

SUMMARY
Micro- and nano-meter size particles have become popular candidates for cancer vaccine adjuvants. However the mechanism by which such particles enhance immune responses remains unclear. Here we report a porous silicon microparticle (PSM)-based cancer vaccine that greatly enhances cross-presentation and activates type I interferon response in dendritic cells. PSM-loaded antigen exhibited prolonged early endosome localization and enhanced cross-presentation through both proteasome- and lysosome-dependent pathways. Phagocytosis of PSM by dendritic cells induced type I interferon responses through a TRIF- and MAVS-dependent pathway. Dendritic cells primed with PSM-loaded HER2 antigen produced robust CD8 T cell-dependent anti-tumor immunity in mice bearing HER2-positive mammary gland tumors. Importantly, this vaccination activated tumor immune microenvironment with elevated levels of intra-tumor type I interferon and MHC-II expression, abundant CD11c+ dendritic cell infiltration, and tumor-specific cytotoxic T cell responses. These findings highlight the potential for PSM as an immune adjuvant to potentiate dendritic cell-based cancer immunotherapy.
INTRODUCTION
Cancer immunotherapy has gained a high level of attention lately thanks to recent FDA-approval of a dendritic cell-based metastatic prostate cancer therapy (Provenge) and checkpoint blockade antibodies (e.g., anti-CTLA4, anti-PD1) for late-stage cancer treatment (Hodi et al., 2010; Postow et al., 2015; Sharma et al., 2011). Despite these advances, complete response to cancer immunotherapy remains sparse in clinic due to multiple factors such as inefficient vaccine delivery, defective antigen cross presentation in tumor tissues, infiltration of suppressive immune cells including regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), and immunosuppressive cytokine milieu (Dougan and Dranoff, 2009). Approaches to reverse the immunosuppressive tumor microenvironment are anticipated to have a significant impact on cancer immunotherapy (Gajewski et al., 2013b).
Innate immunity is a major component of tumor immunity, and proper activation of innate immune cells by recognizing tumor antigens and danger signals from tumor cells ensures efficient adaptive immunity against cancer (Dougan and Dranoff, 2009). Thus, factors bridging innate immunity and adaptive immunity can be targeted for cancer immunotherapy. Dendritic cells (DCs) are the professional antigen presenting cells by surveying and processing antigen to T cells, and the antigen presentation process often requires subcellular antigen delivery and innate immune signaling. It has been previously reported that class I antigen is processed in early endosome and the Toll-like receptor 4 (TLR4)-MyD88 activity is required for proper relocation of the transporter associated with antigen processing (Burgdorf et al., 2008). However, the study was done on soluble antigen cross presentation, and whether the same mechanism can be applied to other forms of antigens such as particulate antigens remains unknown.
Innate immune stimuli such as TLR ligands often serve as immune adjuvants to enhance DC-based immune responses (Coffman et al., 2010). TLR activation stimulates downstream pathways such as NF-κB signaling and MAPK signaling for pro-inflammatory cytokine induction (Kawai and Akira, 2011). These cytokines will further induce expression and translocation of antigen presenting molecules and promote antigen processing. Ironically, too strong TLR stimulation may induce detrimental inflammatory responses (Spaner et al., 2008), which prevents their use in clinic. Besides inflammatory cytokines, type I interferons (IFN-I) also promote DC maturation, antigen cross-presentation, and CD8 T cell clonal expansion (Coffman et al., 2010; Le Bon and Tough, 2008). Furthermore, a recent study reported a pivotal role of IFN-I in anti-tumor immunity by reactivating cross presentation function in intra-tumor DCs (Yang et al., 2014).
Physical properties of antigens and adjuvants may contribute to their immune-stimulating functions. The size, shape, and surface characteristics of an antigen or adjuvant have a significant impact on its immunogenicity (Bachmann and Jennings, 2010). Particulate antigen vaccine might provide advantage over the soluble antigen vaccine by serving as antigen depot and protecting the antigen from enzyme degradation, enabling targeted delivery to specific immune organs and cell types, and stimulating antigen presentation via the desired pathways at controlled release rate (Paulis et al., 2013). For example, alum adjuvant and many nano-size crystal structures can activate inflammasome and promote IL-1β release in DCs, which may facilitate the antigen presentation function of DCs and boost immune responses (Sharp et al., 2009). Nevertheless, the mechanism of action of these particles is still not well understood.
Discoid porous silicon microparticles (PSMs, 1 μm in diameter and 400 nm in height) can carry nano-sized drugs, and have been used for delivery of small molecule drugs and other cancer therapeutics (Chen et al., 2014; Dave et al., 2014; Shen et al., 2013a; Xu et al., 2013). This drug carrier is degradable and biocompatible, and the rate of release of the cargo can be tailored by surface chemical modification (Shen et al., 2013b; Tanaka et al., 2010; Xu et al., 2013). Here we explored the potential of PSM as an adjuvant for cancer vaccine. PSMs loaded with liposomal antigen inside the nanopores were efficiently internalized by DCs and trafficked to early endosomes for efficient cross presentation. Notably, phagocytosis of PSM induced type I interferon responses in DCs. In addition, DCs primed with PSM-loaded HER2 antigen peptide inhibited growth of HER2-overexpressing mammary gland tumors in mice by stimulating stroma cell MHCII expression, Th1 cytokine production, and intra-tumor CD8 T cell cytotoxicity.
RESULTS
Uptake of PSM-loaded antigen by dendritic cells
We analyzed particle uptake by immune cells in mice treated with intravenous injection of PSMs, and found that the particles were preferentially engulfed by the CD11c+ DCs over the F4/80+ macrophages in the peripheral blood (Table S1). Since DCs are the most efficient antigen presenting cells, we explored the possibility of applying PSM to potentiate antigen presentation. We first applied ovalbumin (OVA), a well-characterized model antigen, for in vitro and in vivo immune response studies (Figure S1A). FITC-conjugated OVA (FITC-OVA) was packaged into liposomes and loaded into PSM following a previously described procedure (Shen et al., 2013a; Tanaka et al., 2010; Xu et al., 2013). The PSM-loaded FITC-OVA (PSM/FITC-OVA) was efficiently internalized by DCs as early as 0.5 h post co-incubation in cell culture (Figure 1A), indicating efficient cell uptake of particles. Transmission electron microscopy revealed that the PSM particles were surrounded by phagosome-like vesicular structures 3 h post incubation (Figure 1B). Pretreatment of DCs with either the phagocytosis inhibitor cytochalasin D or the macropinocytosis inhibitor amiloride significantly decreased particle internalization (Figure 1C, 1D), suggesting that both phagocytosis and macropinocytosis were involved in the process. Interestingly, real-time particle tracking revealed that the DC-internalized particles could be transferred to the neighboring cells in vitro (Figure S1D, Movie S1). When PSM-containing DCs were intravenously injected into mice, certain PSM particles parted from the injected cells, and likely phagocytosed by the endogenous cells (Figure S1E).
Figure 1.
