

Abstract
Perivascular epithelioid cell neoplasms (PEComa) are a family of rare mesenchymal tumors with hybrid myo-melanocytic differentiation. Although most PEComas harbor loss of function TSC1/TSC2 mutations, a small subset were reported to carry TFE3 gene rearrangements. As no comprehensive genomic study has addressed the molecular classification of PEComa, we sought to investigate by multiple methodologies the incidence and spectrum of genetic abnormalities and their potential genotype-phenotype correlations in a large group of 38 PEComas. The tumors were located in soft tissue (11 cases) and visceral sites (27) including uterus, kidney, liver, lung and urinary bladder. Combined RNA sequencing and Fluorescence In Situ Hybridization (FISH) analysis identified 9 (23%) TFE3 gene rearranged tumors, with 3 cases showing a SFPQ/PSF-TFE3 fusion and one case a novel DVL2-TFE3 gene fusion. The TFE3-positive lesions showed a distinctive nested/alveolar morphology and were equally distributed between soft tissue and visceral sites. Additionally, novel RAD51B gene rearrangements were identified in 3 (8%) uterine PEComas, which showed a complex fusion pattern and were fused to RRAGB/OPHN1 genes in two cases. Other non-recurrent gene fusions, HTR4-ST3GAL1 and RASSF1-PDZRN3, were identified in 2 cases. Targeted exome sequencing using the IMPACT assay was used to address if the presence of gene fusions are mutually exclusive from TSC gene abnormalities. TSC2 mutations were identified in 80% of the TFE3 fusion-negative cases tested. Co-existent TP53 mutations were identified in 63% of the TSC2 mutated PEComas. Our results showed that TFE3-rearranged PEComas lacked co-existing TSC2 mutations, indicating alternative pathways of tumorigenesis. In summary, this comprehensive genetic analysis significantly expands our understanding of molecular alterations in PEComas and brings forth the genetic heterogeneity of these tumors.
Keywords: PEComa, TFE3, TSC2, RAD51B
INTRODUCTION
Perivascular epithelioid cell (PEC) neoplasms are rare mesenchymal tumors composed of epithelioid and pleomorphic cells with perivascular distribution, that usually express melanocytic and smooth-muscle markers. Apart from tumors arising in soft tissue locations, the PEComa family includes renal angiomyolipoma, clear cell ‘sugar’ tumor of the lung and lymphangioleiomyomatosis, which are often characterized by a benign clinical course.1 Although criteria for malignancy in PEComas have not been clearly established, clinically aggressive or malignant PEComas are typically large and usually show marked nuclear pleomorphism, increased mitoses, necrosis and infiltrative margins.2 Some members of the PEComa family (specifically angiomyolipoma and lymphangioleiomyomatosis) occur in the setting of tuberous sclerosis complex (TSC) syndrome. Furthermore, a high frequency of syndromic and sporadic PEComas have loss of function mutations in TSC1 or TSC2 genes 3, 4, with subsequent activation of the mammalian target of rapamycin (mTOR) pathway5, which has been targeted therapeutically with mTOR inhibitors.6, 7
TFE3 is a member of the MiT family of transcription factors, which also includes MiTF, TFEB, and TFEC.8 TFE3 gene fusions have been demonstrated in several types of neoplasia, such as alveolar soft part sarcoma, resulting in an ASPL-TFE3 fusion9 and pediatric renal cell carcinomas (RCCs), with various TFE3 gene fusions.10 Recently, a small subset of PEComas has been shown to harbor TFE3 gene fusions 11–15, with a single case report showing a SFPQ/PSF-TFE3 gene fusion.16 In this study, we performed a comprehensive genomic characterization by transcriptome analysis of PEComas of various anatomic sites to establish the incidence and spectrum of gene fusions, as well as possible correlations between genetic signature and clinical presentation. Additionally, we sought to investigate if TFE3 rearrangements or other gene fusions abnormalities identified are mutually exclusive from the TSC1/TSC2 loss of functions mutations, to support a dichotomy of genetic alterations in PEComas with obvious therapeutic implications.
MATERIALS AND METHODS
The Department of Pathology files at Memorial Sloan Kettering Cancer Center and the personal consultation files of the corresponding author (CRA) were searched for cases of PEComa between 2000 and 2014. The criteria for the selection included a typical morphology and immunoprofile and available tissue for FISH and/or molecular studies. The study focused mainly on the PEComa group, and most other benign entities included in this family, such as triphasic angiomyolipomas and lymphangioleiomyomatosis were excluded from the study. Hematoxylin and eosin sections and immunohistochemical stains performed at the time of diagnosis were reviewed. The gross and microscopic findings, including tumor size, anatomic location, tumor morphology, mitoses (per 10 high power fields), and presence of necrosis were recorded. Clinical and follow-up data were obtained from the clinical database. The study was approved by the Institutional Review Board (IRB# 02-060 / WA0079-14 MSKCC).
RNA Sequencing
Eleven cases were analyzed by RNA sequencing. Total RNA was prepared for RNA sequencing in accordance with the standard Illumina mRNA sample preparation protocol (Illumina). Briefly, mRNA was isolated with oligo(dT) magnetic beads from total RNA (10 μg) extracted from case. The mRNA was fragmented by incubation at 94°C for 2.5 min in fragmentation buffer (Illumina). To reduce the inclusion of artifactual chimeric transcripts due to random priming of transcript fragments into the sequencing library because of inefficient A-tailing reactions that lead to self ligation of blunt-ended template molecules17, an additional gel size-selection step was introduced prior to the adapter ligation step. The adaptor-ligated library was then enriched by PCR for 15 cycles and purified. The library was sized and quantified using DNA1000 kit (Agilent) on an Agilent 2100 Bioanalyzer according to the manufacturer’s instructions. Paired-end RNA-sequencing at read lengths of 50 or 51 bp was performed with the HiSeq 2000 (Illumina). Across the samples, an average of 47.5M of pair-end reads were generated per sample corresponding to 4.75B bases per sample.
Analysis of RNA Sequencing Results with FusionSeq
All reads were independently aligned with the CASAVA 1.8 software provided by Illumina against the human genome sequence (hg19) and a splice junction library, simultaneously. The splice junction library was generated by considering all possible junctions between exons of each transcript. We considered the University of California, Santa Cruz (UCSC) Known Genes annotation set 18 to generate this library via RSEQtools, a computational method for processing RNA-seq data. 19 The mapped reads were converted into Mapped Read Format 15 and analyzed with FusionSeq 20 to identify potential fusion transcripts. FusionSeq is a computational method successfully applied to paired-end RNA-seq experiments for the identification of chimeric transcripts. 21, 22, 23 Briefly, paired-end reads mapped to different genes are first used to identify potential chimeric candidates. A cascade of filters, each taking into account different sources of noise in RNA-sequencing experiments, was then applied to remove spurious fusion transcript candidates. Once a confident list of fusion candidates was generated, they were ranked with several statistics to prioritize the experimental validation. In these cases, we used the DASPER score (Difference between the observed and Analytically calculated expected SPER): a higher DASPER score indicated a greater likelihood that the fusion candidate was authentic and did not occur randomly. See 20 for further details about FusionSeq.
