

Abstract
Overexposure to the essential metal manganese (Mn) can result in an irreversible condition known as manganism that shares similar pathophysiology with Parkinson’s disease (PD), including dopaminergic (DAergic) cell loss that leads to motor and cognitive impairments. However, the mechanisms behind this neurotoxicity and its relationship with PD remain unclear. Many genes confer risk for autosomal recessive, early-onset PD, including the parkin/PARK2 gene that encodes for the E3 ubiquitin ligase Parkin. Using Caenorhabditis elegans (C. elegans) as an invertebrate model that conserves the DAergic system, we previously reported significantly increased Mn accumulation in pdr-1/parkin mutants compared to wildtype (WT) animals. For the current study, we hypothesize that this enhanced accumulation is due to alterations in Mn transport in the pdr-1 mutants. While no change in mRNA expression of the major Mn importer proteins (smf-1-3) was found in pdr-1 mutants, significant downregulation in mRNA levels of the putative Mn exporter ferroportin (fpn-1.1) was observed. Using a strain overexpressing fpn-1.1 in worms lacking pdr-1, we show evidence for attenuation of several endpoints of Mn-induced toxicity, including survival, metal accumulation, mitochondrial copy number and DAergic integrity, compared to pdr-1 mutants alone. These changes suggest a novel role of pdr-1 in modulating Mn export through altered transporter expression, and provides further support of metal dyshomeostasis as a component of Parkinsonism pathophysiology.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, with a typical age of onset around 60 years of age1. This debilitating disease is characterized by selective dopaminergic (DAergic) cell loss in the substantia nigra pars compacta (SNpc) region of the brain. Hallmark symptoms of PD include bradykinesia, rigidity, tremors and postural instability that are often preceded by emotional instability and cognitive dysfunction. Unfortunately, PD is a progressive and irreversible condition2. Current treatments do not target the molecular origins of PD, warranting further examination into the mechanisms behind its pathophysiology.
Though PD is mostly idiopathic in its etiology, mutations in several genes have been connected to the disease2. For example, homozygous mutations in the PARK2/parkin gene are responsible for nearly 50% of an autosomal recessive, early-onset form of PD3. This gene encodes for an E3 ubiquitin ligase involved in the ubiquitin proteasome system (UPS) that targets substrates for degradation. Mutations in this gene result in impaired ligase activity and substrate binding that can lead to increased protein aggregation4. Parkin knockout animal models show a variety of PD-associated phenotypes, including hypokinetic deficits, DAergic cell loss and increased extracellular dopamine (DA) in the striatum5, 6. Parkin has also been more recently identified as a key regulator of mitophagy, an intracellular autophagic process designed to eliminate damaged mitochondria from the cell7.
Despite the known genetic associations, familial cases often present with heterogeneity in their age-of-onset and symptomatology, in addition to nearly 90% of all PD cases manifesting without genetic disturbances8. The idiopathic component of the disease suggests a contribution of environmental risk factors in the development of PD. One such factor is the heavy metal manganese (Mn), an essential trace element found in many food sources consumed daily by humans. Mn serves as a necessary cofactor for enzymes involved in several critical processes, including reproduction, metabolism, development, and antioxidant responses9. While deficiency is a rare concern, the essentiality of Mn is mirrored by its neurotoxicity upon overexposure. Mn poisoning, or manganism, typically occurs from occupational exposures in industrial settings, such as in welding, where Mn-containing fumes and/or products are abundant10, 11. Mn is also found as an antiknock agent methylcyclopentadienyl manganese tricarbonyl (MMT) in gasoline, but limited studies currently exist on the impact of Mn release from combustion on general human health12, 13. Certain pesticides also contain Mn, making surface runoff from these agricultural uses an additional source of overexposure1. Moreover, Mn toxicity can also affect other susceptible populations, including ill neonates receiving total parenteral nutrition (TPN) that is supplemented with a trace element solution containing Mn. Intravenous TPN administration bypasses the gastrointestinal regulation of Mn absorption, resulting in 100% Mn retention9. Another population at risk of Mn poisoning includes patients suffering from hepatic encephalopathy and/or liver failure, as Mn is excreted from the body through the biliary system14, 15. On the other hand, individuals with iron (Fe) deficiency (e.g., iron deficiency anaemia), a highly prevalent nutritional condition, are at risk for increased Mn body burdens. As Mn shares similar transport mechanisms with Fe, higher Mn levels are often seen in conditions of low Fe levels16.
Tight regulation through an intricate system of transport mechanisms helps maintain proper Mn homeostasis in cells. The divalent metal transporter 1 (DMT1) represents the primary mode of divalent Mn import17. However, Mn efflux remains less understood than Mn import. We previously identified ferroportin (FPN), a well-known iron (Fe) exporter, as facilitating Mn export in cells and mice18. We have previously identified and characterized components of the Mn transport system in the Caenorhabditis elegans (C. elegans) model system. This nematode provides an attractive, alternative system that has a rapid life cycle, short lifespan, and large brood size. Additionally, the well-characterized genome allows for the utilization of various genetic mutants for studies. This nematode also conserves all necessary components of a fully functional DAergic system, allowing for the study of the effects of PD-associated genetic loss on the DAergic system. Our previous studies have identified SMF-1, SMF-2 and SMF-3 as the C. elegans homologs for DMT1, with SMF3 acting as the most DMT1-like homolog in its necessity to regulate Mn uptake19. Thus far, these proteins are the only known Mn importers in the worm. Furthermore, the worm contains 3 homologs for FPN: FPN-1.1, FPN-1.2 and FPN-1.320. As of now, FPN-1.1 is the only known protein that conserves Fe efflux in C. elegans21.