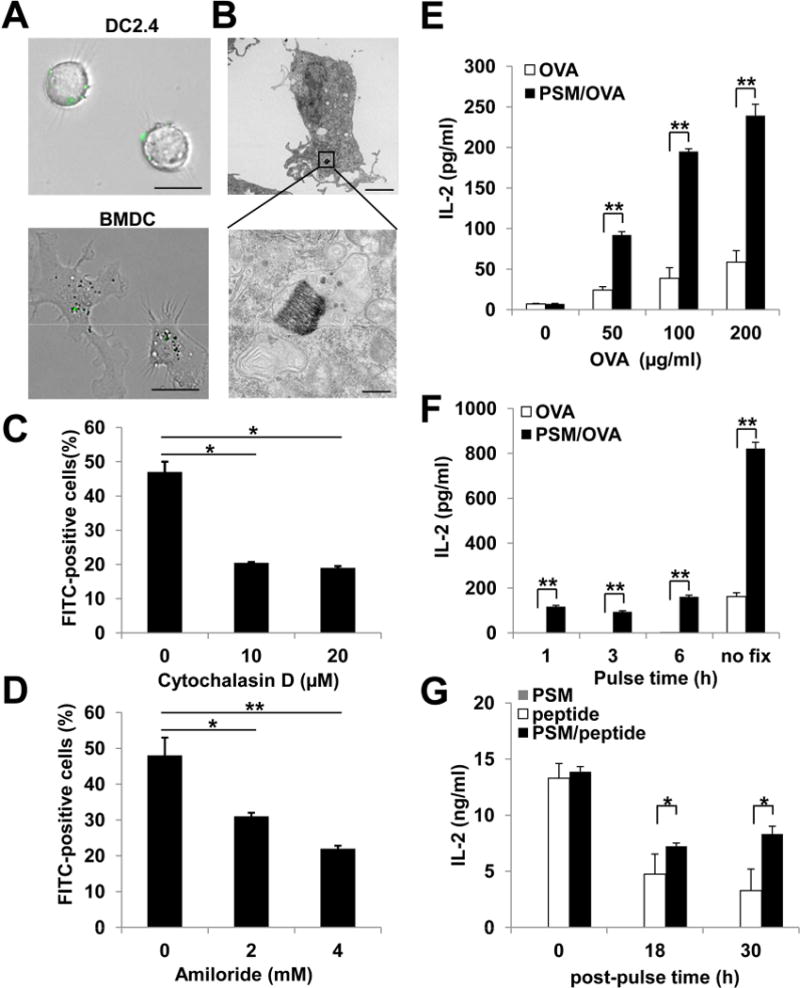
PSM-loaded antigen was efficiently internalized by dendritic cells through phagocytosis and macropinocytosis.
(A) Representative microscopic views of DCs internalizing PSM particles loaded with OVA-FITC nanoliposomes. Upper panel, DC2.4 cells; lower panel, bone marrow-derived DCs. Scale bar, 25 μm.
(B) A representative transmission electron microscopy picture showing the vesicular structure around a PSM/OVA particle inside the DC. Upper panel, 5,000 X; lower panel, 50,000 X. Upper panel scale bar, 4 μm; lower panel bar, 0.5 μm.
(C) DC uptake of PSM/FITC-OVA in the presence of phagocytosis inhibitor cytochalasin D.
(D) DC uptake of PSM/FITC-OVA in the presence of macropinocytosis inhibitor amiloride.
(E) IL-2 production by B3Z cells after co-incubation with DCs pre-treated for 3 h with different concentrations of soluble OVA or PSM/OVA.
(F) IL-2 production by B3Z cells after co-incubation with DCs that were primed with 50 μg/ml soluble OVA or PSM/OVA followed by fixation at different time points.
(G) IL-2 production by B3Z cells after co-incubation with DCs. The DCs were extensively washed after priming with 5 μg/ml soluble peptide or PSM/peptides, and either immediately co-incubated with B3Z cells (0 h), or cultured for 18 to 30 h before T cell co-incubation.
Data are presented as mean ± S.D. *, P<0.05, **, P<0.01. See also Figure S1.
We next assessed T cell response to antigen cross-presentation of PSM-loaded OVA (PSM/OVA) in DCs using an in vitro antigen presentation assay. DCs pulsed with PSM/OVA induced significantly higher levels of IL-2 production in OVA-specific B3Z CD8 T cells than those pulsed with soluble OVA (Figure 1E, 1F, S1B), suggesting that cross-presentation of PSM/OVA was much more efficient than that of soluble OVA in DCs. Efficient cross-presentation of PSM/OVA was not PSM size-dependent, as DCs pulsed with OVA in different size PSM microparticles (diameters 0.6 – 2.5 μm) consistently induced higher levels of IL-2 production in B3Z cells than those pulsed with soluble OVA or liposomal OVA (Figure S1C). Consequently, we chose the 1 μm PSM particles in all further studies. We subsequently tested whether PSM packaging could induce sustained antigen presentation. DCs were pulsed with either soluble or PSM-packaged OVA peptide for 2 h at 37°C, followed by extensive wash, and then either immediately co-cultured with B3Z cells or incubated in medium for 18 or 30 h before B3Z cell co-culture. IL-2 levels in the T cell culture were comparable when DCs were used for co-culture immediately after removal of OVA peptide in either soluble or PSM formulation (Figure 1G). However, T cell activities were significantly higher in co-culture with PSM/peptide-pulsed DCs than soluble peptide-pulsed DCs 18 h and 30 h after peptide removal, indicating that PSM-delivered antigen induced prolonged MHC-peptide presentation to T cells.
Antigen presentation pathway of PSM/OVA antigen
We next checked intracellular antigen trafficking by confocal microscopy. PSM/FITC-OVA particles colocalized with early endosomes (EEA1+) as early as 0.5 h after co-incubation with DCs, and were in the ER (KDEL+) at 3h and 6 h post-incubation. Particles were rarely found in late endosomes (Rab7+) or recycling endosomes (transferring receptor+) (Figure 2A). Interestingly, PSM/FITC-OVA could still be spotted in early endosomes by 6 h, indicating that PSM/OVA could retain in this subcellular organelle for a prolonged time. In comparison, free FITC-OVA showed very weak signal by 6 hour, likely due to quick degradation inside the cells (data not shown).
Figure 2.
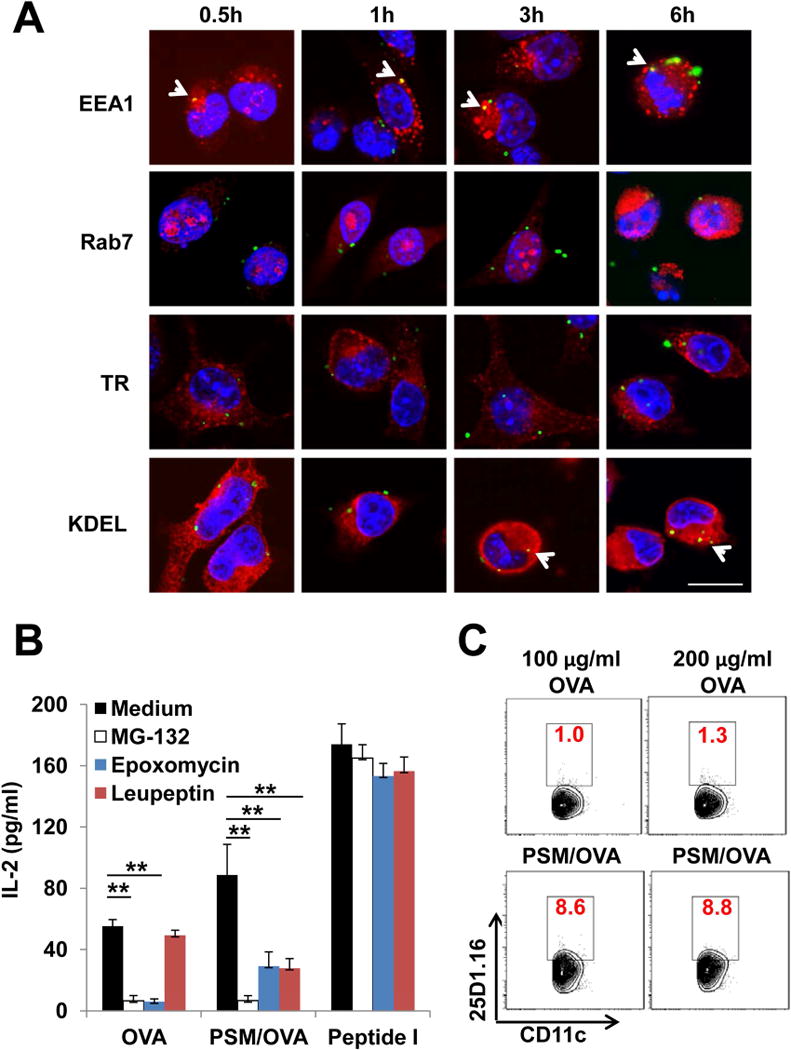
Antigen presentation of PSM-loaded OVA.
