interstitial fluid pressure, which have been demonstrated to effect optimal drug delivery to tumor cells.35 Thus, we measured tumor interstitial fluid pressure one hour after administration of ADH-1 for both DM443 and A375 xenografts. One hour after administration of ADH-1, no significant differences were found in tumor interstitial fluid pressure (Figure 5c,d).
ADH-1 increases drug delivery of regionally infused melphalan but not temozolomide
We have previously reported that systemic ADH-1 treatment prior to LPAM-ILI can augment delivery of regionally LPAM to tumor tissues. 13 Therefore, we tested whether ADH-1 could also enhance delivery of regionally-infused TMZ, which is currently under investigation as an alternative regional chemotherapy agent for treatment of advanced-extremity melanoma. Rats bearing A375 or DM443 xenografts were systemically treated with ADH-1 or saline control and then infused with regional melphalan or TMZ. Consistent with our previous reports, rats treated with ADH-1 prior to ILI had demonstrated an approximate 12- and 16 fold increase in LPAM-DNA adducts (surrogate for LPAM delivery) in DM443 and A375 xenografts respectively (Figure 6a, b). However, immunohistochemical staining of p-H2A.x (surrogate for TMZ delivery) failed to demonstrate any significant changes in the levels of DNA damage in xenografts treated with ADH-1 prior to TMZ-ILI. (Figure 6c, d).
Figure 6. ADH-1 improves delivery of LPAM but not TMZ.

Using our animal model of ILI, rats were pretreated with ADH-1 1 hour prior to infusion of LPAM or TMZ. LPAM and TMZ drug delivery after ILI was assessed using IHC staining of LPAM-DNA adducts (MP5 antibody) and DNA-damage (p-H2A.x antibody) as surrogates for LPAM and TMZ delivery respectively. (a.) Qualitatively, ADH-1 increased LPAM-DNA staining for both xenografts. (b.) LPAM-DNA adduct staining was then quantified digitally as the ratio of brown to total pixels. As compared to saline controls, pretreatment with ADH-1 resulted in an approximate 12- and 16- fold increase in LPAM-DNA adduct formation for both xenografts. (c.) In contrast, pretreatment of ADH-1 prior to TMZ-ILI revealed no significant changes in the amount of DNA damage. (d.) Quantification of IHC staining for p-H2A.x suggest a statistically non-significant decrease in TMZ delivery after ADH-1 treatment.
Discussion
Here, we report that targeting N-cadherin interactions can independently produce dichotomous effects on melanoma tumor growth in vivo (Table 2). When combined with a high dose regional LPAM infusion, the contrasting effects of N-cadherin antagonism on tumor growth were abrogated likely by improved drug delivery to the tumor cells resulting from an increase in vascular permeability. However, with the A375 xenograft, ADH-1 not only augmented tumor growth but failed to increase the cytotoxic effects of TMZ. The net effect led to an ILI-TMZ being more effective than an ILI-TMZ performed in combination with ADH-1. Taken together, these studies and previous work in our laboratory suggest two separate, independent effects of N-cadherin antagonism in modulating regional chemotherapy: (1) ADH-1 may affect tumor growth and sensitivity to some chemotherapy agents by altering AKT activation and (2) ADH-1 can improve the drug delivery of certain cytotoxic agents like LPAM by increasing vascular permeability.
Table 2.
Summary of ADH-1 Drug Effects in a Regional Therapy Model of In-transit Melanoma.
| DM443 | A375 | |
|---|---|---|
| Xenograft growth* | No Change | Increased |
| LPAM-ILI† | Decreased | Decreased |
| TMZ-ILI† | Decreased | Increased |
| AKT Activation | No change | Increased |
| Vascular Permeability | Increased | Increased |
| LPAM Delivery | Increased | Increased |
| TMZ Delivery | No change | No change |
Xenograft growth in rats treated with systemic ADH-1 followed by saline-ILI as compared to control rats treated systemic saline followed by saline-ILI.
Mean growth rate of animals treated with combination therapy (ADH-1 + LPAM-ILI or TMZ-ILI) as compared to the mean growth rate of control animals treated with systemic saline followed by saline-ILI.