The overlap in sites of damage and similar symptomatology between manganism and Parkinsonism has warranted investigations into potential gene-environment interactions. For example, parkin has been shown to selectively protect against Mn-induced DAergic cell death in vitro22, while rats exposed to Mn-containing welding fumes show increased Parkin protein levels23. Our previous study using C. elegans found significantly enhanced Mn accumulation in pdr-1 (parkin homolog) knockout worms compared to WT worms24. With the aforementioned relationships between PD-associated genes and Mn toxicity, we hypothesized that this enhancement is due to an alteration in Mn homeostasis, at the level of transport, in the background of pdr-1 loss. In the present study, while no significant change in mRNA expression of importers was seen, we found a downregulation of fpn-1.1 mRNA. Upon overexpression of this exporter in pdr-1 mutants, we found decreased metal levels that were associated with improved survival and DA-dependent behaviour. Together, our results provide further support for altered metal homeostasis as a component of the pathophysiology seen in Parkinsonism.
Experimental Procedures
Plasmid Constructs
Full-length wildtype (WT) fpn-1.1 with C-terminal FLAG tag was PCR amplified using primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTACATGGCTTGGTTATCCGGAAAAG-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTCACTTGTCATCGTCGTCCTTGTAGTCTTCAAAAGTTGGCGAATCCAAC-3′ from cDNA library which was converted from total RNAs isolated from N2 worms (see below). The plasmid was created with Gateway recombinational cloning (Invitrogen). The above PCR product was initially recombined with the pDONR221 vector to create the pENTRY clone. Next, the fpn-1.1 pENTRY construct was recombined into pDEST-sur-5 vector25, under the promoter of the acetoacetyl-coenzyme A synthetase (sur-5) gene. This plasmid was then used to create transgenic worms.
C. elegans Strains and Strain Construction
C. elegans strains were handled and maintained at 20°C as previously described26. Strains used were: N2, wildtype (Caenorhabditis Genetics Center, CGC) and VC1024, pdr-1(gk448) III (CGC). The MAB326 strain was created by microinjecting Psur-5::fpn-1.1 with pBCN27-R4R3 (Prpl-28::PuroR, Addgene) and Pmyo-3::mCherry (a gift from Dr. David Miller) into VC1024 strain. Over three stable lines were generated and analysed. Representative lines were selectively integrated by using gamma irradiation with an energy setting of 3600 rad.
Preparation of Manganese Chloride (MnCl2)
2 M MnCl2 (> 99.995% purity) (Sigma-Aldrich) stock solutions were prepared in 85 mM NaCl. To prevent oxidation, fresh working solutions were prepared shortly before each experiment. The range of concentrations used in all experiments are based on Mn dose-response curves recently published by our laboratory24.
Mn-Induced Treatments and Lethality Assay
2500 synchronized L1 worms per group were acutely treated with MnCl2 (0–100 mM) in siliconized tubes for 30 minutes. Worms were then pelleted by centrifugation at 7000 rpm for 3 minutes and washed four times with 85 mM NaCl. 30–50 worms were then pre-counted and transferred to OP50-seeded NGM plates in triplicate and blinded. 48 hours post-treatment, the total number of surviving worms was scored as a percentage of the original plated worm count.
TaqMan Gene Expression Assay
Total RNA was isolated via the Trizol method. Briefly, following Mn treatment, 1 mL of Trizol (Life Technologies) was added to each tube containing 20,000 worms resuspended in 100 μl 85 mM NaCl, followed by three cycles of freezing in liquid nitrogen and thawing at 37°C. 200 μL of chloroform was then added to each tube, followed by precipitation using isopropanol and washing with 75% ethanol. Following isolation, 1 μg total RNA was used for cDNA synthesis using the High Capacity cDNA Reverse Transcription Kit (Life Technologies), per manufacturer’s instructions. cDNA samples were stored at 4°C. Quantitative real-time PCR (BioRad CFX96) was conducted in duplicate wells using TaqMan Gene Expression Assay probes (Life Technologies) for each gene, using the gpd-3 (gapdh homolog) housekeeping gene for normalization after determining the fold difference using the comparative 2−ΔΔCt method27. The following probes were used: smf-1 (Assay ID: Ce02496635_g1); smf-2 (Assay ID: Ce02496634_g1); smf-3 (Assay ID: Ce02461545_g1); fpn-1.1 (Assay ID: Ce02414545_m1); and gpd-3 (Assay ID: Ce02616909_gH).
Metal Quantification
Total intraworm metal content was quantified using inductively coupled plasma mass spectrometry (ICP-MS), as previously described24. Briefly, 50,000 synchronized L1 worms were acutely treated with MnCl2. Worms were then pelleted, washed five times with 85 mM NaCl and re-suspended in 1 mL 85 mM NaCl supplemented with 1% protease inhibitor. After sonication, an aliquot was taken for protein normalization using the bicinchoninic acid (BCA) assay kit (Thermo Scientific). Subsequently, the suspension was mixed again, evaporated, and incubated with the ashing mixture (65%HNO3/30%H2O2 (1/1) (both Merck)) at 95 °C for at least 12 h. After dilution of the ash with bidistilled water, metal levels were determined by ICP-MS.
Relative Mitochondrial DNA Copy Number Quantification
Relative mitochondrial DNA copy number was quantified using qPCR methods as previously described28, with slight modifications. Briefly, 1,000 synchronized L1 worms were treated with MnCl2 for 30 minutes, following by several washes. Total genomic DNA was then isolated using a 1X PCR buffer containing 0.1% Proteinase K, and subjected to the following lysis protocol in a thermal cycler (BioRad T100): 65°C for 90 minutes, 95°C for 15 minutes, and then hold at 4°C. Following lysis, DNA was diluted to 3 ng/μl, and real time PCR (BioRad CFX96) using SYBR Green (BioRad) was performed in triplicate with the following primers: nd-1 for mtDNA (forward primer sequence: 5′-AGCGTCATTTATTGGGAAGAAGAC-3′; reverse primer sequence: 5′-AAGCTTGTGCTAATCCCATAAATGT-3′) and cox-4 for nuclear DNA (forward primer sequence: 5′-GCCGACTGGAAGAACTTGTC-3′; reverse primer sequence: 5′-GCGGAGATCACC TTCCAGTA-3′). The PCR reaction consisted of: 2μL of template DNA, 1μL each of mtDNA and nucDNA primer pairs (400nM final concentration each), 12.5μL SYBR Green PCR Master Mix and 8.5μL H2O. The following protocol was used: 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds and 62°C for 60 seconds. The mitochondrial DNA content relative to nuclear DNA was calculated using the following equations: ΔCT = (nucDNA CT – mtDNA CT), where relative mitochondrial DNA content = 2 × 2ΔCT.