(A) Subcellular transport of antigen. DCs were incubated with PSM/FITC-OVA for 0.5 h, and then washed extensively to remove unbound particles. The cells were cultured for an additional 0.5 h, 1 h, 3 h, or 6 h before fixation followed by immunostaining with antibodies recognizing specific organelles (EEA1 for early endosome, Rab7 for late endosome, TR for recycling endosome, and KDEL for the ER). Scale bar, 25 μm.
(B) IL-2 production by B3Z cells after co-incubation with DCs treated with 100 μg/ml OVA or PSM/OVA in the presence of proteosome inhibitors (MG-132, epxomycin) or lysosome inhibitor (leupeptin). Peptide I (100 ng/ml) served as the positive control.
(C) OVA cross-presentation by BMDCs. BMDCs were incubated with OVA or PSM/OVA for 16 h, followed by harvest and labeling with the anti-CD11c antibody identifying DCs and the 25D1.16 antibody recognizing OVA257–264/H-2Kb complex on DC surface. Percentages of 25D1.16 staining positive populations in DCs were shown in red numbers.
Data are presented as mean ± S.D. *, P<0.05, **, P<0.01. See also Figure S2.
To identify route(s) of antigen processing of the PSM-packaged vaccine, we treated DCs with proteasome and lysosome inhibitors before co-incubation with soluble OVA or PSM/OVA, and measured induction of IL-2 expression. Proteasome inhibitors (MG-132 and epoxomycin), but not the lysosome inhibitor leupeptin, completely blocked class I antigen presentation of soluble OVA (Figure 2B). In contrast, PSM/OVA-induced IL-2 production was inhibited by all inhibitors, suggesting that both the proteasome and lysosome pathways were involved in antigen presentation of PSM/OVA. Since the transporter associated with the antigen processing (TAP) protein is an essential component of class I antigen presentation pathway (Trombetta and Mellman, 2005), we tested antigen processing in DCs isolated from TAP1 knockout mice (TAP−/− DCs), As expected, class I antigen presentation of PSM/OVA was abolished in TAP−/− DCs (Figure S2A). On the contrary, class II antigen presentation of PSM/OVA was comparable between WT and TAP−/− cells (Figure S2B). Thus, PSM/OVA cross-presentation by DCs requires TAP-dependent antigen processing.
Since OVA antigen processing resulted in surface expression of OVA peptide bound class I MHC molecules, we probed the processed antigen with antibody 25D1.16 that specifically recognized the MHCI-peptide (SIINFEKL-H-2Kb) complex. Indeed, more 25D1.16-positive cells could be identified in PSM/OVA-primed DCs than in soluble OVA-primed DCs (Figure 2C), thus confirming enhanced antigen processing and presentation of PSM loaded OVA by DCs.
These observations indicate that, after internalization by DCs through phagocytosis/macropinocytosis, PSM/OVA leaves the phagosome, and the released antigen is processed through the proteasome pathway. The processed antigen then binds to MHC molecules in the ER and is transported to the cell surface. Alternatively, the antigen can be processed directly by endosomal proteases in early endosomes, and presented to the cell surface in the form of an antigen peptide-MHC complex. In either case, the TAP protein is required for loading antigen peptide onto MHC molecules (Figure S2C).
PSM-induced innate immune responses
We reasoned that the enhanced cross-presentation by PSM loaded antigen might be associated with activation of innate immunity in addition to early endosome delivery. While alum adjuvant can activate inflammasome and induce IL-1β maturation and release in DCs, PSM did not induce IL-1β release in LPS-primed DCs (Figure S3A). Neither PSM nor antigen-loaded PSM changed mRNA or protein levels of pro-inflammatory cytokines (Figures 3A and S3B). In addition, there was almost no change in expression of surface co-stimulatory molecules (CD80, CD86, and CD40) after PSM treatment (Figure 3B).
Figure 3.
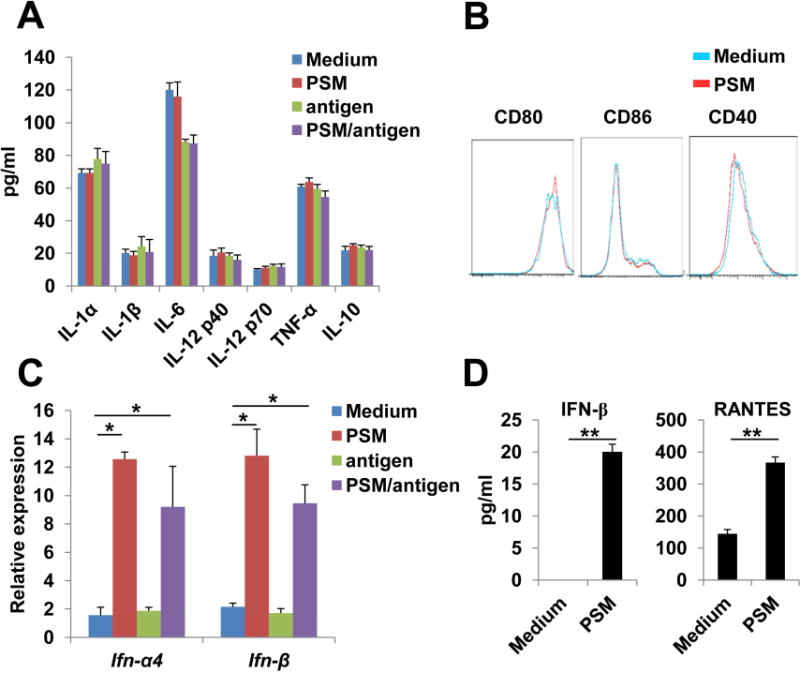
PSM induced IFN-I signaling in DCs.
(A) Protein levels of pro-inflammatory cytokines in culture media of BMDCs 24 h after incubation with PSM, free antigen, or PSM/antigen. Cell culture medium served as the negative control.
(B) Expression pattern of co-stimulatory molecules on the surface of BMDCs 24 h after incubation with PSM.
(C) QPCR analysis on mRNA levels of the Ifn-α4 and Ifn-β genes in BMDCs 5 h after co-incubation with PSM, free antigen, or PSM/OVA antigen.
(D) ELISA assay for IFN-β and RANTES in culture media of BMDCs 24 h after incubation with PSM.
Data are presented as mean ± S.D. *, P<0.05, **, P<0.01. See also Figure S3.
As one important arm of innate immune responses to viral infection, IFN-I response is required for optimal T cell activation by promoting cross-presentation and stimulating CD8 T cell clonal expansion (Le Bon and Tough, 2008; Ng and Gommerman, 2013). We analyzed expression profile of IFN-I-related genes, and found that both empty PSM and antigen-loaded PSM, but not free antigen, induced a significant increase in IFN-α4 and IFN-β expression (Figure 3C). ELISA results confirmed that PSM-treated DCs exhibited a modest but significant increase in IFN-β production, and also secreted a high level of RANTES, an IFN-I regulated chemokine (Lapteva and Huang, 2010) (Figure 3D).
PSM-induced IFN-I response is dependent on TRIF and MAVS signaling
To determine whether the IFN-I responses elicited by PSM was dependent on any known TLR- or RLR-induced IFN-I pathways, we knocked down expression of key adaptor molecules in these pathways, then analyzed PSM-induced IFN-I responses in DCs (Figure 4A–4D). PSM retained the ability to induce IFN-I response with suppressed expression of MyD88 and STING. In contrast, IFN-I induction was abolished when TRIF or MAVS was knocked down, suggesting that PSM induced IFN-I response through TRIF- and MAVS-dependent pathways. We next applied bone marrow-derived dendritic cells (BMDCs) from gene knockout mice to validate the results from shRNA-based studies. IFN-I induction by PSM was completely inhibited in Trif−/− BMDCs, but not in StingGt/Gt (Sauer et al., 2011) or Myd88−/−BMDCs (Figure 4E–4H). Consistently, PSM treatment enhanced RANTES production in BMDCs from WT, Myd88−/−, StingGt/Gt, and Tlr3−/− mice; however, RANTES production was inhibited in Trif−/− BMDCs (Figure 4I). Although TLR4, TLR9, and TLR3 deficiency abolished DC response to LPS, CpG, and polyI:C respectively (Figure S4A), PSM-induced IFN-I response was not affected (Figure S4B), suggesting a TLR-independent mechanism for PSM-induced IFN-I responses.