We have previously shown that blocking N-cadherin interactions with the pentapeptide ADH-1 prior to ILI with LPAM results in increased staining of LPAM-DNA adducts in tumors analyzed by immunohistochemistry.13 Although exhibiting a dichotomous effect on tumor growth, the N-cadherin targeted agent ADH-1 increased vascular permeability without altering interstitial tumor pressure in both xenografts one hour after a single injection. How might the increased vascular permeability resulting from disrupting N-cadherin binding interactions improve the efficacy of LPAM but not TMZ? Tumor angiogenesis is notoriously rapid and abnormal, resulting in tumor vasculature that is saccular, tortuous and leaky resulting in abnormal pressure gradients as well as inefficient drug delivery 36. The enhanced permeability and retention (EPR) effect, which results from this abnormal vasculature, states that drug-transporting macromolecules preferentially extravasate and accumulate into tumor tissues as opposed to normal tissues where intact tight-junctions of endothelial cells prevent leakage of such drug carriers 37. Furthermore, the increase in tumor vascular permeability occurred in the absence of tumor interstitial fluid pressure changes. Thus, if the increased drug delivery seen in our study is indeed manifested by the EPR effect, it would be not be hindered from an increase in IFP that is often seen with increased tumor vascular permeability.38 Sixty to ninety percent of all melphalan is bound to plasma proteins.39, 40 TMZ, on the other hand, has lower affinity of binding to plasma proteins, with only 12% to 16% bound to plasma proteins.41 Thus, TMZ being less protein bound, may be less efficient as compared to LPAM at accumulating in the interstitium after the approximate 30% increase in vascular permeability seen in this study. This theory is supported by our immunohistochemical studies which suggested ADH-1 to increase LPAM but not TMZ delivery to tumor cells.
Another potential mechanism may be related to the distinct mechanisms by which LPAM and TMZ enter cells and manifest their cytotoxic effects. LPAM is actively transported into cells 42, 43 whereas TMZ is lipophilic and not known to undergo any specialized membrane transport processes.44 Therefore, it is conceivable that ADH-1 mediated alterations in vascular permeability may allow greater accumulation of drugs into the interstitium, providing active transporters more time to uptake LPAM while not affecting the passive diffusion through which TMZ or its active metabolite MTIC enter tumor cells.
The ability of ADH-1 to both inhibit and augment melanoma tumor growth in vivo appears to be independent of its effect on the tumor vasculature, suggesting N-cadherin to have heterogeneous roles in regulating melanoma tumor signaling potentially dependent on the magnitude of other molecular and signaling alterations within the tumor. In order to provide insight into the differential effects of ADH-1, we performed proteomic analysis of ADH-1 treated xenografts by western blotting and RPPA. This analysis demonstrated that despite the increased growth observed, treatment of the A375 xenografts with ADH-1 was associated with decreased expression of activation-specific markers of multiple signaling pathways, including the JAK-STAT, RAS-RAF-MEK-ERK and NFκB pathways (Figure 6B). In contrast, while treatment with ADH-1 appeared to also inhibit the PI3K-AKT pathway in the sensitive DM443 xenograft, we observed increased expression of multiple activation- (phospho-) specific and total protein levels of critical effectors of this pathway in the resistant A375 xenograft. ADH-1 treatment also resulted in decreased expression of PTEN in the A375 xenograft, which has been shown to correlate with increased activation/phosphorylation of multiple kinases in this pathway, particularly AKT, in melanoma previously.22 It should be noted, however, that the alterations seen in these signaling pathways after ADH-1 treatment may be purely associative and may not necessarily be a direct result ADH-1 binding to N-cadherin.