Glutathione Quantification
Total intracellular glutathione levels (reduced and oxidized GSH) have been determined using the “enzymatic recycling assay”, as previously described29. Briefly, whole worm extracts were prepared out of 40,000 L1 worms acutely exposed to MnCl2. This was followed by washes with 85 mM NaCl and sonication of the pellet in 0.12 mL ice-cold extraction buffer (1% Triton X-100, 0.6% sulfosalicylic acid) and 1% protease inhibitor in KPE buffer (0.1 M potassium phosphate buffer, 5 mM EDTA). After centrifugation at 10,000 rpm for 10 minutes at 4°C, the supernatant was collected, with an aliquot reserved for protein normalization using the BCA assay. Total intracellular GSH was quantified by measuring the change in absorbance per minute at 412 nm by a microplate reader (FLUOstar Optima microplate reader, BMG Labtechnologies) after reduction of 5,5′-dithio-2-nitrobenzoic acid (DTNB, Sigma-Aldrich). Hydrogen peroxide was used as a positive control.
Basal Slowing Response Assay
This assay of dopaminergic integrity was performed as previously described30, with slight modifications. Briefly, 2500 synchronized L1 worms were acutely treated in siliconized tubes with MnCl2 for 30 minutes. Following washes with 85 mM NaCl, treated worms were transferred to seeded NGM plates. 48 hours after treatment, 60 mm NGM plates with seeded with bacteria spread in a ring (inner diameter of ~1 cm and an outer diameter of ~3.5 cm) in the center of the plate. Two seeded and two unseeded plates per group were kept at 37°C overnight, and allowed to cool to room temperature before use. Once Mn-treated animals reached the young adult stage, animals were washed at least two times with S basal buffer and then transferred to the central clear zone of the ring-shaped bacterial lawn (5–10 worms per plate) in a drop of S basal buffer that was delicately absorbed from the plate using a Kimwipe. After a five-minute acclimation period, the number of body bends in a 20-second interval was scored for each worm on the plate. Data are presented as the change (Δ) in body bends per 20-second interval between worms transferred to unseeded plates and those with bacterial rings. Worms lacking cat-2 (the homolog for tyrosine hydroxylase) were used as a positive control, as these worms are impaired in bacterial mechanosensation30. General locomotion was assessed using the number of body bends/20 seconds of the group transferred to unseeded plates.
Statistics
Dose-response lethality curves and all histograms were generated using GraphPad Prism (GraphPad Software Inc.). A sigmoidal dose-response model with a top constraint at 100% was used to draw the lethality curves and determine the respective LD50 values, followed by a one-way ANOVA with a Dunnett post-hoc test to compare all strains to their respective control strains. Two-way ANOVAs were performed on TaqMan gene expression, metal content, total GSH, relative mtDNA copy number and basal slowing response data, followed by Bonferroni’s multiple comparison post-hoc tests.
Results
pdr-1 mutants show alterations in mRNA expression of Mn exporter-, but not importer-related genes
We previously reported a statistically significant increase in Mn accumulation in pdr-1 mutants vs. WT worms24. To test whether this enhancement was due to a change in transcription of Mn importer and/or exporter genes, we performed quantitative reverse transcription PCR (qRT-PCR) to examine smf-1,2,3 (Fig. 1A–1C) and fpn-1.1 gene expression (Fig. 1D), respectively, following acute Mn exposure. Two-way ANOVA analysis showed no overall effect of Mn treatment on transcription of any of the genes tested. However, while pdr-1 mutants showed no significant changes in smf-1,2,3 (the importers) mRNA expression (Fig. 1A–1C), a significant genotype difference (p<0.0001) was noted in fpn-1.1 (the exporter) between pdr-1 mutants and WT animals. Post-hoc analysis revealed a significant fpn-1.1 downregulation at 0 and 2.5 mM MnCl2 (Fig 1D).
Fig. 1. pdr-1 mutants show alterations in mRNA expression of Mn exporter, but not importer, genes.
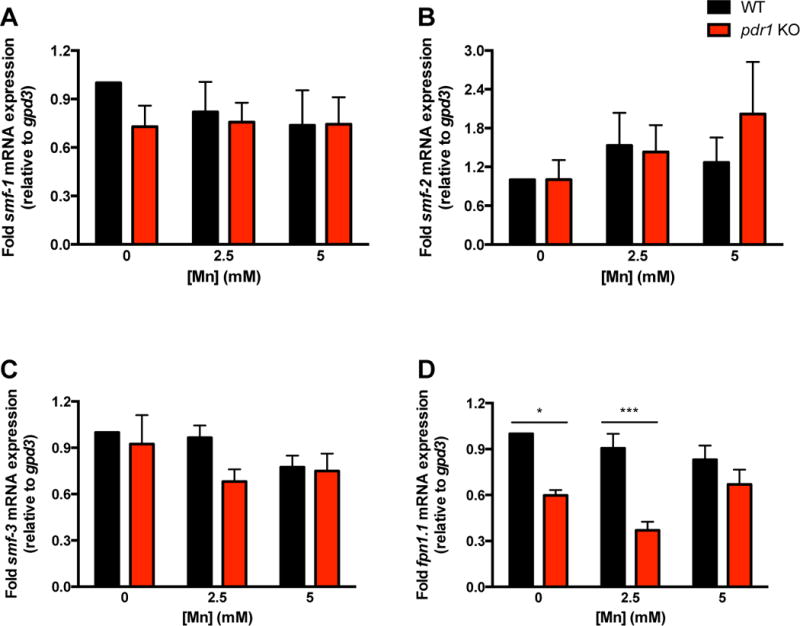
(A-D) smf-1,2,3 and fpn-1.1 mRNA expression after an acute, 30 min treatment of L1 worms with 0, 2.5 and 5 mM MnCl2. Relative gene expression was determined by qRT-PCR. (A) smf-1 mRNA expression in N2 (WT) and pdr-1 KO animals. (B) smf-2 mRNA expression in N2 (WT) and pdr-1 KO animals. (C) smf-3 mRNA expression in N2 (WT) and pdr-1 KO animals. (D) fpn-1.1 mRNA expression in N2 (WT) and pdr-1 KO animals. (A-D) Data are expressed as mean values + SEM of at least five independent experiments in duplicates normalized to the untreated wildtype and relative to gpd3 mRNA. Statistical analysis by two-way ANOVA: (A) interaction, ns; genotype, ns; concentration, ns; (B) interaction, ns; genotype, ns; concentration, ns; (C) interaction, ns; genotype, ns; concentration, ns; (D) interaction, ns (trend level, p=0.0639); genotype, p<0.0001; concentration, ns. *p < 0.05, ***p < 0.001 vs. respective wildtype worms.