Figure 4.
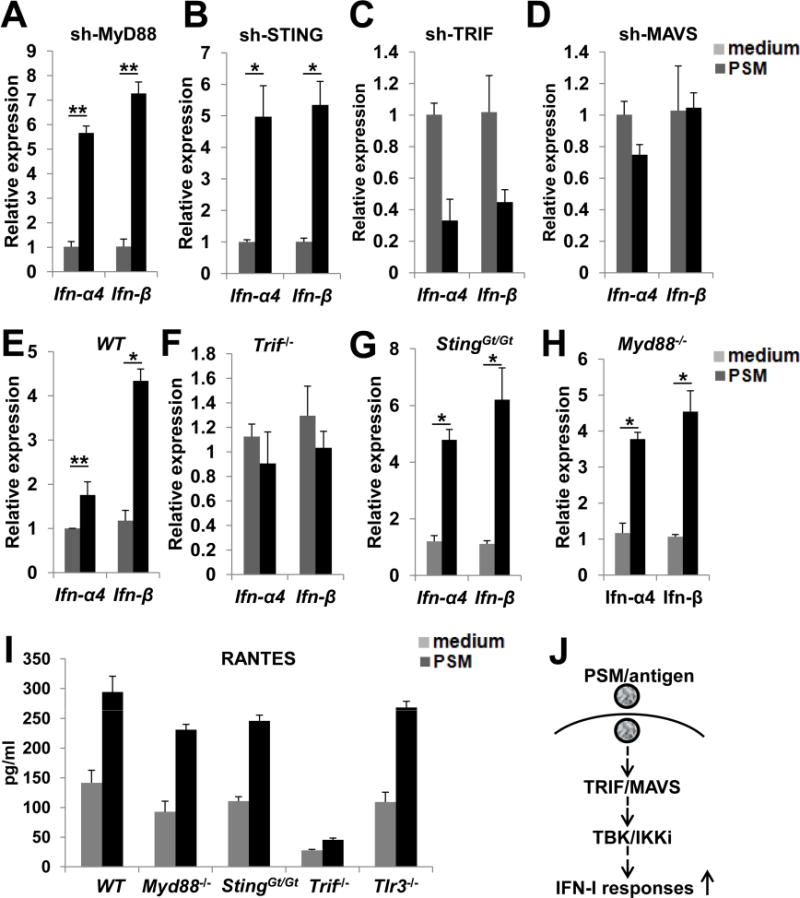
TRIF and MAVS mediated PSM induced IFN-I activation.
(A–D) PSM-induced changes of Ifn-α4 and Ifn-β gene expression in BMDCs infected with lentiviruses carrying shRNAs targeting the indicated genes.
(E–H) PSM-induced changes of Ifn-α4 and Ifn-β gene expression in BMDCs isolated from wild-type or gene knockout mice.
(I) PSM-induced RANTES production in BMDCs isolated from wild-type or gene knockout mice.
(L) Schematic view of PSM-induced signaling pathway that mediates IFN-I activation.
Data are presented as mean ± S.D. *, P<0.05, **, P<0.01. See also Figure S4.
Since PSM uptake and cross-presentation requires phagocytosis of PSM/antigen, we examined whether phagocytosis is required for PSM-induced IFN-I. Pretreatment with the phagocytosis inhibitor cytochalasin D suppressed IFN-I induction by PSM (Figure S4C). We also analyzed downstream kinase signaling of phagocytosis. PI3K inhibition has no effect on IFN-I induction (Figure S4D). In comparison, inhibition of the key kinases (TBK1/IKKi) for IFN-I signaling by BX795 markedly inhibited IFN-I induction (Figure S4E). We further analyzed activation status of MAPKs and NF-κB signaling in BMDCs after PSM/antigen priming. Except a slight increased level of ERK phosphorylation, PSM priming did not strongly activate JNK, p38, AKT, or NF-κB signaling (Figure S4F).
Taken together, phagocytosis of PSM-loaded antigen by DCs activates TRIF and MAVS-dependent signaling and the downstream TBK1/IKKi to elicit IFN-I responses (Figure 4J).
Anti-tumor efficacy of dendritic cells primed with PSM/HER2 antigen peptide
Given the critical role of efficient cross-presentation and CD8 T cell cytotoxicity in anti-tumor immunity, we next tested whether enhanced cross presentation and activated IFN-I in DCs by PSM/antigen could be translated into enhanced anti-tumor activity in animal tumor models. Balb/c mice were inoculated with TUBO tumor cells which were originally derived from mammary gland tumor of Balb-neuT transgenic mice (overexpressing rat neu under MMTV promoter) (Lucchini et al., 1992). When tumors reached a palpable size, the mice were treated i.v. with PSM, PSM/p66 (p66 is a class I HER2 antigen peptide) (Nagata et al., 1997), p66-primed DCs (DC+p66), or PSM/p66-primed DCs (DC+PSM/p66). While PSMs had no impact on tumor growth, direct PSM/p66 administration inhibited tumor growth significantly (Figure 5A). Mice that received DC+p66 showed comparable tumor growth inhibition as those in the PSM/p66 treatment group. The most significant effect was observed in mice treated with DC+PSM/p66, demonstrated by the nearly complete tumor growth inhibition and extended animal survival (Figure 5A, S5A). The effect on tumor growth was specific for HER2 peptide p66, as treatment with DC only or DCs primed with PSM-TRP-2 (a non-related antigen) could not inhibit tumor growth (Figure S5B). In addition, the anti-tumor efficacy correlated with intra-tumor HER2-specific CD8 T cell frequency (Figure S5C). In a separate study, we evaluated tumor prevention effect from PSM/p66. Balb-neuT mice started to develop spontaneous tumors at 15 weeks, and all mice developed tumors by week 17 (Figure S5D). There were on average over 5 tumors per mouse by week 20 (Figure 5B). Mice treated with PSM/p66 or DC+PSM/p66 developed significantly less number of tumors and exhibited longer tumor latency than the non-vaccinated mice (Figure 5B, S5D). The result indicates that PSM-loaded HER2 peptide served as an anti-HER2 vaccine and prevented mammary gland tumor development in the transgenic mice.
Figure 5.
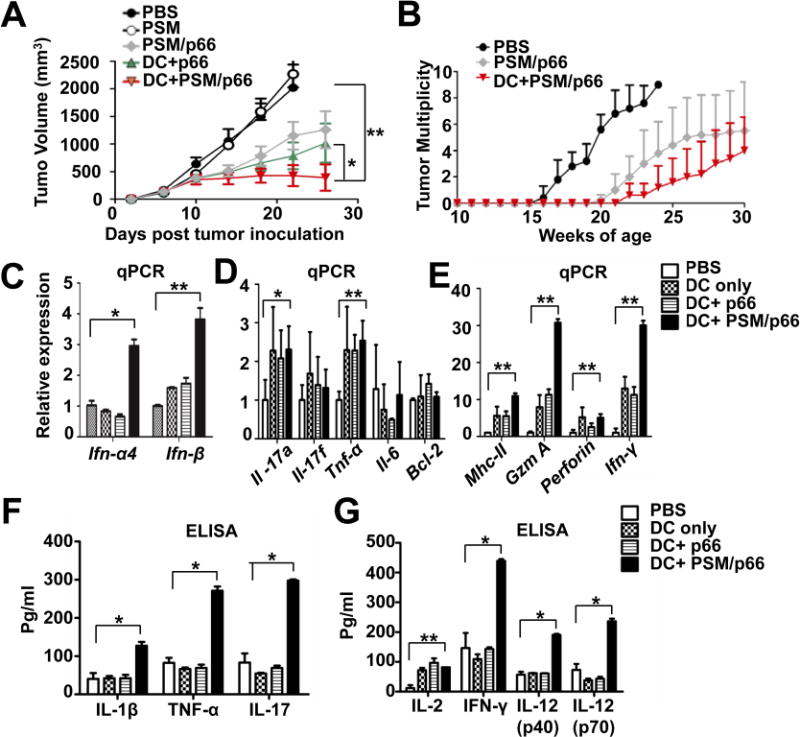
PSM/HER2 DC vaccine inhibited mouse mammary gland tumor growth.