The “double-edged sword” effect reported here after N-cadherin antagonism has been seen in other settings. For instance, angiogenesis inhibitors in preclinical studies have been shown to eventually increase local invasion and metastasis by selecting for a tumor clonal population with hypoxia tolerance and promoting rescue angiogenesis.45, 46 More recently, PLX4032 (vemurafenib), a novel inhibitor of the activating V600E BRAF mutation seen in approximately 60% of melanomas, has been shown to be markedly effective in a phase III trial of metastatic melanoma. For patients harboring the V600E BRAF mutation, 48% treated with PLX4032 responded as compared to only 5% of patients treated with the standard chemotherapeutic agent darcabazine.47 Despite high responses to PLX4032, 21% of patients eventually developed cutaneous squamous cell carcinomas after initiating treatment. Further research suggests that BRAF V600E mutant inhibitors such as PLX4032 paradoxically activate downstream signaling (i.e. MEK-ERK) in both transformed and non-transformed cells lacking the BRAF mutation, and may be responsible for the development of cutaneous squamous cell carcinomas.48–50
Given that ADH-1has already been studied in combination with LPAM-ILI in phase I and II clinical trials, this study reports findings of significant clinical relevance. With these trials, no evidence of rapid growth or disease progression was observed but patients only received 2 doses of ADH-1 as part of these studies. Here, we report the response of ADH-1 amongst different melanoma xenografts is variable, highlighting the need for further investigation to delineate the biological underpinnings for responders and non-responders. Although ADH-1 may significantly improve delivery of small cytotoxic agents, its disruption of N-cadherin binding, may have variable effects on tumor growth based upon the alterations in association with AKT activation. As future trials may be planned targeting N-cadherin, it will be important to understand factors that predict how tumor cells will respond to this targeted intervention. In addition, clinicians will have to be vigilant for potential detrimental tumor augmenting effects that may occur which are not always identified in preclinical studies that focus on small numbers of tumors.
Supplementary Material
Acknowledgments
Financial support for this manuscript was from the Duke University Melanoma Research Fund and a VA Merit Review Grant (Dr Tyler). Drs. Padussis, Lidsky, and Turley received salary support from NIH grant 5T32CA093245. Dr. Turley also receivied financial support from the NIH loan repayment program. Dr. Dewhirst received financial support from NIH grant CA40355 and Dr. Fontanella received support from the Department of Defense Grant BC083195.
Footnotes
Conflicts of Interest: Dr. Tyler received grant support from Adherex Technologies and is also a co-inventor on a patent entitled “Cancer treatment methods using cadherin antagonists in combination with anticancer agents”. Dr. Tyler has material transfer agreements with Bayer, Schering, and Genta pharmaceuticals. Adherex Technologies funded the phase I and II clinical trials of systemic ADH-1 and regional melphalan. Bayer provided drug only (sorafenib) for a phase I trial of systemic sorafenib and regional melphalan. Schering is funding a phase I trial of intra-arterial temozolomide for regionally advanced melanoma.
References
- 1.Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006;355(1):51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–7. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 4.Balch CM, Houghton AN, Peterson LJ. Cutaneous melanoma. In: Devita VT, Hellman S, Rosenberg SA, editors. Cancer: principles and practice of oncology. Philadelphia: JB Lippincott; 1993. p. 1612. [Google Scholar]
- 5.Pawlik TM, Ross MI, Johnson MM, et al. Predictors and natural history of in-transit melanoma after sentinel lymphadenectomy. Ann Surg Oncol. 2005;12(8):587–96. doi: 10.1245/ASO.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 6.Turley RS, Raymond AK, Tyler DS. Regional treatment strategies for in-transit melanoma metastasis. Surg Oncol Clin N Am. 2011;20(1):79–103. doi: 10.1016/j.soc.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beasley GM, Caudle A, Petersen RP, et al. A multi-institutional experience of isolated limb infusion: defining response and toxicity in the US. J Am Coll Surg. 2009;208(5):706–15. doi: 10.1016/j.jamcollsurg.2008.12.019. discussion 715–7. [DOI] [PubMed] [Google Scholar]