Overexpression of fpn-1.1 in pdr-1 mutants suppresses Mn-induced lethality
In addition to enhanced Mn accumulation, pdr-1 mutants showed a leftward shift in the Mn dose-response survival curve, with WT worms exhibiting a LD50 of 10.43 mM24. To determine whether downregulation of fpn-1.1 may have played a role in exacerbating Mn-induced lethality of pdr-1 mutants, we overexpressed fpn-1.1 in the pdr-1 mutant background. Upon Mn exposure, pdr-1 mutants overexpressing fpn-1.1 (pdr-1 KO; fpn-1.1 OVR) exhibited a rightward shift in the dose-response curve of compared to pdr-1 mutants alone (Fig. 2A). The LD50 of pdr-1 KO; fpn-1.1 OVR animals (10.84 mM) relatively normalized to previously published WT levels, while pdr-1 mutants alone show a LD50 of 7.416 mM (Table, 2B). Two-way ANOVA analysis showed a significant interaction effect (p=0.0064) between both genotype and treatment (p<0.0001).
Fig. 2. Overexpression of fpn-1.1 in pdr-1 mutants rescues Mn-induced lethality.
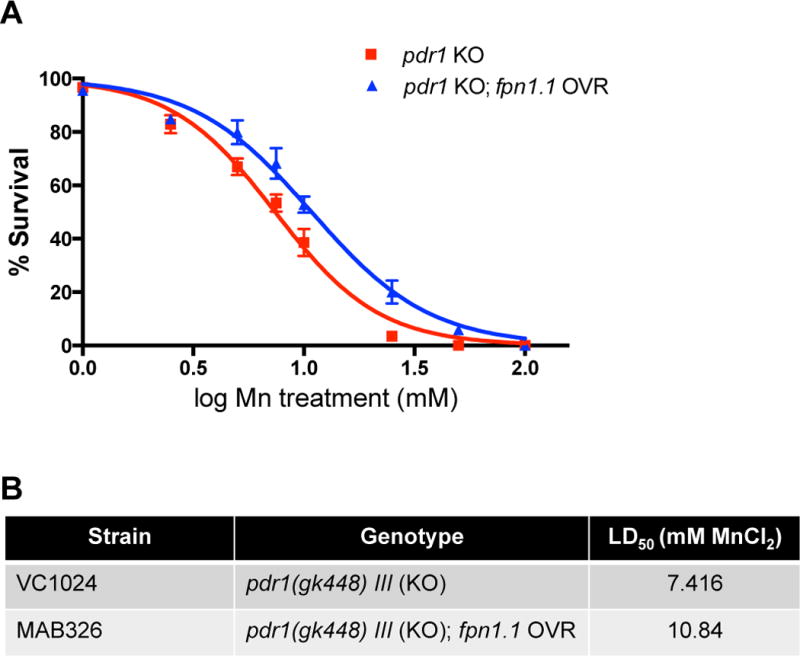
(A,B) Dose-response survival curves following acute Mn exposure. All values were compared to untreated worms set to 100% survival and plotted against the logarithmic scale of the used Mn concentrations. (A) pdr-1 KO animals and pdr-1 mutants overexpressing fpn-1.1 (pdr-1 KO; fpn-1.1 OVR) were treated at the L1 stage for 30 min with increasing MnCl2 concentrations. (B) The respective LD50 concentrations (mM MnCl2) for both genotypes. Data are expressed as mean values + SEM from at least five independent experiments. Statistical analysis by two-way ANOVA: interaction, p=0.0064; genotype, p<0.0001; concentration, p<0.0001.
Overexpression of fpn-1.1 in pdr-1 mutants decreases levels of pro-oxidant metals
Upon noting the improved survival in pdr-1 KO; fpn-1.1 OVR animals, we hypothesized that this attenuation in Mn-induced toxicity is associated with a decrease in redox active metal accumulation. Using inductively coupled plasma mass spectrometry (ICP-MS), we measured intraworm concentrations of various metals, including Mn, iron (Fe), zinc (Zn) and copper (Cu) following acute Mn exposure. To our surprise, Mn levels remained relatively similar between strains, though two-way ANOVA analysis revealed a significant treatment effect (p=0.0165). However, endogenous Fe levels were significantly decreased in pdr-1 KO; fpn-1.1 OVR animals compared to pdr-1 KOs alone revealed as a significant genotype effect by ANOVA (Fig. 3B, p=0.0092). No significant changes were seen in Zn levels (Fig. 3C). However, similar to Fe, Cu levels were significantly decreased in pdr-1 KO; fpn-1.1 OVR animals compared to pdr-1 KOs (Fig. 3D, p=0.0256), with no post-hoc level differences. In summary, Mn levels stayed relatively the same, while Fe and Cu were both significantly decreased in pdr-1 KO; fpn-1.1 OVR animals. These results indicate that the improved survival is probably due to decreased levels of Fe and Cu, and suggests that fpn-1.1 may prefer Fe and Cu as substrates over Mn.
Fig. 3. Overexpression of fpn-1.1 in pdr-1 mutants decreases levels of highly pro-oxidant metals.