In addition, RNA seq data was analyzed for gene mutation calls. Samtools mpileup (http://samtools.sourceforge.net/mpileup.shtml) was used to generate BCF file from aligned BAM file and only potential variants were reported. Downstream Variant Filter from Samtools was applied and variants were excluded if quality score was < 40 or RNAseq read coverage was < 20. The Variant Effect Predictor tool provided by Ensembl (http://useast.ensembl.org/info/docs/tools/vep/index.html) was used to detect variants with missense mutations in the 340 genes from the IMPACT panel. Potential missense locations were compared to NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp/). Sanger PCR validation was performed for novel mutations or those occurring with a frequency < 1%.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
An aliquot of the RNA extracted above from frozen tissue (Trizol Reagent; Invitrogen; Grand Island, NY) was used to confirm the novel fusion transcript identified by FusionSeq. RNA quality was determined by Eukaryote Total RNA Nano Assay and cDNA quality was tested for PGK housekeeping gene (247 bp amplified product). Three microgram of total RNA was used for cDNA synthesis by SuperScript ® III First-Strand Synthesis Kit (Invitrogen, Carlsbad, CA). RT-PCR was performed using the Advantage-2 PCR kit (Clontech, Mountain View, CA) for 30 cycles at a 64.5°C annealing temperature. Primers used are listed in Supplementary Table 1. Amplified products were purified and sequenced by Sanger method.
DNA PCR
Genomic DNA was isolated either from fresh-frozen or archival paraffin tissue, as described previously (Antonescu et al., 2003). Targeted PCR / Long Range PCR was performed for the gene fusion for 30–35 cycles at a 64.5°C annealing temperature. Primers used are listed in Supplementary Table 1.
Fluorescence In Situ Hybridization (FISH)
FISH on interphase nuclei from paraffin-embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking genes that were identified as potential fusion partners in the RNA-seq experiment. BAC clones were chosen according to UCSC genome browser (http://genome.ucsc.edu), see Supplementary Table 2. The BAC clones were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution, as previously described.24 The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems, Newton, MA). A positive score was interpreted when at least 20% of the nuclei showed a break-apart signal. Nuclei with incomplete set of signals were omitted from the score.
Targeted Exome Sequencing
We profiled 11 PEComas for genomic alterations in 340 key cancer-associated genes using our IMPACT assay (Integrated Mutation Profiling of Actionable Cancer Targets). This assay utilizes solution phase hybridization-based exon capture and deep-coverage massively parallel DNA sequencing.25 Custom oligonucleotides were designed to capture all protein-coding exons and select introns of commonly implicated oncogenes, tumor suppressor genes, and members of pathways deemed actionable by targeted therapies. Tumors and patient-matched normal were run in parallel for every case. Barcoded sequence libraries were prepared according to manufacturers’ protocols (New England Biolabs, Ipswich, MA; Kapa Biosystems, Wilmington, MA) using 36–250 ng of genomic DNA as input. Libraries were pooled and input to a single exon capture reaction as previously described.26 To prevent off-target hybridization, a pool of blocker oligonucleotides complementary to the full sequences of all barcoded adaptors was spiked in to a final total concentration of 10 micromolar. DNA was subsequently sequenced on an Illumina HiSeq 2500 to generate paired-end 100-bp reads. Sequence data were demultiplexed using CASAVA, and reads were aligned to the reference human genome (hg19) using the Burrows-Wheeler Alignment tool.27 Local realignment and quality score recalibration were performed using the Genome Analysis Toolkit (GATK) according to GATK best practices.28 We achieved a mean unique sequence coverage of 792x per tumor. Sequence data were analyzed to identify three classes of somatic alterations: single-nucleotide variants, small insertions/deletions (indels), and copy number alterations. Single-nucleotide variants and indels were called using muTect and SomaticIndelDetector, respectively.28, 29 The mean sequence coverage was calculated using the DepthOfCoverage tool in GATK and was used to compute copy number as described previously.22 Increases and decreases in the coverage ratios (tumor:normal) were used to infer amplifications and deletions, respectively.
RESULTS
Pathologic Features and Ancillary Findings
Thirty-eight PEComa cases were selected for the study (Table 1). There were 23 females and 15 males, with ages ranging from 24–79 years (median age – 56 years). Eleven tumors were located in the soft tissue, including thigh, calf, pelvis, buttock, retroperitoneum, intra-abdominal, back, mediastinum, paraspinal and peri-rectal. Other visceral sites included uterus, 11 cases (4 primary and 7 recurrences); GI/Liver and pancreas, 7; kidney, 6; and one each in urinary bladder, lung and brain.
Table 1.
Clinical Information, Genetic alterations and Follow-up.
| Case | Age Sex | Site | Immunohistochemistry | Fusion/Rearrangement | TSC2 mutation | TP53 mutation | Follow up | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SMA | Desmin | HMB45 | TFE3 | Duration | LR | DR | Status | ||||||
| 1 | 36/F | thigh | − | + | + | PSF-TFE3 | na | na | |||||
| 2α β | 47/F | pelvis | F+ | F+ | + | PSF-TFE3 | 51 | + | NED | ||||
| 3 | 33/F | rectum | − | − | + | PSF-TFE3 | 26 | NED | |||||
| 4 α β | 58/F | calf | F+ | DVL2-TFE3 | 16 | + | AWD | ||||||
| 5 | 69/M | pancreas | F+ | − | − | + | TFE3 | na | na | ||||
| 6 α β | 40/F | uterus | + | + | TFE3 | 12 | NED | ||||||
| 7 | 61/F | buttock | + | − | + | TFE3 | na | na | |||||
| 8 | 56/M | lung | − | − | − | + | TFE3 | na | na | ||||
| 9 | 62 / F | Buttock | + | − | − | + | TFE3 | na | na | ||||
| 10 α β # | 58/F | uterus (i-abd R) | + | F+ | RAD51B-RRAGB/OPHN1 | p.E393* | T150_P151 del | 18 | + | DOD | |||
| 11 | 44/F | uterus (i-abd R) | + | + | + | − | RAD51B-RRAGB/OPHN1 | 38 | + | + | NED | ||
| 12 | 66/F | uterus (lung met) | + | + | RAD51B | 16 | + | NED | |||||
| 13 α β | 65/F | uterus (i-abd R) | + | + | + | HTR4-ST3GAL1 | 17 | + | NED | ||||
| 14 α | 44/M | urinary bladder | + | − | + | − | RASSF1-PDZRN3 | 13 | NED | ||||
| 15 α β # | 56/M | small bowel | + | + | F+ | p.Y648* | p.Y200C | 60 | + | NED | |||
| 16^ | 37/F | Liver | + | − | + | p.W358X | 44 | NED | |||||
| 17 | 69/M | Kidney | F+ | F+ | + | 108 | + | + | DOD | ||||
| 18 | 44/M | Kidney | + | 91 | + | + | DOD | ||||||
| 19 α | 75/M | Liver | + | + | 2 | NED | |||||||
| 20 | 43/F | Abdomen | − | F+ | na | na | |||||||
| 21 | 65/M | Back | − | − | + | 13 | NED | ||||||
| 22 β | 57/M | Kidney | F+ | p.AKIVSDRNL 1700del | p.G244R | 94 | + | + | DOD | ||||
| 23 | 59/M | Mediastinum | + | + | + | F+ | na | na | |||||
| 24 | 61/M | Paraspinal | + | F+ | na | na | |||||||
| 25 β # | 54/F | uterus (i-abd R) | − | − | + | p.Q1605X | p.H380fs | 20 | + | DOD | |||
| 26 β | 79/M | Liver | + | F+ | F+ | + | D1598fs*23 | p.R283P | 7 | DOO | |||
| 27 α | 49/F | kidney | + | + | F+ | 21 | NED | ||||||
| 28 α | 36/M | stomach | + | − | + | 24 | NED | ||||||
| 29 | 57/M | RP | + | − | F+ | 25 | + | + | AWD | ||||
| 30 | 42/F | uterus (i-abd R) | − | 19 | + | NED | |||||||
| 31 | 68/F | uterus (i-abd R) | F+ | 28 | + | NED | |||||||
| 32 | 71/F | uterus | + | F+ | 9 | NED | |||||||
| 33 α # | 78/F | kidney (lung met) | − | − | F+ | p.Q1010* | 140 | DOD | |||||
| 34 | 48/F | uterus | + | − | F+ | 36 | NED | ||||||
| 35 β | 58/F | peri-rectal | + | + | − | 3 | + | NED | |||||
| 36 | 43/M | brain | + | − | F+ | − | 17 | + | DOD | ||||
| 37 | 59/F | uterus | + | + | F+ | 5 | NED | ||||||
| 38 β | 24/F | kidney (lung met) | − | − | + | F+ | p.D1696fs | 18 | + | NED | |||
Previously reported by Dickson et al. (2013);
Cases analyzed by RNA sequencing
Cases analyzed by targeted exome sequencing (IMPACT);
Cases with mutations confirmed by Sanger sequencing
RP, retroperitoneal; i-abd R, intra-abdominal recurrence; met, metastasis.