(A) Inhibition of primary TUBO tumor growth. TUBO cells were inoculated into the mammary gland fat pads. After 4 days, mice (n = 8/group) were treated i.v. with PBS, PSM, PSM/p66, DC+p66, or DC+PSM/p66, and tumor growth was monitored in the next 4 weeks. Tumor volume is presented as mean ± SEM.
(B) Tumor multiplicity in Balb-neuT mice with different treatments. The Balb-neuT transgenic mice were divided into 3 groups (n = 5), and treated twice (weeks 6 and 8) i.v. with the indicated agents. Number of tumor nodules per mouse was counted on weekly basis.
(C–E) Quantitative RT-PCR measurement on mRNA levels of IFN-I, pro-inflammatory cytokines, and cytotoxic T cell markers in tumor tissues from different treatment groups.
(F and G) ELISA measurement on intra-tumor cytokine levels in mice with different treatments.
Data are represented as mean ± S.D. except tumor volume. *, P<0.05, **, P<0.01. See also Figure S5.
PSM/antigen-primed DC elicited immunostimulating tumor microenvironment
We analyzed intra-tumor cytokine levels in the post-treatment TUBO tumors, since anti-tumor immunity is often dependent on the immune-stimulating tumor microenvironment (Coussens et al., 2013). Consistent with the cell-based study (Figure 3C), quantitative RT-PCR (qPCR) analysis revealed significantly increased intra-tumor IFN-I levels in vaccinated mice (Figure 5C). Levels of pro-inflammatory cytokines were mixed. While expression of TNF-α and IL-17a increased in the vaccinated mice, IL-17F, IL-6 and the anti-apoptotic molecule Bcl-2 remained unchanged (Figure 5D). On the other hand, expression of the antigen presentation molecule MHC-II significantly increased in vaccinated groups (Figure 5E). Moreover, levels of cytotoxic T cell markers (granzyme and perforin) and IFN-γ also increased after vaccination, with granzyme and IFN-γ levels being 2–3 folds as high in the DC+PSM/p66 treatment group as in the DC or DC+p66 treatment groups (Figure 5E). Interestingly, all DC vaccines induced intra-tumoral expression of the Treg marker gene Foxp3 (Figure S5E), suggesting vaccination induced Treg expansion and tolerance (Ambrosino et al., 2006). To confirm the qPCR results, we isolated single cells from tumor samples, grew them in culture, and measured cytokine levels in cell growth media by ELISA. We detected significantly increased levels of key pro-inflammatory cytokines such as IL-1β, IL-17 and TNF-α in tumor cells from the DC+PSM/p66 treatment group over the other groups (Figure 5F). In addition, Th1 cytokines such as IFN-γ, IL-12p40 and IL-12p70 were the highest in the DC+PSM/p66 treatment group (Figure 5G). As Balb/c mice are prone to Th2 response (Mills et al., 2000), we observed dramatic increase in level of the Th2 cytokine IL-4 after DC and DC+p66 treatments (Figure S5F). However, DC+PSM/p66 treatment did not induce IL-4 production. Thus, PSM/p66 priming could obviate the strong Th2 response associated with DC treatment in Balb/c mice. These results suggest that DC+PSM/p66 induced a pro-inflammatory environment in tumor tissues and shifted an otherwise predominant Th2 response in Balb/c mice to a Th1 response, which may contribute to the anti-tumor immunity.
PSM/HER2-primed dendritic cell vaccine activated cytotoxic T cell immunity against tumor
We further analyzed intra-tumor cell population that might contribute to the intra-tumor cytokine milieu. Western blot analysis of tumor tissue lysates revealed comparable MHC-I protein levels among the treatment groups; however, MHC-II expression dramatically increased in tumor from the DC+PSM/p66 group (Figure 6A). Immunohistochemical (IHC) staining confirmed a high level of MHCII positive cells on the edge of tumor nodules in the DC+PSM/p66 group (Figure 6B). Immunofluorescent staining and flow cytometry analysis revealed markedly increased percentage of CD11c+MHCII+ cells in tumors from the DC+PSM/p66 group over other groups (Figure 6C, 6D).
Figure 6.
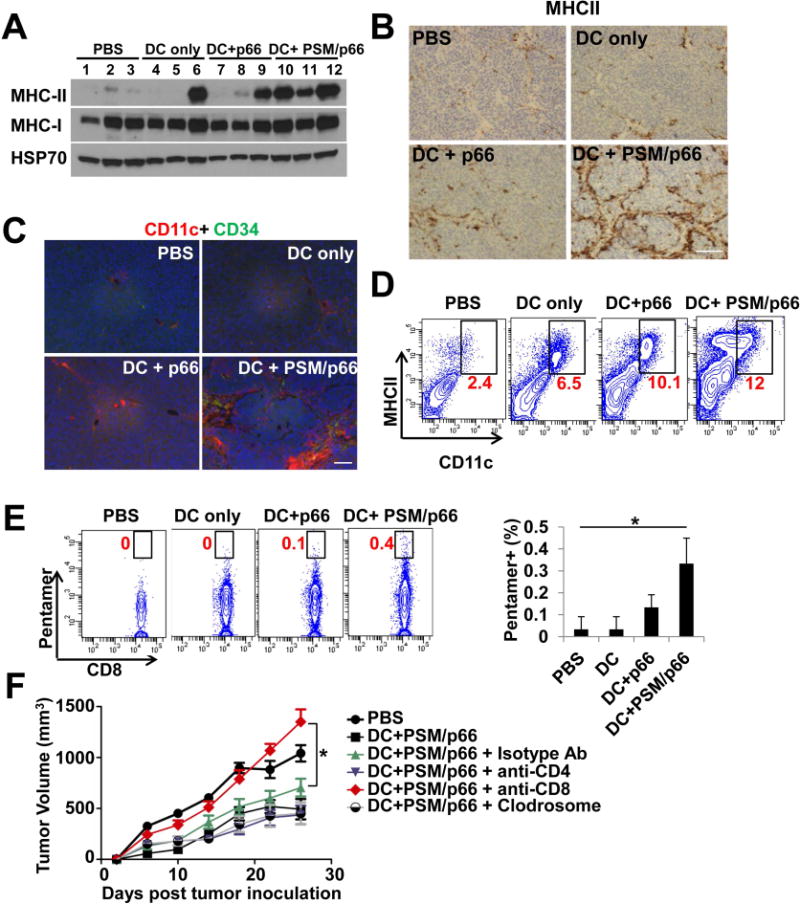
DCs primed with PSM/HER2 elicited CD8 T cell-mediated anti-tumor immunity.
(A) Western blot analysis on MHCII protein levels in TUBO tumor tissues from post-treatment Balb/c mice. Tumor tissues were harvested 10 days after treatment.
(B) Immunohistochemical staining of MHCII in TUBO tumor tissues from post-treatment Balb/c mice. Scale bar, 100 μm.
(C) Immunofluorescent staining of CD11c and CD34 (vascular marker) in TUBO tumor tissues from post-treatment Balb/c mice. Scale bar, 50 μm.