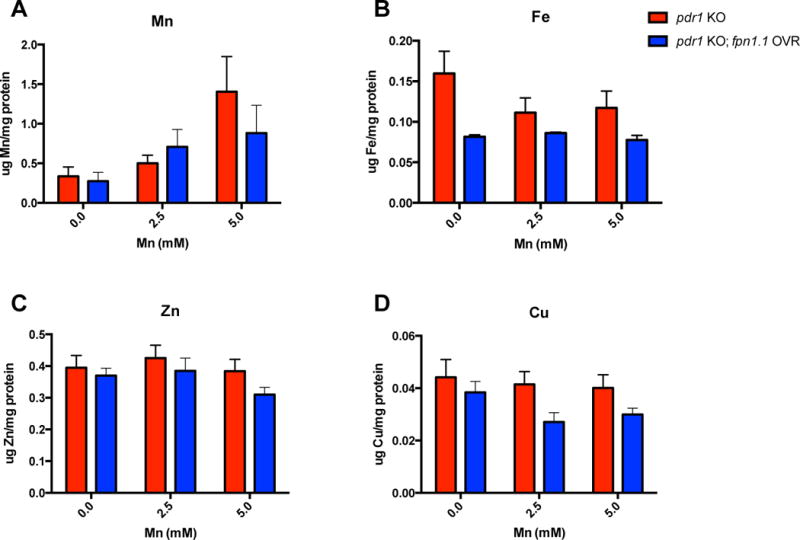
(A-D) Intraworm metal concentrations following acute, 30 min MnCl2 treatment (0, 2.5 and 5 mM) at the L1 stage, as quantified by ICP-MS/MS. (A) Mn content (μg Mn/mg protein) in pdr-1 KO and pdr-1 KO; fpn-1.1 OVR animals. (B) Iron (Fe) content (μg Fe/mg protein) in pdr-1 KO and pdr-1 KO; fpn-1.1 OVR animals. (C) Zinc (Zn) content (μg Zn/mg protein) in pdr-1 KO and pdr-1 KO; fpn-1.1 OVR animals. (D) Copper (Cu) content (μg Cu/mg protein) in pdr-1 KO and pdr-1 KO; fpn-1.1 OVR animals. (A-D) Data are expressed as mean values + SEM from at least six independent experiments and normalized to total protein content. Statistical analysis by two-way ANOVA: (A) interaction, ns; genotype, ns; concentration, p=0.0165; (B) interaction, ns; genotype, p=0.0092; concentration, ns; (C) interaction, ns; genotype, ns; concentration, ns; (D) interaction, ns; genotype, p=0.0256; concentration, ns.
Overexpression of fpn-1.1 in pdr-1 mutants improves mitochondrial integrity and antioxidant response
Increased Mn levels in pdr-1 KOs (vs. WT animals) have been noted concurrently with significantly increased basal levels of reactive oxygen species (ROS) and depleted basal levels of total glutathione (GSH)24, suggesting an overall exacerbated environment of oxidative stress in pdr-1 KO animals. Therefore, we next sought to determine whether the significant decrease in Fe and Cu levels (Fig. 3) and improvement in survival of pdr-1 KO;fpn-1.1 OVR animals (Fig. 2) were associated with improved defence mechanisms against oxidative stress. This was investigated using two measures: relative mitochondrial DNA (mtDNA) copy number and total GSH levels. Alterations in mtDNA copy number have been associated with aging and degenerative processes30. Moreover, parkin has been shown to regulate mitochondrial turnover to maintain proper mitochondrial integrity7. Using a quantitative PCR (qPCR) technique, we found pdr-1 KO animals had a significantly elevated mtDNA copy number relative to WT animals, whereas pdr-1 KO;fpn-1.1 OVR animals exhibited levels similar to WT animals (Fig. 4A); two-way ANOVA analysis reveals a significant genotype effect (p=0.0116), though significance was not reached at the post-hoc level. Moreover, we previously published the basal depletion of total GSH in pdr-1 KOs compared to WT controls. Given the reversal of increased mtDNA copy number in pdr-1 KO;fpn-1.1 OVR animals, we examined whether there was a similar rescue of GSH depletion. While statistical significance wasn’t reached, there was a slight increase in GSH levels in pdr-1 KO;fpn-1.1 OVR animals relative to pdr-1 KOs (Fig. 4C, p=0.09). In both measures, Mn treatment itself did not significantly affect the outcomes.
Fig. 4. Overexpression of fpn-1.1 in pdr-1 mutants improves mitochondrial integrity and antioxidant response.
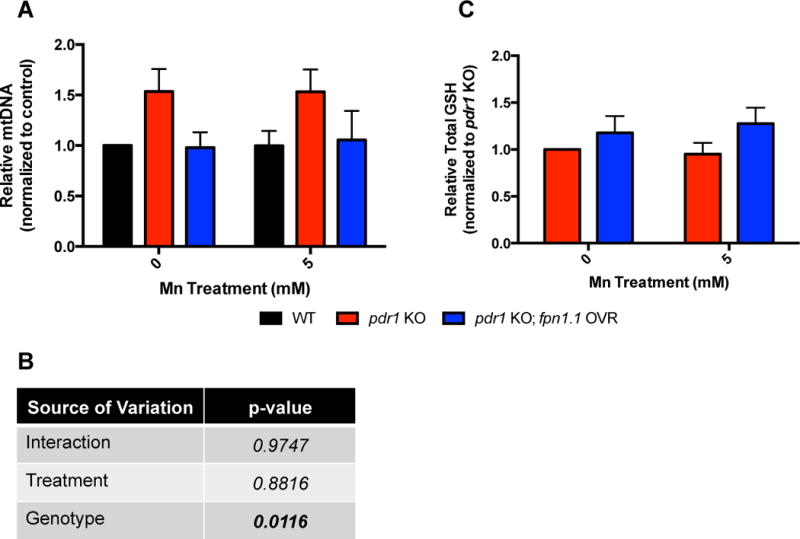
(A) Relative mitochondrial DNA (mtDNA) copy number in pdr-1 KO and pdr-1 KO; fpn-1.1 OVR animals following an acute, 30 min treatment with 0 and 5 mM MnCl2. Relative gene expression was determined by qPCR. (B) Two-way ANOVA analysis of data in 4A showing genotype significance in mtDNA copy number. (C) Total glutathione (GSH) levels of pdr-1 KO and pdr-1 KO; fpn-1.1 OVR animals following an acute, 30 min treatment with 0 and 5 mM MnCl2. (A) Relative mtDNA copy number is expressed as a ratio of nd-1 (mtDNA marker) to cox-4 (nuclear DNA marker). Data are expressed as mean values + SEM of at least five independent experiments in duplicates normalized to the untreated N2 wildtype values. (C) Data are expressed as mean values + SEM of at least five independent experiments in duplicates, normalized to total protein content and relative to untreated pdr-1 KO values. Statistical analysis by two-way ANOVA: (A) interaction, ns; genotype, p=0.0116; concentration, ns; (B) interaction, ns; genotype, ns (trend level, p=0.09); concentration, ns.