Transcriptome Analysis Identifies Novel Gene Fusions
RNA Sequencing and FusionSeq analysis was performed on 11 cases. Gene fusion candidates were identified in 5 cases. Two of the cases showed a TFE3 associated gene fusion with one showing a SFPQ/PSF-TFE3 gene fusion (previously reported by Tanaka et al.) and the other showing a novel DVL2-TFE3 gene fusion. Three novel non-TFE3 gene fusions were identified including RAD51B-RRAGB/OPHN1, HTR4-ST3GAL1 and RASSF1-PDZRN3. Six of the cases showed no evidence of gene fusions. These gene fusions were validated by the FISH technique and RT-PCR in all of 5 cases. FISH was also used to screen all of the remaining study cases for possible recurrent gene fusions.
TFE3 gene rearrangements are seen in a subset of PEComa with distinct nested morphology
FISH analysis for TFE3 was performed on all study cases and identified TFE3 gene rearrangements in 9 (23%) PEComa cases. There were 7 females and 2 males, ranging in age from 33–69 years (median – 56 years). This group spanned broad anatomic sites, including 5 soft tissue, 1 uterine, 2 gastrointestinal and 1 lung. Tumor size, available in 4 cases, ranged from 1–8 cm. All except one case showed a strikingly similar epithelioid morphology with abundant clear to granular cytoplasm, arranged in a nested to alveolar pattern (Fig. 1, Supplementary Table 3). One case (Case 8) showed spindle to ovoid cells in sheet-like arrangement with scattered pleomorphic cells (Fig. 1I). Mitotic activity was low in all cases, with most cases showing 0–1 mitosis per 10 high power fields (HPFs). Immunohistochemically, 6 of the 9 cases showed positivity for HMB45 (Fig. 1) and all 5 cases tested showed strong and diffuse TFE3 positivity (including the 3 cases that were negative for HMB45) (Fig. 1). SMA positivity was seen in 4 cases and desmin was negative in all except one case.
Figure 1. Morphologic appearance of TFE3 fusion positive PEComas.
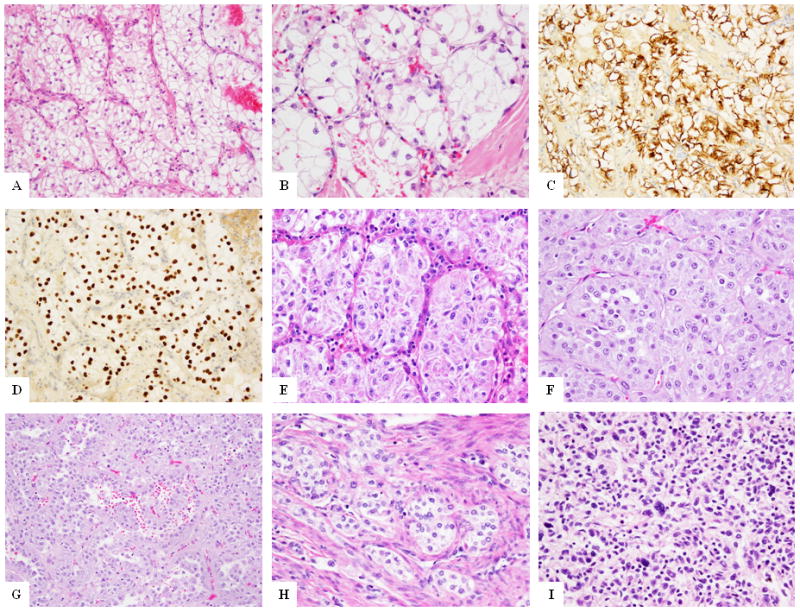
PEComa with PSF-TFE3 gene fusion (Case 1) at low (A) and higher magnification (B) showing the characteristic epithelioid cells in a nested architecture. HMB45 (C) and TFE3 (D) immunostains show diffuse positivity of the tumor cells. PEComa with PSF-TFE3 gene fusion (Case 3) showing similar morphology (E) of epithelioid cells with clear to granular cytoplasm in a nested appearance. PEComa with DVL2-TFE3 gene fusion (Case 4) showing epithelioid cells in a nested pattern (F) and other areas of pseudo-papillary arrangement (G). Uterine PEComa (Case 6) showing nested epithelioid cells in infiltrating the myometrium (H). Pulmonary PEComa (Case 8) showing spindle to ovoid cells with scattered atypical cells (I).
SFPQ/PSF-TFE3 is the most frequent gene fusion in PEComa
SFPQ/PSF-TFE3 fusion was identified by RNA seq in Case 2. Based on this result, all TFE3-positive tumors were then screened for SFPQ/PSF by FISH. Three out of 8 (38%) cases showed SFPQ/PSF rearrangements by FISH, all occurring in the soft tissue (pelvis, thigh, and calf) and showed a similar morphology as described above.
Novel DVL2-TFE3 gene fusion identified in soft tissue PEComa
DVL2-TFE3 fusion was identified in one case (Case 4) by RNA seq method (Fig. 2). DVL2 (disheveled segment polarity protein 2) is located on chromosome 17 (17p13.1). Experimental validation by RT-PCR confirmed a transcript composed of DVL2 exon 4 fused to TFE3 exon 7. FISH break-apart assay for TFE3 and DVL2 confirmed rearrangements in both genes. DNA PCR further confirmed the intronic break showing DVL2 exon 5 fused to TFE3 intron 6, with an intercalated small fragment of anti-parallel sequence of TFE3 exon 6 and TFE3 intron 5 (Fig. 2). None of the other TFE3-rearranged PEComas, analyzed by FISH, showed DVL2 gene rearrangement.