(D) Flow cytometry analysis on CD11c- and MHCII-double positive cells isolated from post-treatment TUBO tumor tissues. Percentage of CD11c- and MHCII-double positive cells in each sample was labeled in red.
(E) Enhanced HER2-specific CD8 T cells in post-treatment TUBO tumors. Left: Flow cytometry analysis on CD8 and p66-pentamer positive tumor-infiltrating lymphocytes in post-treatment tumor tissues. Right: Quantification of percentage of p66-pentamer positive CD8 T cells in tumor-infiltrating lymphocytes.
(F) Effect of immune cell depletion on inhibition of TUBO tumor growth by DC+PSM/p66 treatment. Balb/c mice inoculated with TUBO tumor (n = 8/group) were treated with a control isotype antibody, anti-CD4 antibody, anti-CD8 antibody, or clodrosome to deplete specific cell types. Mice were then treated with DC+PSM/p66, and tumor growth was monitored in the next 4 weeks. Tumor volume is presented as mean ± SEM.
Data are represented as mean ± S.D. except tumor volume. *, P<0.05, **, P<0.01.
As the executor of T cell-mediated anti-tumor immunity, HER2-specific CD8 T cells had the highest level in the DC+PSM/p66 treated tumors (Figure 6E). To determine which type(s) of immune cells were required for the anti-tumor immunity, we depleted CD4 T cells, CD8 T cells, or macrophages in mice bearing TUBO tumors, and then treated mice with DC+PSM/p66 (Figure 6F). CD8 T cell depletion completely abolished anti-tumor immunity. In contrast, depletion of CD4 T cells or macrophages did not impair the anti-tumor immunity, neither did treatment with an isotype control antibody. These results indicate that CD8 T cells are essential for DC+PSM/p66-induced anti-tumor activity.
DISCUSSION
It has been suggested that phagocytosis of particulate antigens or adjuvants not only facilitates their uptake by APCs, but also triggers innate immune activation by interaction with innate immune receptors (Fric et al., 2014). In the current study, we have demonstrated that phagocytosis was required both for cross-presentation and IFN-I induction in PSM/antigen-primed DCs, reinforcing the dual role of phagocytosis in antigen uptake and innate immune activation.
IFN-I response is elicited in DCs after exposure to microbial or tumor-derived DNA and RNA. Binding of nucleotides activates innate immune receptors such as TLRs and RLRs, and subsequently recruits individual adaptor proteins and the key kinases TBK1 and IKKi, leading to IRF3 or IRF7 phosphorylation, nuclear translocation and activation of IFN-I gene transcription (Prinz and Knobeloch, 2012). It is generally accepted that MyD88 is required for TLR7/8/9-induced IFN-I signaling, while TRIF is needed for TLR3/4-induced IFN-I signaling. The intracellular RNA and DNA sensors rely on adaptor molecules MAVS and STING for signaling transduction (Prinz and Knobeloch, 2012). Interestingly, membrane fusion can directly activate IFN-I response through STING-dependent signaling without activating DNA or RNA sensors, suggesting that physical fusion sometimes serves as “danger signal” to elicit innate immune response (Holm et al., 2012). However, membrane fusion is unlikely the primary factor for PSM-induced IFN-I response, as knockdown or knockout of STING in DCs did not abolish PSM-induced IFN-I responses. It is intriguing that both TRIF and MAVS are essential for PSM-induced IFN-I response, since they seem to mediate different upstream signals from TLRs and RLRs. However, MAVS was initially identified as a downstream adaptor of TRIF in intracellular poly(I:C) induced IFN-I signaling (Xu et al., 2005), and both TRIF and MAVS are required for DDX1/DDX21/DHX36 complex-induced IFN-I activation (Zhang et al., 2011). Therefore, a TRIF-MAVS axis may exist for IFN-I induction in response to intracellular DNA/RNA molecules or phagocytosed PSM particles. Alternatively, the particulate adjuvant may activate downstream signaling by altering membrane lipid structures, as in the case with alum (Flach et al., 2011). Thus, further studies are warranted to understand the underlying mechanism on PSM activation of IFN-I signaling. It is noteworthy that PSM induced moderate IFN-I expression. Such a level of IFN-I is competent enough for stimulating cross-presentation and CD8 T cell expansion, as demonstrated in lymphotoxin receptor-mediated IFN-I induction (Summers deLuca et al., 2011).
In the context of tumor microenvironment, although endogenous immunity against cancer still exists, many aspects of anti-tumor immunity have been crippled during tumor development and thus require exogenous boosting treatment (Gajewski et al., 2013a). Increased level of IFN-I has been reported as favorably correlating with clinical immune responses against cancer (Fuertes et al., 2011), and intra-tumor IFN-I levels predict clinical responses of breast cancer patients to anthracycline-based chemotherapy (Sistigu et al., 2014). Exogenous therapeutic antibody coupled with IFN-β also enhances anti-tumor activity by activating the cross-presentation capacity of endogenous dendritic cells (Yang et al., 2014). Therefore, targeting DCs and re-activating their cross-presentation capacity are a promising strategy for cancer immunotherapy. Dendritic cell-based immunotherapy can be done by administration of exogenously prepared DCs or injection of antigen and adjuvants targeting endogenous DCs. Mechanistically, injected DCs may present antigen directly to T cells, or serve as a cellular delivery vector and transfer the antigen to endogenous DCs (Kleindienst and Brocker, 2003; Yewdall et al., 2010). It is tempting to speculate that the PSM/antigen-loaded DCs may transfer PSM/antigen to endogenous DCs and promote their anti-tumor immunity. Our data support such a scenario, as we observed inter-cellular exchange of PSMs between DCs in vitro, and PSMs released from injected DCs in vivo.
Previous reports have also identified anti-tumor immunity accompanied with increased intra-tumoral MHC-II expression after treatment with the agonist anti-CD40 antibody, which was attributed to activated innate immune cells, mainly macrophages (Beatty et al., 2011; O’Sullivan et al., 2012). In the current study, PSM/antigen-primed DC treatment also drastically increased MHC-II expression in tumor tissues. Nevertheless, depletion of macrophages in the TUBO tumor model did not have a significant impact on DC-based therapeutic effect. Instead, increased tumor infiltration of CD11c+ cells accompanied with MHC-II upregulation was observed in mice treated with PSM/HER2 primed DCs, demonstrating the critical role of activated DCs in anti-tumor activity. These findings are in line with a recent report describing activation of intra-tumoral dendritic cells by IFN-I and enhanced antibody-mediated tumor immunity (Yang et al., 2014).
CD8 T cells are regarded as the key player in anti-tumor activity (Dougan and Dranoff, 2009). We have shown that depletion of CD8 T cells abrogates anti-tumor activity of PSM/antigen primed DC vaccine. This result is in line with previous reports using the same CD8 epitope HER2 peptide that anti-tumor immunity is mainly dependent on CD8 T cell cytotoxic killing activity (Assudani et al., 2008; Nava-Parada et al., 2007). Another important observation is that vaccination also induced FOXP3 expression in tumor tissues. Treg infiltration is a major barrier for inducing effective anti-tumor immune responses both in human clinical trials (Gajewski et al., 2013b) and in murine tumor models such as the Balb-neuT transgenic mice (Ambrosino et al., 2006). Transient Treg ablation by antibody or genetic manipulation has shown antitumor effect by itself (Bos et al., 2013; Marabelle et al., 2013). This may explain why CD4 antibody depletion did not affect anti-tumor immunity of DCs primed with PSM/antigen, as CD4 antibody may also deplete Tregs in addition to T help cells. Based on the current study, a combinational treatment with PSM/antigen-primed DC vaccine and transient Treg ablation may further improve the outcomes of anti-tumor vaccines.