Overexpression of fpn-1.1 in pdr-1 mutants improves the DA-dependent basal slowing response
Loss of parkin is connected to PD-associated DAergic neurodegeneration, and we previously published similar results of pdr-1 KOs showing increased DAergic neurodegeneration vs. WT worms with fluorescence microscopy24. Consequently, we investigated whether the visual effects of DAergic neurodegeneration persisted to alter a behavioral outcome of DAergic integrity. The basal slowing response is a DA-dependent behavior that affects the mechanosensation needed for proper food sensing in C. elegans, as worms slow their movement when encountering a bacterial lawn. Worms lacking cat-2, the homolog for tyrosine hydroxylase, are defective in this response from the loss of dopamine synthesis and do not slow down31. Thus, the changes (Δ) in number of body bends between plates with and without bacteria reflect the integrity of DAergic neurons. Using this paradigm, pdr-1 KO animals exhibited a significantly defective basal slowing response vs. WT animals (p<0.001) that was analogous to that of cat-2 mutants (Fig. 5). The pdr-1 KO;fpn-1.1 OVR animals showed a partial rescue of the response, without reaching statistical significance. However, in the presence of Mn treatment, pdr-1 KO;fpn-1.1 OVR fully restored the response to WT levels, with the changes (Δ) in number of body bends being significantly higher than pdr-1 KOs alone (p<0.01). To ensure that these effects were not due to general locomotion differences, we compared the number of body bends per group on plates without bacterial lawns; there were no significant differences between all groups (data not shown).
Fig. 5. Overexpression of fpn-1.1 in pdr-1 mutants improves the DA-dependent basal slowing response.
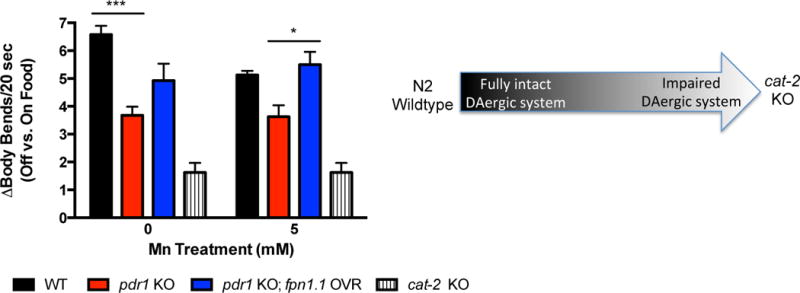
Behavioral data are expressed as the change (Δ) in body bends per 20 seconds between treated (5 mM MnCl2) and untreated WT, pdr-1 KO and pdr-1 KO; fpn-1.1 OVR animals placed on plates without food vs. plates with food. Schematic shows the spectrum of change, with N2 wildtype animals possessing a higher change in body bends (i.e., a fully intact DAergic system) to cat-2 mutants possessing a smaller, almost negligible change in body bends (i.e., an impaired DAergic system). cat-2 KO animals were used as a positive control. Statistical analysis by two-way ANOVA: interaction, ns (trend level, p=0.0872); genotype, p<0.0001; concentration, ns. ***p<0.001 vs. untreated WT, *p<0.05 vs. pdr-1 KO.
Discussion
The relationship between genetic mutations and the contribution of environmental risk factors in the development of PD has yet to be clearly defined. In the present study, the C. elegans model system was utilized to investigate alterations in Mn homeostasis and toxicity in animals lacking pdr-1/parkin, a genetic risk factor for PD. We previously published evidence that animals lacking pdr-1 show high sensitivity to an acute Mn exposure, with decreased survival and significantly elevated Mn accumulation compared to WT animals24. The present study aimed to determine whether the enhanced Mn concentrations were due to altered expression of Mn transporters in C. elegans to affect Mn homeostasis.
Parkin’s role in regulating metal homeostasis has only recently begun to be investigated. Previous in vitro evidence has shown that parkin can modulate levels of the 1B isoform of DMT1 through ubiquitination32. Moreover, Drosophila studies show that both pharmacological (BCS/BPD) or genetic (increased expression of the metal responsive transcription factor 1, MTF-1) chelation of redox-active metals decreases oxidative stress, improves reduced lifespan and normalizes metal concentrations in parkin mutant flies33, 34. Therefore, parkin’s regulation of metal homeostasis and its role as an E3 ligase raise the possibility of parkin-mediated regulation of Mn-responsive proteins. The C. elegans system represents a ideal model to study this possibility, as PDR-1 conserves its ligase activity35, and their genome contains less E3 ligases36 to minimize the possible compensatory mechanisms seen in vertebrate knockout models.
The enhanced Mn accumulation in pdr-1 mutant animals may be a selective phenotype of this particular genetic background. Notably, our previous studies using methylmercury (MeHg) exposure do not show the same accumulation phenotypes in pdr-1 KO’s37. Moreover, Aboud et al. showed increased oxidative stress in response to Mn exposure in neuroprogenitor cells from patients possessing parkin mutations, despite exhibiting reduced Mn accumulation38, which is opposite to our ICP-MS findings. This discrepancy may be due to their human data arising from isolated neuroprogenitors, whereas the current study assesses whole-worm Mn levels. Nonetheless, such studies provide further support of alterations in neuronal Mn biology in the presence of parkin mutations. This may be true for other PD genetic risk factors as well, as recent studies have highlighted the role of another PD-linked gene known as PARK9, which encodes for the P-type ATPase ion pump ATP13A2. Evidence shows that this protein modulates Zn homeostasis39, with previous evidence indicating that this protein can also transport Mn40. However, our findings with the pdr-1 mutant background show no differences in Zn accumulation, providing further support for a selective relationship between Parkin and Mn homeostasis.