Figure 2. PEComa with DVL2-TFE3 gene fusion.
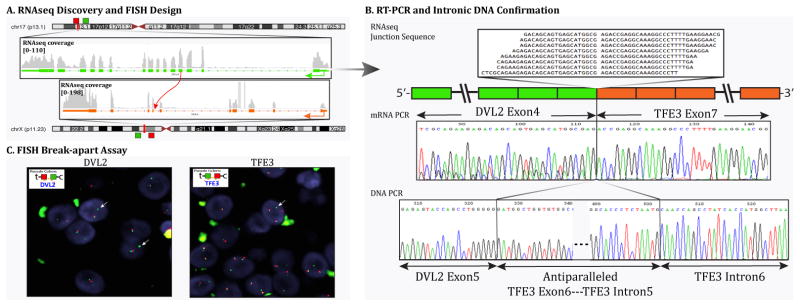
(Case 4).
(A) Schematic representation of the fusion of DVL2 located on 17p13.1 with TFE3 on Xp11.2, resulting in a t(X;17)(p11.2;p13.1) translocation. (B) RT-PCR validation showing fusion of the DVL2 exon 4 to TFE3 exon 7 (top right), followed by DNA PCR confirming the fusion of DVL2 exon 5 with TFE3 intron 6 with anti-parallel sequence of TFE3 exon 6 and TFE3 intron 5 in-between (bottom right). (C) FISH break-apart assays showing unbalanced rearrangements of DVL2 (arrows) with loss of telomeric signal (red) and trisomy of Xp11.2 locus with TFE3 rearrangements (arrows) (t-telomeric; c-centromeric).
Five of the TFE3-rearranged PEComas lacking a fusion partner were also analyzed by FISH for PRCC gene abnormalities, one of the known TFE3gene partners in renal cell carcinomas.30 However, no PRCC gene rearrangements were identified.
Novel recurrent RAD51B gene associated fusions identified in uterine PEComa
RNA sequencing and Fusion seq analysis in one uterine case (Case 10) identified two fusion transcript candidates, both involving RAD51B gene on 14q23-24.2 locus. (Fig. 3) One of the fusion partners was RRAGB on Xp11.21 locus, while the other was the OPHN1 gene on Xq12 locus. Both RAD51B-RRAGB and RAD51B-OPHN1 fusions were confirmed and validated by FISH, RT-PCR and DNA-PCR methodologies. For the RAD51B-RRAGB fusion, RT-PCR showed fusion of the RAD51B exon 8 to RRAGB exon 2 with an intervening 120bp intronic sequence of Xp11.21. FISH break-apart assays confirmed rearrangements in both RAD51B and RRAGB genes. For the RAD51B-OPHN1 fusion, RT-PCR confirmed the fusion transcript between RAD51B exon 3 with OPHN1 exon 17 (Fig. 3). FISH fusion assay showed the fused signal of RAD51B and OPHN1 genes (Fig. 3). Long range DNA PCR confirmed the intronic break with intron 3 of RAD51B fused to intron 16 of OPHN1.
Figure 3. RAD51B-associated gene fusion in uterine PEComa (Case 10).

(A) Schematic representation of the two fusion transcript candidates identified by RNAseq, involving the RAD51B locus on 14q24.1 with RRAGB located on xp11.2, resulting in a t(x;14)(p11.2;q24.1) translocation, and the other involving the RAD51B with OPHN1 gene on xq12 resulting in a t(x;14)(q12;q24.1); (B) Fusion candidates were validated by RT-PCR showing fusion of the RAD51B exon 8 to RRAGB exon 2 with an intervening intronic sequence of Xp11.21, and the fusion of RAD51B exon 3 with OPHN1 exon 17. (C) FISH break-apart assays showing unbalanced rearrangement of RAD51B gene (arrows) with loss of the telomeric signal (upper left). FISH break-apart assay showing unbalanced rearrangement of RRAGB gene (arrows) on Xp11.21 with loss of the telomeric signal (lower left); FISH for OPHN1 gene on Xq12 showing loss of one copy of signals, indicating a larger deletion at this locus (upper right). The RAD51B-OPHN1 fusion assay (lower right) showing fusion of the RAD51B (red) with a small fragment of OPHN1 (green) gene (arrows), suggesting a deletion in the OPHN1 gene locus. (t-telomeric; c-centromeric) (D) Bar chart showing increased expression of RRAGB in PEC10 compared to other PEComas. The dot plot further shows the differential exon expression of RRAGB (case 9, after the exon 2 breakpoint) compared to other PEComas.
FISH for RAD51B abnormalities was performed on 25 additional TFE3-negative PEComas and identified 2 additional positive cases, both originating from the uterus. One of the RAD51B-positive PEComa (case 11) was similarly associated with rearrangements in both RRAGB and OPHN1 genes by FISH. In the remaining RAD51B-rearranged PEComa (case 12) no gene partner was identified.
Morphologically, these 3 cases showed varied morphology. Case 10 showed a cellular epithelioid neoplasm, displaying a solid and sheet-like arrangement, with minimal cytoplasm and nuclei with prominent nucleoli, somewhat reminiscent of a small blue round cell tumor (Fig. 4A). Case 11 showed epithelioid cells arranged in small nests with focal areas of sclerosis (Fig. 4B). Case 12 showed spindle cells with scattered pleomorphism in a partially sclerotic background with focal areas showing hemangiopericytoma-like vascular pattern (Fig. 4C). Strikingly, all 3 cases showed increased mitotic activity of >10 mitoses/10 HPFs. Necrosis was identified in one case. Furthermore, a consistent pattern of smooth muscle immunoprofile was discerned in all 3 cases, with reactivity for both SMA and desmin. Additionally all 3 cases were positive for HMB45 and two of the tested cases also showed MITF positivity.
Figure 4. Morphologic spectrum of PEComas with non-TFE3 fusions.