- 8.Padsis J, Turley R, Tyler D. Pharmacotherapy of regional melanoma therapy. Expert Opin Pharmacother. 2010;11(1):79–93. doi: 10.1517/14656560903428003. [DOI] [PubMed] [Google Scholar]
- 9.Hsu MY, Wheelock MJ, Johnson KR, et al. Shifts in cadherin profiles between human normal melanocytes and melanomas. J Investig Dermatol Symp Proc. 1996;1(2):188–94. [PubMed] [Google Scholar]
- 10.Hsu MY, Meier FE, Nesbit M, et al. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000;156(5):1515–25. doi: 10.1016/S0002-9440(10)65023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perotti A, Sessa C, Mancuso A, et al. Clinical and pharmacological phase I evaluation of Exherin (ADH-1), a selective anti-N-cadherin peptide in patients with N-cadherin-expressing solid tumours. Ann Oncol. 2009;20(4):741–5. doi: 10.1093/annonc/mdn695. [DOI] [PubMed] [Google Scholar]
- 12.Mariotti A, Perotti A, Sessa C, et al. N-cadherin as a therapeutic target in cancer. Expert Opin Investig Drugs. 2007;16(4):451–65. doi: 10.1517/13543784.16.4.451. [DOI] [PubMed] [Google Scholar]
- 13.Augustine CK, Yoshimoto Y, Gupta M, et al. Targeting N-cadherin enhances antitumor activity of cytotoxic therapies in melanoma treatment. Cancer Res. 2008;68(10):3777–84. doi: 10.1158/0008-5472.CAN-07-5949. [DOI] [PubMed] [Google Scholar]
- 14.Beasley GM, McMahon N, Sanders G, et al. A phase 1 study of systemic ADH-1 in combination with melphalan via isolated limb infusion in patients with locally advanced in-transit malignant melanoma. Cancer. 2009;115(20):4766–74. doi: 10.1002/cncr.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beasley GM, Riboh JC, Augustine CK, et al. Prospective multicenter phase II trial of systemic ADH-1 in combination with melphalan via isolated limb infusion in patients with advanced extremity melanoma. J Clin Oncol. 2011;29(9):1210–5. doi: 10.1200/JCO.2010.32.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko SH, Ueno T, Yoshimoto Y, et al. Optimizing a novel regional chemotherapeutic agent against melanoma: hyperthermia-induced enhancement of temozolomide cytotoxicity. Clin Cancer Res. 2006;12(1):289–97. doi: 10.1158/1078-0432.CCR-05-0210. [DOI] [PubMed] [Google Scholar]
- 17.Grubbs EG, Ueno T, Abdel-Wahab O, et al. Modulation of resistance to regional chemotherapy in the extremity melanoma model. Surgery. 2004;136(2):210–8. doi: 10.1016/j.surg.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 18.Attis MG, Burchette JL, Selim MA, et al. Differential expression of N-cadherin distinguishes a subset of metastasizing desmoplastic melanomas. Hum Pathol. 2006;37(7):899–905. doi: 10.1016/j.humpath.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Tilby MJ, Styles JM, Dean CJ. Immunological detection of DNA damage caused by melphalan using monoclonal antibodies. Cancer Res. 1987;47(6):1542–6. [PubMed] [Google Scholar]
- 20.Lo HW, Antoun GR, Ali-Osman F. The human glutathione S-transferase P1 protein is phosphorylated and its metabolic function enhanced by the Ser/Thr protein kinases, cAMP-dependent protein kinase and protein kinase C, in glioblastoma cells. Cancer research. 2004;64(24):9131–8. doi: 10.1158/0008-5472.CAN-04-0283. [DOI] [PubMed] [Google Scholar]
- 21.Augustine CK, Toshimitsu H, Jung SH, et al. Sorafenib, a multikinase inhibitor, enhances the response of melanoma to regional chemotherapy. Mol Cancer Ther. 2010;9(7):2090–101. doi: 10.1158/1535-7163.MCT-10-0073. [DOI] [PubMed] [Google Scholar]
- 22.Davies MA, Stemke-Hale K, Lin E, et al. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15(24):7538–7546. doi: 10.1158/1078-0432.CCR-09-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gopal YN, Deng W, Woodman SE, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer research. 2010;70(21):8736–47. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23(7):1473–82. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 25.Dhawan P, Singh AB, Ellis DL, et al. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-kappaB and tumor progression. Cancer research. 2002;62(24):7335–42. [PubMed] [Google Scholar]