Contrary to in vitro evidence of parkin-mediated control of a DMT isoform, we observed no significant changes in expression of the smf genes, especially with smf-3 being the most DMT1-like homolog19. Instead, significant downregulation of fpn-1.1 was observed in pdr-1 KOs compared to WT animals. These findings suggest that the loss of pdr-1 in C. elegans results in increased Mn accumulation from defective export, rather than from impaired uptake. Notably, we recently identified a novel role for SLC30A10 in Mn export that is associated with heightened risk for PD. However, no homologs for this protein are expressed in C. elegans41. Thus, for the present study, given the downregulation of fpn-1.1 mRNA in pdr-1 mutants, we investigated whether overexpression of the only known Mn exporter in C. elegans would result in a rescue of pdr-1 mutant phenotypes.
Mn uptake is modulated by a variety of proteins, including: DMT1, the transferrin receptor (TfR), the choline transporter, the citrate transporter, the magnesium transporter HIP14, ATP13A2, the solute carrier 39 family of zinc transporters, and calcium channels10. Among these, DMT1 has been given the most attention, as it is not only the primary mode of uptake, but is also associated with parkinsonism. Increased DMT1 expression has been found in the SNpc of PD patients, as well as in SNpc of MPTP mouse models42. Elevated DMT1 mRNA expression and DAergic neurotoxicity was also seen in rats exposed to Mn-containing welding fumes43. Moreover, specific polymorphisms in DMT1 have been found in a Chinese population suffering from PD44. These studies highlight altered metal homeostasis in the etiology of Parkinsonism. Interestingly, the overexpression of FPN in our pdr-1 mutants altered not only Mn, but Fe and Cu levels to a greater extent. We were not surprised to observe a treatment effect for Mn, as this was the only exogenous treatment administered to the nematodes. However, we did expect to see a greater decrease in Mn concentrations. It is possible that FPN’s affinity for Fe is greater than that of Mn, as the differential binding affinities have yet to be determined. Moreover, as FPN has not been shown to export Cu, the decrease in Cu levels may be a secondary effect of lowered intracellular Fe due to increased Fe efflux. Fe-deficiency anaemia has been associated with copper deficiencies, though the mechanism remains unknown45, 46. Though future studies are needed to further elucidate FPN’s transport profile in C. elegans, the rescue of the pdr-1 mutant phenotype through FPN overexpression supports our hypothesis that metal dyshomeostasis in the background of pdr-1 loss may be due to alterations in transporter expression.
In addition to the well-characterized toxicity of Mn resulting in Parkinsonian symptoms, enhanced iron accumulation in the SN is often seen in PD brains42, 47, with pharmacological Fe chelation showing potential therapeutic value48, 49. Moreover, Mn treatment has been recently shown to disrupt general metal homeostasis in WT C. elegans, with excess Mn resulting in altered Fe and Cu levels50. Though the authors of this study used slightly higher Mn concentrations (10–30 mM) than the present study, this was most likely due to the use of older worms treated for 24 hours, rather than larval stage worms acutely treated for 30 minutes. However, as their lowest dose (10 mM) is within the range of the doses used in the present study, similar findings were seen with higher Mn concentrations (30 mM) corresponding with comparatively lower Fe and Cu levels overall50. The results in the present study provide further support of the interplay between metals, as exogenous Mn treatment results in the alteration of endogenous metal concentrations that may alter vital downstream processes. It is possible that the combined effects of decreased Fe and Cu levels, rather than the moderate to slight decrease in Mn levels, results in the amelioration of the pdr-1 KO phenotypes. Moreover, the connection between Cu and a mutant parkin background is further supported by recent human data showing increased Cu sensitivity in neuroprogenitors from patients carrying PARK2 mutations51.
The recently discovered role of parkin as a mediator of mitophagy has introduced the potential significance of mitochondrial integrity in Parkinsonism52; loss of parkin could result in the accumulation of defective mitochondria to increase cellular oxidative stress. This could explain the significant increase in relative mtDNA copy number in pdr-1 KO animals as a measure that could equate with increased mitochondrial mass in pdr-1 KOs. This data corresponds with our previously published findings that pdr-1 KOs exhibit significantly increased ROS levels24. Notably, it seems controversial in the literature whether increased mtDNA copy number is protective or damaging in degenerative processes53, 54. However, increased mtDNA copy number has been associated with aging, as well as a response to increased oxidative stress30. Therefore, the increased copy number may also be in response to increased oxidative stress from enhanced Mn accumulation to compensate for damaged mitochondria. This may be especially true due to the preferential accumulation of Mn in mitochondria55. Consequently, the beneficial alterations in Mn, Fe and Cu in pdr-1 KO;fpn-1.1 OVR animals would then help to reverse this effect by decreasing metal-induced oxidative stress. Additionally, the increase in the antioxidant GSH in pdr-1 KO;fpn-1.1 OVR animals vs. pdr-1 KO animals is modest, though it does not reach statistical significance. This may represent a slight improvement in the overall handling of oxidative stress. It has been previously shown that neurons treated with increasing Fe concentrations show depletion in GSH content56. This is similar to the elevation in GSH content of pdr-1 KO;fpn-1.1 OVR animals that also exhibit decreased Fe accumulation. However, we are limited in the present study, as we have been unsuccessful in using the microplate assay format to measure both GSSG and GSH. While pdr-1 KO;fpn-1.1 OVR and pdr-1 KO animals show no difference in gcs-1 (homolog for the glutamate-cysteine ligase responsible for catalysing GSH synthesis) mRNA expression (data not shown), future studies should be done to determine whether this change in GSH is due to more reduced vs. oxidized forms of GSH.