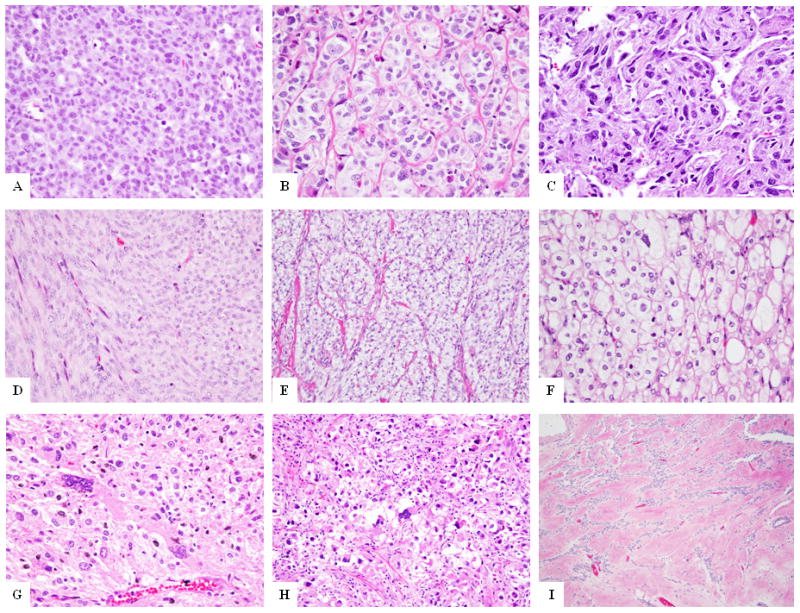
RAD51B-rearranged tumors (A-C): epithelioid cells in a sheet-like arrangement (A, case 10); or nested pattern (B, case 11), or spindle cells in fascicles in a hemangiopericytoma-like branching vascular pattern (C, case 12); (D) PEComa with HTR4-ST3GAL1 fusion (Case 13) showing predominantly spindle cells in fascicles, reminiscent of smooth muscle neoplasm; (E) PEComa with RASSF1-PDZRN3 gene fusion (Case 14) showing epithelioid to spindle cells with clear cytoplasm in a nested and fascicular pattern; (F-I) PEComas with no known gene fusions: epithelioid cells with clear cytoplasm in sheets (F, case 32), spindle to epithelioid cells and scattered pleomorphic cells (G, case 16); epithelioid neoplasm with scattered pleomorphic cells (H, case 19), and fascicles of spindle cells in a densely sclerotic background (I, case 28).
Additional non-recurrent HTR4-ST3GAL1 and RASSF1-PDZRN3 gene fusions in PEComa
RNA sequencing on two additional PEComa cases (Cases 13 & 14) identified novel HTR4-ST3GAL1 and RASSF1-PDZRN3 gene fusions. Both of these gene fusions were validated by RT-PCR and FISH analysis. Case 13 was a pelvic PEComa composed morphologically of intersecting fascicles of spindle cells, more reminiscent of a smooth muscle neoplasm (Fig. 4D). Immunohistochemically, the tumor expressed SMA, desmin, MITF and HMB45 (focally). Case 14 occurred in the urinary bladder wall and displayed a mixed spindle and epithelioid morphology with cells arranged in fascicles and a partially nested pattern (Fig. 4E). Lesional cells showed moderate clear cytoplasm and spindle to round nuclei with prominent nucleoli. Immunohistochemically, the tumor expressed SMA, MITF and HMB45 and was negative for desmin.
FISH analysis on the remaining PEComa cases failed to identify additional cases with these gene fusions, indicating that these are most likely non-recurrent genetic events.
PEComas with no identifiable gene fusions
In a majority of PEComas (24 of 38, 63%), no gene fusions were identified. These tumors were located at various anatomic sites, including soft tissue and viscera (GI, GYN and renal). Tumor morphology (Fig. 4F–4I) was predominantly that of epithelioid cells with clear to granular cytoplasm arranged in nests and cords (17 of 24 cases). Some of the cases (3 of 24) showed a mixed spindle and epithelioid phenotype and other cases (4 of 24) showed a purely spindled morphology, similar to a smooth muscle tumor. One case (Case 27) showed a sclerosing pattern with spindle cells in a densely sclerotic background (Fig. 4I). Nuclear pleomorphism was noted in 11 of 24 cases.
Co-existent TSC2 and TP53 Mutations Identified in TFE3 Fusion-Negative PEComas by Targeted Exome Sequencing
Targeted Exome Paired-End Sequencing analysis using the IMPACT assay was performed on a group of 11 PEComa cases with matched normal tissue available. The 11 cases tested included 3 TFE3 rearranged PEComa, 1 PEComa with RAD51B-RRAGB/OPHN1 fusion, 1 PEComa with HTR4-ST3GAL1, and 6 PEComas with no known gene fusions. One fusion-negative PEComa (case 16) had the TSC2 gene sequenced as part of a prior study (Dickson et al., 2013). In one other fusion negative PEComa (Case 33), RNA seq data was analyzed for possible mutations. Overall, 8 of 13 (62%) cases tested showed TSC2 gene mutations (Table 1). Upon excluding the TFE3 fusion-positive PEComas, the incidence of TSC2 gene abnormalities was higher with 8 of 10 (80%) cases showing mutations. Interestingly, 5 of the 8 (63%) cases with TSC2 mutations also showed co-existent TP53 mutations. The only case of RAD51B fusion-positive PEComa (case 10) tested showed both TSC2 and TP53 gene mutations. None of the 3 TFE3-rearranged PEComas tested showed TSC1/TSC2 or TP53 mutations. Mutations in 4 of the 8 positive cases were also validated using Sanger sequencing methodology (Table 1).
Treatment
Treatment history was available on 29 cases of the study. Sixteen cases were treated by surgical resection only. One case (Case 2) received post-op radiation therapy following local recurrence. Twelve cases received systemic therapy with cytotoxic chemotherapy (including gemcitabine, docetaxel, doxorubicin, ifosfamide, cyclophosphamide, irinotecan, dacarbazine, paclitaxel), tyrosine kinase inhibitors (pazopanib, sunitinib and sorafenib), and mTOR inhibitors (sirolimus and everolimus). In one case of PEComa in the liver in a 37 year old woman (case 16), treatment with an mTOR inhibitor (everolimus) was given in the neo-adjuvant setting with good clinical and pathologic response (previously reported in Dickson et al. 2013).6 In all other cases, systemic therapy was given for recurrent or relapsed disease. Of note, some patients in this series were treated in the era before the discovery of the activity of mTOR inhibitors in PEComa and thus were never treated with this class of drugs.
In the TSC2 mutation positive group of 8 cases, mTOR inhibitors were used in 4 cases. One case (case 16) received the drug in a neoadjuvant setting, as described above and showed a good clinical response. In another case (case 22), treatment showed initial response followed by progression of disease. No significant response was seen in 2 other cases.
Clinical follow-up
Follow-up information was available on 30 of the 38 study cases (Table 1) with duration ranging from 2–140 months. Ten patients developed local recurrence (LR) and 10 patients developed distant recurrence (DR), including 5 patients who developed both LR and DR. At the time of last follow-up, 20 patients had no evidence of disease (NED), 2 were alive with disease (AWD), 7 had died of disease (DOD) and 1 had died of other causes (DOO).
In the TFE3-fusion positive group, follow-up information was available on 4 patients ranging from 12–51 months, of which 2 developed LR. At last follow-up, 3 patients were NED and 1 was AWD.
In the RAD51B fusion positive group, follow-up was available on all the 3 cases, ranging from 16–38 months. All 3 patients developed DR of which one developed both LR and DR. At last follow-up, 2 patients were NED and one had DOD.
In PEComas with TSC2 gene mutations, follow-up was available in all of the 8 cases, ranging from 7–140 months. Two patients developed LR and 4 patients developed DR. At last follow-up, 4 patients DOD, 1 patient DOO and 3 were NED. Looking exclusively at the subset of cases having both TSC2 and TP53 mutations, follow-up ranged from 7–94 months with 1 patient developing LR and 3 patients developing DR. At last follow-up, 3 patients DOD, 1 patient DOO, and 1 was NED.