In conclusion, we have shown that PSM/antigen-primed DC vaccination produced a strong anti-tumor immunity by inducing efficient antigen cross-presentation and eliciting IFN-I response. Phagocytosis of PSM/antigen generated prolonged early endosome antigen localization and simultaneously activated IFN-I signaling, thus providing another example how phagocytosis and innate immune signaling cooperate to enhance cross-presentation. We have also revealed a specific intracellular pathway mediating PSM-induced IFN-I signaling, illustrating the essential functions of TRIF and MAVS in response to particulate antigen. In addition, we have demonstrated the role of immuno-competent tumor microenvironment in PSM-potentiated DC vaccine. These findings further highlight the advantage of particulate vaccine and the IFN-I signaling in stimulating the tumor immune microenvironment for successful immunotherapy.
EXPERIMENTAL PROCEDURES
Mice
Balb-neuT transgenic mice were kindly provided by Dr. Guido Forni (University of Torin, Italy) and Dr. Larry Pease (Mayo Clinic, Minnesota). C57BL/6 and Balb/c mice were ordered from Charles River or Jackson Laboratory. B6.129S2-Tap1tm1Arp/J (Tap−/−), B6N.129S1-Tlr3tm1flv/J (Tlr3−/−), C57BL/6J-Tmem173gt/J (StingGt/Gt) mouse lines were ordered from The Jackson Laboratory. Tlr4−/−, Tlr9−/−, Trif−/− and Myd88−/− mouse lines were maintained as previously described (Duggan et al., 2011; Millien et al., 2013). All animal experiments were approved by Houston Methodist Research Institute IACUC.
Preparation of antigen-encapsulated liposomes and loading into PSM
Liposomes were made as previously described (Tanaka et al., 2010; Xu et al., 2013). Briefly, protein or peptide antigen were dissolved in H2O, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) was prepared in t-butanol at 20mg/ml, and 0.1% Tween-20 was prepared in water, then mixed together into t-butanol by vortex for 1 minute. The samples were then freeze-dried in a lyophilizer. To load the liposomes into PSM particles (1 μm in diameter and 400 nm in height), 120 μL H2O was added to liposome powder and followed by brief sonication. The liposomes in H2O were then added to dry PSM particles followed by brief sonication.
In vitro antigen presentation assay
The in vitro antigen presentation assay was performed as previously described (Mukai et al., 2011). Briefly, DC2.4 cells or BMDCs isolated from C57BL/6 mice or TAP−/− mice were seeded in a 96-well flat-bottom culture plate at a density of 2×105 cells/well and cultured for 12 h at 37°C. Each well was washed once with PBS, and then the cells were pulsed with soluble OVA, PSM/OVA, OVA-derived MHC class I epitope peptide (OVA257–264; SIINFEKL), or MHC class II epitope peptide (OVA323-339; ISQAVHAAHAEINEAGR; Peptide 2.0) for indicated time points, and immediately fixed with 1% paraformaldehyde. The cells were then co-cultured with 2×105 B3Z cells or 2×105 DOBW cells for 18 h at 37°C. The response of stimulated B3Z or DOBW cells was assessed by determining the amount of IL-2 released into the culture medium using a murine IL-2 ELISA KIT (Ebioscience). For the inhibition assay, BMDCs were pretreated with 10 mM MG-132 (EMD chemicals, San Diego, CA), 1 mM epoxomicin (EMD chemicals, San Diego, CA), or 40 mM leupeptin (EMD chemicals, San Diego, CA) for 1 h at 37°C before antigen pulsing. The antigen pulsing media also contained each inhibitor.
Confocal laser scanning microscopy
DC2.4 cells or BMDCs were seeded on eight-well chamber slides at a density of 1×105 cells/well and cultured for 24 h at 37°C. Each well was washed twice with PBS, and the cells were pulsed with FITC-OVA or PSM/FITC-OVA for 20 min. Cells were washed twice with PBS and incubated for an additional 30 min, 1, 3, or 6 hours in culture media at 37°C. Cells were washed with PBS, fixed with 4% paraformaldehyde, and then stained with antibodies against specific markers of individual organelles. Antibodies used to detect the early endosome, late endosome, early and recycling endosome, and endoplasmic reticulum (ER) were rabbit anti-EE Ag 1 (EEA1) Ab (ab74906; Abcam, Cambridge, MA), rabbit anti-Rab7 Ab (ab50313; Abcam), rat anti-transferrin receptor (TfR) Ab (ab22391; Abcam), and rabbit anti-KDEL Ab (ab50601; Abcam), respectively. Cells were then washed with PBS and incubated with Alexa 594-conjugated anti-rabbit IgG Ab (A11037; Invitrogen) or Alexa 594-conjugated anti-rat IgG Ab (A21209; Invitrogen) for 1 h at room temperature. The samples were embedded with Prolong Gold anti-fade reagent with DAPI (Invitrogen) and analyzed by confocal laser scanning microscopy (Olympus IX81).
Real-Time PCR
Total RNA was isolated with Trizol reagent (Life Technologies) and used for cDNA synthesis with a reverse transcription kit (Roche). Real-time PCR was performed with SYBR green select Master Mix on a Stepone Plus Real-Time PCR system (Life Technologies). The expression of individual genes was calculated by a ΔΔCT method and normalized to the expression of β-actin.
Cytokine measurement
Cytokine release from tumor cells or splenocytes were measured by using Bio-Plex Pro Mouse cytokine 23-plex assay (Bio-Rad) or individual cytokine ELISA kits from Ebioscience or PBL Assay Science.
ShRNA-mediated gene knockdown
The pLKO.1 lentiviral vectors encoding shRNAs were described previously (Zhang et al., 2011) or purchased from Sigma. To produce lentiviral particles, the lentiviral vectors were transfected into HEK293T cells along with packaging plasmids psPAX2 and pMD2. Dendritic cells were infected with the lentiviruses as previously described (Zhang et al., 2011), and knockdown efficiency was confirmed by Real-time PCR.
Tumor studies
TUBO cells (1×106 cells/mouse) suspended in cold Matrigel™/PBS (1:1) were implanted into mammary gland fat pads of female Balb/c mice (age of 6–10 weeks). On day 4 after tumor inoculation (tumor reached palpable size), animals were randomly divided into groups (eight mice per group) and received i.v. injections of treatments, and the mice were monitored for tumor size and mouse survival. Tumor volume was recorded as Width X Length2/2. For intra-tumor cytokine and protein analysis, mice were sacrificed on day 14 after tumor inoculation, and the tumors were harvested for immune cell typing, RNA and protein analysis by flow cytometry, qPCR and Western blot analysis, respectively. In the tumor prevention experiment on Balb-neuT mice, the groups of mice (five mice per group) were treated by i.v. injection of vaccines at age of 6 and 8 weeks old, and then the tumor incidence, tumor multiplicity, and mouse survival was monitored for 30 weeks. In the depletion experiments, anti-CD4 (GK1.5) and anti-CD8 (2.43) mAbs were used for in vivo depletion of T-cell subsets. Clodrosomes (Encapsula NanoSciences LLC) was used to deplete macrophages. Animals were injected i.p. with 200 μg of anti-CD4 and anti-CD8 mAb or 200 μl of clodrosomes twice per week for 3 weeks, starting 1 week before inoculation of the tumor cells (0.5×106 cells per mouse).
Statistical analyses
Statistical significance was determined by Student’s t test to evaluate the P value. The relationship between two variables was tested using regression analysis, and P < 0.05 was considered significant. Survival analysis was analyzed by the Log-rank test to compare Kaplan-Meier survival curves.