- 26.Slipicevic A, Holm R, Nguyen MT, et al. Expression of activated Akt and PTEN in malignant melanomas: relationship with clinical outcome. American journal of clinical pathology. 2005;124(4):528–36. doi: 10.1309/YT58WWMTA6YR1PRV. [DOI] [PubMed] [Google Scholar]
- 27.Ruth MC, Xu Y, Maxwell IH, et al. RhoC promotes human melanoma invasion in a PI3K/Akt-dependent pathway. J Invest Dermatol. 2006;126(4):862–8. doi: 10.1038/sj.jid.5700211. [DOI] [PubMed] [Google Scholar]
- 28.Sinnberg T, Lasithiotakis K, Niessner H, et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J Invest Dermatol. 2009;129(6):1500–15. doi: 10.1038/jid.2008.379. [DOI] [PubMed] [Google Scholar]
- 29.Hardt M, Chantaravisoot N, Tamanoi F. Activating mutations of TOR (target of rapamycin) Genes to cells: devoted to molecular & cellular mechanisms. 2011;16(2):141–51. doi: 10.1111/j.1365-2443.2010.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rieger-Christ KM, Lee P, Zagha R, et al. Novel expression of N-cadherin elicits in vitro bladder cell invasion via the Akt signaling pathway. Oncogene. 2004;23(27):4745–53. doi: 10.1038/sj.onc.1207629. [DOI] [PubMed] [Google Scholar]
- 31.Luo Y, Radice GL. N-cadherin acts upstream of VE-cadherin in controlling vascular morphogenesis. J Cell Biol. 2005;169(1):29–34. doi: 10.1083/jcb.200411127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabascio C, Muratori E, Mancuso P, et al. Assessing tumor angiogenesis: increased circulating VE-cadherin RNA in patients with cancer indicates viability of circulating endothelial cells. Cancer Res. 2004;64(12):4373–7. doi: 10.1158/0008-5472.CAN-04-0265. [DOI] [PubMed] [Google Scholar]
- 33.Yamaoka-Tojo M, Tojo T, Kim HW, et al. IQGAP1 mediates VE-cadherin-based cell-cell contacts and VEGF signaling at adherence junctions linked to angiogenesis. Arterioscler Thromb Vasc Biol. 2006;26(9):1991–7. doi: 10.1161/01.ATV.0000231524.14873.e7. [DOI] [PubMed] [Google Scholar]
- 34.Hatanaka K, Simons M, Murakami M. Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am J Physiol Heart Circ Physiol. 2011;300(1):H162–72. doi: 10.1152/ajpheart.00650.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tong RT, Boucher Y, Kozin SV, et al. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64(11):3731–6. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 36.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 37.Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjugate chemistry. 2010;21(5):797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 38.Torosean S, Flynn B, Axelsson J, et al. Nanoparticle uptake in tumors is mediated by the interplay of vascular and collagen density with interstitial pressure. Nanomedicine: nanotechnology, biology, and medicine. 2012 doi: 10.1016/j.nano.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greig NH, Sweeney DJ, Rapoport SI. Melphalan concentration dependent plasma protein binding in healthy humans and rats. Eur J Clin Pharmacol. 1987;32(2):179–85. doi: 10.1007/BF00542192. [DOI] [PubMed] [Google Scholar]
- 40.Melphalan [Clinical Pharmacology web site] 2009 Available at: www.clinicalpharmacology.com.
- 41.Darkes MJM, Plosker GL, Jarvis B. Temozolomide: A Review of its Use in the Treatment of Malignant Gliomas, Malignant Melanoma, and Other Advanced Cancers. Am J Cancer. 2002;1(1):55–80. [Google Scholar]
- 42.Goldenberg GJ, Lam HY, Begleiter A. Active carrier-mediated transport of melphalan by two separate amino acid transport systems in LPC-1 plasmacytoma cells in vitro. The Journal of biological chemistry. 1979;254(4):1057–64. [PubMed] [Google Scholar]
- 43.Begleiter A, Lam HY, Grover J, et al. Evidence for active transport of melphalan by two amino acid carriers in L5178Y lymphoblasts in vitro. Cancer research. 1979;39(2 Pt 1):353–9. [PubMed] [Google Scholar]
- 44.Ma J, Pulfer S, Li S, et al. Pharmacodynamic-mediated reduction of temozolomide tumor concentrations by the angiogenesis inhibitor TNP-470. Cancer research. 2001;61(14):5491–8. [PubMed] [Google Scholar]
- 45.Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer cell. 2009;15(3):220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ebos JM, Lee CR, Cruz-Munoz W, et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer cell. 2009;15(3):232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chapman PB, Hauschild A, Robert C, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. New England Journal of Medicine. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.