DISCUSSION
TFE3-gene fusions were initially reported in PEComa by Tanaka et al. who reported a gastrointestinal PEComa with an SFPQ/PSF-TFE3 gene fusion.16 Subsequently, Argani et al. 11 reported a series of 29 PEComa, of which 4 (13%) cases showed TFE3 gene rearrangements by FISH. Two of these 4 cases were tested for PSF-TFE3 fusion by RT-PCR but were negative. None of the other recent studies of PEComa 31, 32 have identified TFE3 gene fusions as recurrent events in 8 cases tested by FISH. The goal of our study was to provide a molecular characterization of PEComas of different clinical presentations by applying the latest transcriptome analysis, a highly sensitive method best suited for novel fusion gene discovery. The cases were first screened for the known TFE3 and SFPQ/PSF gene rearrangements by FISH, followed by RNA sequencing in the negative cases with available frozen tissue. Additional targeted sequencing for detection of somatic DNA mutations (i.e. TSC1/TSC2 loss of function mutations) was applied focusing mainly on the fusion-positive cohort, in order to test the hypothesis of mutually exclusive abnormalities from the TFE3-related fusions or alternative translocations. This comprehensive genomic analysis identified 9 (23%) cases of TFE3-gene fusion positive PEComas, of which 3 showed a SFPQ/PSF-TFE3 gene fusion. This gene fusion emerges as the most prevalent genetic event among TFE3-rearranged PEComas, interestingly all occurring in soft tissue PEComas. The SFPQ/PSF-TFE3 gene fusion results from a t(X;1)(p11.2;p34) translocation, which has been previously reported in a gastrointestinal PEComa16, as well as in a small subset (1.2%) of TFE3-fusion associated renal cell carcinomas.33 An additional novel DVL2-TFE3 fusion was identified by RNA sequencing in a soft tissue PEComa, resulting from a t(X;17)(p11.2;p13.1) translocation. As expected from most other TFE3-related fusions, which result in TFE3 oncogenic activation, DVL2-TFE3 fusion also demonstrated high levels of TFE3 mRNA by transcriptome analysis. DVL2 encodes a member of the dishevelled (dsh) protein family, which may play a role in the signal transduction pathway mediated by multiple Wnt proteins.34
Similar to prior reports of TFE3-associated PEComa11, 16 and other TFE3-fusion positive neoplasms, such as alveolar soft part sarcoma and Xp11.2 renal cell carcinoma, the TFE3-rearranged PEComa in our study showed similar morphologic findings of a distinctively epithelioid nested neoplasm, with abundant clear to granular eosinophilic cytoplasm and relatively monotonous nuclei with low mitotic activity. Only one TFE3-positive tumor showed a more spindle cell to ovoid morphology with scattered pleomorphic cells. In contrast to Argani et al. 11, who reported a mean age of 23.6 years for TFE3 fusion positive PEComas as opposed to 53 years for TFE3 fusion negative PEComas, our study did not reveal such an age difference between these two genetically distinct PEComa subsets (50 versus 57 years mean age).
In addition to the TFE3-rearranged subgroup, our findings further point out to another genetic subset, characterized by recurrent RAD51B gene fusions seen only in uterine PEComas. Of the three positive cases identified, two showed complex RAD51B-RRAGB/OPHN1 gene fusions. No partner gene was identified in one remaining case. Interestingly, RAD51B associated fusions, RAD51B-HMGIC, have been previously identified in a subset of uterine leiomyoma.35, 36 The shared RAD51B gene abnormalities in uterine leiomyomas and uterine PEComas (expressing desmin) raise questions regarding a morphologic spectrum or possible common pathogenesis between these two neoplasms in the uterus. The distinction between uterine leiomyosarcomas and PEComas has been quite challenging. Not surprisingly, all 3 RAD51B-rearranged PEComas were initially interpreted as leiomyosarcomas. Only after the recurrent disease was examined or melanocytic markers tested, the lesions were recognized as uterine PEComas. To complicate issues further, studies have pointed out an immunoprofile overlap between leiomyosarcoma and PEComa of the GYN tract, showing HMB45 reactivity (i.e. ‘leiomyosarcomas with HMB45 positivity’).37, 38 In our study, one of the three tumors with RAD51B fusion also showed TSC2 and TP53 mutations, further supporting the classification of these tumors as PEComas. Further studies are needed in order to investigate if these observations point to a common disease spectrum or merely overlapping immunophenotype of two genetically distinct pathologic entities. RAD51B-PLAG1 gene fusion has also been reported in a case of lipoblastoma.39 The protein encoded by this gene is a member of the RAD51 protein family, with important function in the DNA repair by homologous recombination. This protein has been shown to form a stable heterodimer with the family member RAD51C, which further interacts with the other family members, such as RAD51, XRCC2, and XRCC3. RAD51B overexpression was found to cause cell cycle G1 delay and cell apoptosis, suggesting a role of this protein in sensing DNA damage.40 The morphology of the three RAD51B-positive PEComas were relatively different from each other and from the TFE3 rearranged group, with only one of the three cases showing a nested morphology.
Our study included a broad spectrum of clinical presentations in an attempt to identify potential correlations between the genetic signatures and anatomic locations in PEComas. The largest subset was originating from the GYN tract (11 cases), which showed a wide variability in morphologic features, including tumors composed of nests of epithelioid cells with clear cytoplasm, while others displayed a mixed spindle and epithelioid phenotype, with rare cases showing a pure spindle cell pattern reminiscent of smooth muscle tumors. This morphologic spectrum is in keeping with other studies that have focused on PEComa from the gynecologic tract 2, 32 or from other visceral site, such as the GI tract.41 Similar to PEComas from other sites, only a subset of uterine PEComas show TFE3 associated gene fusions. In the report by Argani et al. (2010) only one of the 4 TFE3 fusion positive cases was from the uterus, this being the only reported case so far. Schoolmeester et al. (2014) tested 3 uterine PEComas for TFE3 gene rearrangements by FISH, but none were found to be positive. In our study, 1 of the 9 (11%) TFE3 fusion positive PEComa was from the uterus. Additionally, the novel RAD51B gene fusion identified in our study was seen exclusively in uterine PEComa. Our study findings indicate that PEComas of the uterus/gynecologic tract, similar to the soft tissue and other visceral PEComas, show phenotypic and genotypic heterogeneity.
Pan et al.42, using comparative genomic hybridization, identified losses of chromosome 16p involving the region of TSC2 gene in PEComa. Apart from the 16p deletions, they also identified losses in chromosome 17p involving the TP53 gene. This study showed similar alterations in both renal and extrarenal PEComa. Subsequently, the same group3, using LOH analysis, identified LOH of the TSC2 locus and mTOR activation in PEComa. Qin et al.4, studying a group of renal angiomyolipomas, identified TSC2 mutations in 7 of 8 cases tested, with most of the mutations representing deletions. No mutations were identified in the TSC1 gene. Malinowska et al.43, compared 4 cases each of TFE3 fusion positive and TFE3 negative PEComas, and reported that TSC2 alterations were exclusively identified in PEComas lacking TFE3 gene fusions and postulated that TFE3-rearranged PEComas have a different pathogenetic mechanism that does not involve the TSC2 gene through mutation or allelic loss. Our study, using next generation targeted exome sequencing, identified TSC2 mutations in 8 of 11 (72%) PEComas lacking TFE3 rearrangements. A subset of 5 cases showed concurrent TP53 mutations. No TSC1 mutations were identified. Given that TSC2 deletions are also found in the benign triphasic angiomyolipomas, it appears that TSC2 loss is an early event in PEComa family and additional secondary genetic alterations determine the biology and behavior of the tumor.