Supplementary Material
Acknowledgments
We thank Drs. K. L. Rock at University of Massachusetts, C.V. Harding at Case Western Reserve University, W. Wei at Wayne State University, G. Forni at University of Torin, Italy, and L. Pease at Mayo Clinic for providing cell lines and transgenic mice, Mr. Matthew Landry for providing graphic support, and Advanced Cellular and Tissue Microscope and Flow Cytometry Core Facilities at Houston Methodist Research Institute for technical support. The authors acknowledge financial support from the following sources: Department of Defense grants W81XWH-09-1-0212, W81XWH-12-1-0414, National Institute of Health grants U54CA143837 and U54CA151668, the CPRIT grant RP121071 from the state of Texas, and the Ernest Cockrell Jr. Distinguished Endowed Chair.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ambrosino E, Spadaro M, Iezzi M, Curcio C, Forni G, Musiani P, Wei WZ, Cavallo F. Immunosurveillance of Erbb2 carcinogenesis in transgenic mice is concealed by a dominant regulatory T-cell self-tolerance. Cancer Res. 2006;66:7734–7740. doi: 10.1158/0008-5472.CAN-06-1432. [DOI] [PubMed] [Google Scholar]
- Assudani D, Cho HI, DeVito N, Bradley N, Celis E. In vivo expansion, persistence, and function of peptide vaccine-induced CD8 T cells occur independently of CD4 T cells. Cancer Res. 2008;68:9892–9899. doi: 10.1158/0008-5472.CAN-08-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210:2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol. 2008;9:558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave B, Granados-Principal S, Zhu R, Benz S, Rabizadeh S, Soon-Shiong P, Yu KD, Shao Z, Li X, Gilcrease M, et al. Targeting RPL39 and MLF2 reduces tumor initiation and metastasis in breast cancer by inhibiting nitric oxide synthase signaling. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1320769111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol. 2009;27:83–117. doi: 10.1146/annurev.immunol.021908.132544. [DOI] [PubMed] [Google Scholar]
- Duggan JM, You D, Cleaver JO, Larson DT, Garza RJ, Guzman Pruneda FA, Tuvim MJ, Zhang J, Dickey BF, Evans SE. Synergistic interactions of TLR2/6 and TLR9 induce a high level of resistance to lung infection in mice. J Immunol. 2011;186:5916–5926. doi: 10.4049/jimmunol.1002122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach TL, Ng G, Hari A, Desrosiers MD, Zhang P, Ward SM, Seamone ME, Vilaysane A, Mucsi AD, Fong Y, et al. Alum interaction with dendritic cell membrane lipids is essential for its adjuvanticity. Nat Med. 2011;17:479–487. doi: 10.1038/nm.2306. [DOI] [PubMed] [Google Scholar]
- Fric J, Zelante T, Ricciardi-Castagnoli P. Phagocytosis of Particulate Antigens – All Roads Lead to Calcineurin/NFAT Signaling Pathway. Front Immunol. 2014;4:513. doi: 10.3389/fimmu.2013.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013a;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, Spranger S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013b;25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol. 2012;13:737–743. doi: 10.1038/ni.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Kleindienst P, Brocker T. Endogenous dendritic cells are required for amplification of T cell responses induced by dendritic cell vaccines in vivo. J Immunol. 2003;170:2817–2823. doi: 10.4049/jimmunol.170.6.2817. [DOI] [PubMed] [Google Scholar]
- Lapteva N, Huang XF. CCL5 as an adjuvant for cancer immunotherapy. Expert Opin Biol Ther. 2010;10:725–733. doi: 10.1517/14712591003657128. [DOI] [PubMed] [Google Scholar]
- Le Bon A, Tough DF. Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev. 2008;19:33–40. doi: 10.1016/j.cytogfr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Lucchini F, Sacco MG, Hu N, Villa A, Brown J, Cesano L, Mangiarini L, Rindi G, Kindl S, Sessa F, et al. Early and multifocal tumors in breast, salivary, harderian and epididymal tissues developed in MMTV-Neu transgenic mice. Cancer Lett. 1992;64:203–209. doi: 10.1016/0304-3835(92)90044-v. [DOI] [PubMed] [Google Scholar]
- Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123:4980. doi: 10.1172/JCI64859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millien VO, Lu W, Shaw J, Yuan X, Mak G, Roberts L, Song LZ, Knight JM, Creighton CJ, Luong A, et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science. 2013;341:792–796. doi: 10.1126/science.1240342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–6173. [Google Scholar]
- Mukai Y, Yoshinaga T, Yoshikawa M, Matsuo K, Yoshikawa T, Niki K, Yoshioka Y, Okada N, Nakagawa S. Induction of endoplasmic reticulum-endosome fusion for antigen cross-presentation induced by poly (gamma-glutamic acid) nanoparticles. J Immunol. 2011;187:6249–6255. doi: 10.4049/jimmunol.1001093. [DOI] [PubMed] [Google Scholar]
- Nagata Y, Furugen R, Hiasa A, Ikeda H, Ohta N, Furukawa K, Nakamura H, Kanematsu T, Shiku H. Peptides derived from a wild-type murine proto-oncogene c-erbB-2/HER2/neu can induce CTL and tumor suppression in syngeneic hosts. J Immunol. 1997;159:1336–1343. [PubMed] [Google Scholar]
- Nava-Parada P, Forni G, Knutson KL, Pease LR, Celis E. Peptide vaccine given with a Toll-like receptor agonist is effective for the treatment and prevention of spontaneous breast tumors. Cancer Res. 2007;67:1326–1334. doi: 10.1158/0008-5472.CAN-06-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D, Gommerman JL. The Regulation of Immune Responses by DC Derived Type I IFN. Front Immunol. 2013;4:94. doi: 10.3389/fimmu.2013.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Uppaluri R, Andrews DM, Ngiow SF, Teng MW, et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med. 2012;209:1869–1882. doi: 10.1084/jem.20112738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulis LE, Mandal S, Kreutz M, Figdor CG. Dendritic cell-based nanovaccines for cancer immunotherapy. Curr Opin Immunol. 2013;25:389–395. doi: 10.1016/j.coi.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015 doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Knobeloch KP. Type I interferons as ambiguous modulators of chronic inflammation in the central nervous system. Front Immunol. 2012;3:67. doi: 10.3389/fimmu.2012.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11:805–812. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O’Hagan DT, Petrilli V, Tschopp J, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870–875. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Rodriguez-Aguayo C, Xu R, Gonzalez-Villasana V, Mai J, Huang Y, Zhang G, Guo X, Bai L, Qin G, et al. Enhancing chemotherapy response with sustained EphA2 silencing using multistage vector delivery. Clin Cancer Res. 2013a;19:1806–1815. doi: 10.1158/1078-0432.CCR-12-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Xu R, Mai J, Kim HC, Guo X, Qin G, Yang Y, Wolfram J, Mu C, Xia X, et al. High capacity nanoporous silicon carrier for systemic delivery of gene silencing therapeutics. ACS Nano. 2013b;7:9867–9880. doi: 10.1021/nn4035316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014 doi: 10.1038/nm.3708. advance online publication. [DOI] [PubMed] [Google Scholar]
- Spaner DE, Foley R, Galipeau J, Bramson J. Obstacles to effective Toll-like receptor agonist therapy for hematologic malignancies. Oncogene. 2008;27:208–217. doi: 10.1038/sj.onc.1210905. [DOI] [PubMed] [Google Scholar]
- Summers deLuca L, Ng D, Gao Y, Wortzman ME, Watts TH, Gommerman JL. LTbetaR signaling in dendritic cells induces a type I IFN response that is required for optimal clonal expansion of CD8+ T cells. Proc Natl Acad Sci U S A. 2011;108:2046–2051. doi: 10.1073/pnas.1014188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, Han HD, Shahzad MM, Liu X, Bhavane R, et al. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70:3687–3696. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Xu R, Huang Y, Mai J, Zhang G, Guo X, Xia X, Koay EJ, Qin G, Erm DR, Li Q, et al. Multistage Vectored siRNA Targeting Ataxia-Telangiectasia Mutated for Breast Cancer Therapy. Small. 2013;9:1799–1808. doi: 10.1002/smll.201201510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, Fu YX. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell. 2014;25:37–48. doi: 10.1016/j.ccr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdall AW, Drutman SB, Jinwala F, Bahjat KS, Bhardwaj N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS One. 2010;5:e11144. doi: 10.1371/journal.pone.0011144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu YJ. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011;34:866–878. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.