In conclusion, this study significantly expands our understanding of molecular alterations (Figure 5) in PEComas and brings forth the morphologic and genetic heterogeneity of these tumors. TFE3-rearrangements are seen in 23% of all PEComa cases studied and spanned a broad spectrum of locations. Among this group, the SFPQ/PSF-TFE3 emerges as the most prevalent fusion, accounting for about one third of cases, with rare tumors exhibiting an alternative DVL2-TFE3 fusion. The expected mechanism of tumorigenesis is through oncogenic activation of TFE3, likely by the acquisition of a strongly expressed promoter. Although our study was not designed for determining an accurate prevalence of TSC1/TSC2 gene mutations in PEComa cohorts, we wanted to address the specific question of mutually exclusive abnormalities within the fusion-positive group. Thus, our targeted NGS corroborated with mutation calls from the RNA sequencing, was able to exclude the presence of TSC1/TSC2 and p53 mutations in the TFE3-positive cohort. This finding not only adds to the speculation that TFE3-rearranged tumors have a different pathogenesis and most likely represent a distinct subgroup of PEComas, but also points to the potential differences in therapeutic targeting. Based on prior case reports 6, 7, 44, the current reflex treatment for most PEComas consists of mTOR inhibitors, which may not be as effective in this genetic subgroup. However, this hypothesis would have to be tested in a larger patient cohort, since the response of TFE3-rearranged tumors to mTOR inhibitors is largely unknown. Alternative drugs, such as crizotinib and tivantinib (MET inhibitors) are being explored in other TFE3-overexpressed sarcomas (i.e. alveolar soft part sarcomas).45
Figure 5. Schematic pie chart representing the genetic findings in PEComas.
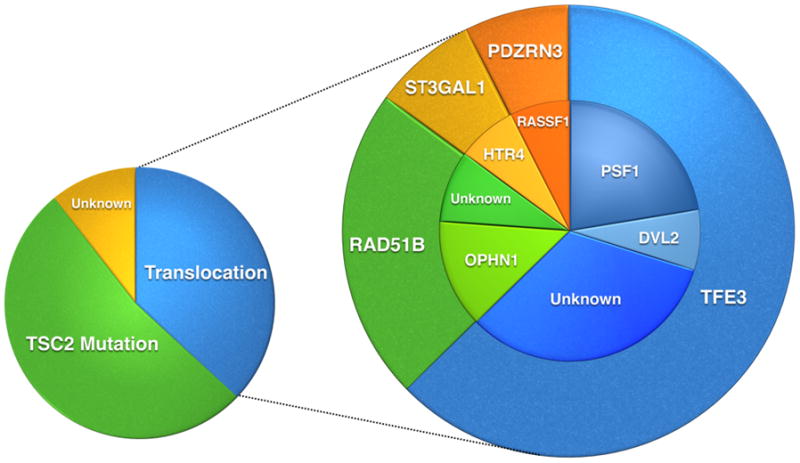
The larger pie chart demonstrates the different translocations identified in PEComas. The smaller pie on the left shows the different genetic subgroups in PEComa with a predominant group showing TSC2 mutations, a second group with translocations, and a smaller group with unknown genetic alterations.
In contrast, TSC2 mutations and co-existing TP53 mutations were identified in a subset of tumors sharing a spindle/pleomorphic morphology spanning soft tissue and visceral anatomic sites, with possibly more aggressive behavior. Given that TSC2-mutated, TFE3 fusion negative PEComas from both soft tissue and visceral locations show similar morphology and genetic alterations, it is probably more appropriate to regard these tumors as a single group rather than as site-specific entities.
Supplementary Material
Acknowledgments
Supported in part by: P01CA47179 (SS, CRA), P50 CA 140146-01 (SS, CRA), Cycle for Survival.
The authors thank Milagros Soto for editorial assistance and Allyne Manzo for assistance with composite figures. The authors also thank the following physicians who contributed cases and kindly provided follow-up information where possible: Dr. Cristina Hajdu, New York, NY; Dr. Adnan Hasanovic, New York, NY; Dr. Frank-Uwe Breuer, New Hyde Park, NY and Dr. Michael Goldfischer, Hackensack, NJ. The authors also thank and acknowledge the efforts of Agnès Viale and the Genomics Core Laboratory at MSKCC where RNA sequencing was performed.
Footnotes
Conflict of interest: none
References
- 1.Fletcher C, Bridge JA, Hogendoorn PC, et al. WHO Classification of Tumours of Soft Tissue and Bone. IARC; Lyon: 2013. [Google Scholar]
- 2.Folpe AL, Mentzel T, Lehr HA, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol. 2005;29(12):1558–75. doi: 10.1097/01.pas.0000173232.22117.37. [DOI] [PubMed] [Google Scholar]
- 3.Pan CC, Chung MY, Ng KF, et al. Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): genetic evidence for the relationship of PEComa with angiomyolipoma. J Pathol. 2008;214(3):387–93. doi: 10.1002/path.2289. [DOI] [PubMed] [Google Scholar]
- 4.Qin W, Bajaj V, Malinowska I, et al. Angiomyolipoma have common mutations in TSC2 but no other common genetic events. PLoS One. 2011;6(9):e24919. doi: 10.1371/journal.pone.0024919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenerson H, Folpe AL, Takayama TK, et al. Activation of the mTOR pathway in sporadic angiomyolipomas and other perivascular epithelioid cell neoplasms. Hum Pathol. 2007;38(9):1361–71. doi: 10.1016/j.humpath.2007.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickson MA, Schwartz GK, Antonescu CR, et al. Extrarenal perivascular epithelioid cell tumors (PEComas) respond to mTOR inhibition: clinical and molecular correlates. Int J Cancer. 2013;132(7):1711–7. doi: 10.1002/ijc.27800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol. 2010;28(5):835–40. doi: 10.1200/JCO.2009.25.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haq R, Fisher DE. Biology and clinical relevance of the micropthalmia family of transcription factors in human cancer. J Clin Oncol. 2011;29(25):3474–82. doi: 10.1200/JCO.2010.32.6223. [DOI] [PubMed] [Google Scholar]
- 9.Ladanyi M, Lui MY, Antonescu CR, et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20(1):48–57. doi: 10.1038/sj.onc.1204074. [DOI] [PubMed] [Google Scholar]
- 10.Argani PAC, Illei PB, et al. Primary renal neoplasms with ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol. 2001;159:179–192. doi: 10.1016/S0002-9440(10)61684-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Argani P, Aulmann S, Illei PB, et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol. 2010;34(10):1395–406. doi: 10.1097/PAS.0b013e3181f17ac0. [DOI] [PubMed] [Google Scholar]
- 12.Williamson SR, Bunde PJ, Montironi R, et al. Malignant perivascular epithelioid cell neoplasm (PEComa) of the urinary bladder with TFE3 gene rearrangement: clinicopathologic, immunohistochemical, and molecular features. Am J Surg Pathol. 2013;37(10):1619–26. doi: 10.1097/PAS.0b013e318293729d. [DOI] [PubMed] [Google Scholar]
- 13.Lee SE, Choi YL, Cho J, et al. Ovarian perivascular epithelioid cell tumor not otherwise specified with transcription factor E3 gene rearrangement: a case report and review of the literature. Hum Pathol. 2012;43(7):1126–30. doi: 10.1016/j.humpath.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 14.Liu F, Zhang R, Wang ZY, et al. Malignant perivascular epithelioid cell tumor (PEComa) of cervix with TFE3 gene rearrangement: a case report. Int J Clin Exp Pathol. 2014;7(9):6409–14. [PMC free article] [PubMed] [Google Scholar]
- 15.Shen Q, Rao Q, Xia QY, et al. Perivascular epithelioid cell tumor (PEComa) with TFE3 gene rearrangement: Clinicopathological, immunohistochemical, and molecular features. Virchows Arch. 2014;465(5):607–13. doi: 10.1007/s00428-014-1655-x. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka M, Kato K, Gomi K, et al. Perivascular epithelioid cell tumor with SFPQ/PSF-TFE3 gene fusion in a patient with advanced neuroblastoma. Am J Surg Pathol. 2009;33(9):1416–20. doi: 10.1097/PAS.0b013e3181a9cd6c. [DOI] [PubMed] [Google Scholar]
- 17.Quail MA, Kozarewa I, Smith F, et al. A large genome center’s improvements to the Illumina sequencing system. Nat Methods. 2008;5(12):1005–10. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu F, Kent WJ, Clawson H, et al. The UCSC Known Genes. Bioinformatics. 2006;22(9):1036–46. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 19.Habegger L, Sboner A, Gianoulis TA, et al. RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics. 2011;27(2):281–3. doi: 10.1093/bioinformatics/btq643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sboner A, Habegger L, Pflueger D, et al. FusionSeq: a modular framework for finding gene fusions by analyzing paired-end RNA-sequencing data. Genome Biol. 2010;11(10):R104. doi: 10.1186/gb-2010-11-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanas MR, Sboner A, Oliveira AM, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med. 2011;3(98):98ra82. doi: 10.1126/scitranslmed.3002409. [DOI] [PubMed] [Google Scholar]
- 22.Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44(4):461–6. doi: 10.1038/ng.1107. [DOI] [PubMed] [Google Scholar]
- 23.Mosquera JM, Sboner A, Zhang L, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer. 2013;52(6):538–50. doi: 10.1002/gcc.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49(12):1114–24. doi: 10.1002/gcc.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Won HH, Scott SN, Brannon AR, et al. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. J Vis Exp. 2013;(80):e50710. doi: 10.3791/50710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagle N, Berger MF, Davis MJ, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2(1):82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sidhar SK, Clark J, Gill S, et al. The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum Mol Genet. 1996;5(9):1333–8. doi: 10.1093/hmg/5.9.1333. [DOI] [PubMed] [Google Scholar]
- 31.Llamas-Velasco M, Mentzel T, Requena L, et al. Cutaneous PEComa does not harbour TFE3 gene fusions: immunohistochemical and molecular study of 17 cases. Histopathology. 2013;63(1):122–9. doi: 10.1111/his.12145. [DOI] [PubMed] [Google Scholar]
- 32.Schoolmeester JK, Howitt BE, Hirsch MS, et al. Perivascular epithelioid cell neoplasm (PEComa) of the gynecologic tract: clinicopathologic and immunohistochemical characterization of 16 cases. Am J Surg Pathol. 2014;38(2):176–88. doi: 10.1097/PAS.0000000000000133. [DOI] [PubMed] [Google Scholar]
- 33.Kauffman EC, Ricketts CJ, Rais-Bahrami S, et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol. 2014;11(8):465–75. doi: 10.1038/nrurol.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee YN, Gao Y, Wang HY. Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, -2, and -3. Cell Signal. 2008;20(2):443–52. doi: 10.1016/j.cellsig.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schoenmakers EF, Huysmans C, Van de Ven WJ. Allelic knockout of novel splice variants of human recombination repair gene RAD51B in t(12;14) uterine leiomyomas. Cancer Res. 1999;59(1):19–23. [PubMed] [Google Scholar]
- 36.Takahashi T, Nagai N, Oda H, et al. Evidence for RAD51L1/HMGIC fusion in the pathogenesis of uterine leiomyoma. Genes Chromosomes Cancer. 2001;30(2):196–201. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1078>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 37.Simpson KW, Albores-Saavedra J. HMB-45 reactivity in conventional uterine leiomyosarcomas. Am J Surg Pathol. 2007;31(1):95–8. doi: 10.1097/01.pas.0000213346.57391.70. [DOI] [PubMed] [Google Scholar]
- 38.Hurrell DP, McCluggage WG. Uterine leiomyosarcoma with HMB45+ clear cell areas: report of two cases. Histopathology. 2005;47(5):540–2. doi: 10.1111/j.1365-2559.2005.02147.x. [DOI] [PubMed] [Google Scholar]
- 39.Deen M, Ebrahim S, Schloff D, et al. A novel PLAG1-RAD51L1 gene fusion resulting from a t(8;14)(q12;q24) in a case of lipoblastoma. Cancer Genet. 2013;206(6):233–7. doi: 10.1016/j.cancergen.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 40.Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol. 2011;22(8):898–905. doi: 10.1016/j.semcdb.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 41.Doyle LA, Hornick JL, Fletcher CD. PEComa of the gastrointestinal tract: clinicopathologic study of 35 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2013;37(12):1769–82. doi: 10.1097/PAS.0b013e31829caab3. [DOI] [PubMed] [Google Scholar]
- 42.Pan CC, Jong YJ, Chai CY, et al. Comparative genomic hybridization study of perivascular epithelioid cell tumor: molecular genetic evidence of perivascular epithelioid cell tumor as a distinctive neoplasm. Hum Pathol. 2006;37(5):606–12. doi: 10.1016/j.humpath.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Malinowska I, Kwiatkowski DJ, Weiss S, et al. Perivascular epithelioid cell tumors (PEComas) harboring TFE3 gene rearrangements lack the TSC2 alterations characteristic of conventional PEComas: further evidence for a biological distinction. Am J Surg Pathol. 2012;36(5):783–4. doi: 10.1097/PAS.0b013e31824a8a37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gennatas C, Michalaki V, Kairi PV, et al. Successful treatment with the mTOR inhibitor everolimus in a patient with perivascular epithelioid cell tumor. World J Surg Oncol. 2012;10:181. doi: 10.1186/1477-7819-10-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stacchiotti S, Marrari A, Dei Tos AP, et al. Targeted therapies in rare sarcomas: IMT, ASPS, SFT, PEComa, and CCS. Hematol Oncol Clin North Am. 2013;27(5):1049–61. doi: 10.1016/j.hoc.2013.07.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.