Finally, we previously reported that pdr-1 KO animals show an exacerbation of DAergic neurodegeneration compared to WT animals24. Currently, conflicting findings exist on the effects of Mn on DAergic neurodegeneration in C. elegans50, 57. However, this may be due to differences in treatment paradigms and doses. Additionally, fluorescence microscopy is a common technique to assess degeneration; however, microscopy for GFP visualization remains a mostly qualitative readout of cell death. Accordingly, we focused on an output parameter of an intact DAergic system by assaying a DA-dependent behavioural measure. The basal slowing response (BSR) is a well-known feeding response that requires DA and affects mechanosensation to properly recognize food sources (bacteria) in C. elegans31. Similar to our previous results24, Mn treatment itself in WT animals did not result in a statistically significant decrease in BSR, though a slight decline was apparent. However, while pdr-1 KOs show impairment in this response, the rescue of BSR deficits by pdr-1 KO;fpn-1.1 OVR animals normalizes to the WT response. These data suggest that the overexpression of FPN normalizes DAergic integrity in the background of pdr-1 loss. The effect of Mn on BSR in pdr-1 KO and pdr-1 KO;fpn-1.1 OVR animals is negligible. This may be due to the complete loss of pdr-1 resulting in a “ceiling effect,” such that the addition of Mn exposure does not further exacerbate the basal differences. However, the BSR in pdr-1 KO;fpn-1.1 OVR animals fully normalizes to WT levels upon treatment.
Notably, we cannot relate the improvement in BSR to the improved survival of pdr-1 KO;fpn-1.1 OVR animals, as it has been previously reported that ablation of DAergic neurons in nematodes does not affect overall survival58. However, the relationship between metals and dopamine toxicity is well known. Dopamine itself is a strong oxidant that can undergo an auto-oxidation process to produce highly damaging intermediates, which makes a strong argument for the vulnerability of DA-specific brain areas to toxins and other oxidants59. Mn has been shown to catalyse dopamine oxidation60, while Fe has been shown to specifically bind neuromelanin found in DAergic neurons61. Thus, the pdr-1 KO; fpn-1.1 OVR animals may show improvement in the DA-dependent BSR due to the lower bioavailability of Mn, Fe and Cu that would otherwise participate in directly enhancing DA oxidation and/or indirectly producing damaging free radicals in an already susceptible cell type.
Conclusion
In conclusion, the present study provides further support for altered metal homeostasis as a critical component of PD pathophysiology. Using the genetically tractable C. elegans system, we show a novel role of pdr-1/parkin in modulating metal homeostasis following an acute Mn exposure, affecting metal efflux. Though human mutations in FPN have not yet been associated with PD, our findings demonstrate the importance and specificity of PD genetics (e.g. loss of pdr-1/parkin) in interacting with environmental factors to exacerbate physiological processes that may lead to cell death. Future studies should focus on potential therapeutic routes that help understand the interplay between pdr-1/parkin-mediated mitochondrial dynamics and enhanced efflux of redox-active metals like Mn, Fe and Cu as a strategy against Mn-induced Parkinsonism.
Acknowledgments
This work was funded by the NIH grant R01 ES10563, the Josef Schormüller Award and the DFG (BO 4103/1-1). We would also like to thank the Miller laboratory (Vanderbilt University Medical Center) for sharing resources. Lastly, we would like to thank the laboratories of Drs. Bill Valentine, Keith Erikson and Joel Meyer for scientific communications.
References
- 1.ATSDR. US Department of Health and Human Services, Public Service. 2008 [Google Scholar]
- 2.Lees AJ, Hardy J, Revesz T. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 3.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 4.Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM. Human molecular genetics. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- 5.Sang TK, Chang HY, Lawless GM, Ratnaparkhi A, Mee L, Ackerson LC, Maidment NT, Krantz DE, Jackson GR. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:981–992. doi: 10.1523/JNEUROSCI.4810-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. The Journal of biological chemistry. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 7.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blesa J, Phani S, Jackson-Lewis V, Przedborski S. Journal of biomedicine & biotechnology. 2012;2012:845618. doi: 10.1155/2012/845618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aschner JL, Aschner M. Molecular aspects of medicine. 2005;26:353–362. doi: 10.1016/j.mam.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aschner M, Erikson KM, Herrero Hernandez E, Tjalkens R. Neuromolecular medicine. 2009;11:252–266. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuschl K, Mills PB, Clayton PT. International review of neurobiology. 2013;110:277–312. doi: 10.1016/B978-0-12-410502-7.00013-2. [DOI] [PubMed] [Google Scholar]
- 12.Bhuie AK, Ogunseitan OA, White RR, Sain M, Roy DN. The Science of the total environment. 2005;339:167–178. doi: 10.1016/j.scitotenv.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Finkelstein MM, Jerrett M. Environmental research. 2007;104:420–432. doi: 10.1016/j.envres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Zeron HM, Rodriguez MR, Montes S, Castaneda CR. Journal of trace elements in medicine and biology: organ of the Society for Minerals and Trace Elements. 2011;25:225–229. doi: 10.1016/j.jtemb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Klos KJ, Ahlskog JE, Josephs KA, Fealey RD, Cowl CT, Kumar N. Archives of neurology. 2005;62:1385–1390. doi: 10.1001/archneur.62.9.1385. [DOI] [PubMed] [Google Scholar]
- 16.Smith EA, Newland P, Bestwick KG, Ahmed N. Journal of trace elements in medicine and biology: organ of the Society for Minerals and Trace Elements. 2013;27:65–69. doi: 10.1016/j.jtemb.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Garrick MD, Dolan KG, Horbinski C, Ghio AJ, Higgins D, Porubcin M, Moore EG, Hainsworth LN, Umbreit JN, Conrad ME, Feng L, Lis A, Roth JA, Singleton S, Garrick LM. Biometals: an international journal on the role of metal ions in biology, biochemistry, and medicine. 2003;16:41–54. doi: 10.1023/a:1020702213099. [DOI] [PubMed] [Google Scholar]
- 18.Yin Z, Jiang H, Lee ES, Ni M, Erikson KM, Milatovic D, Bowman AB, Aschner M. Journal of neurochemistry. 2010;112:1190–1198. doi: 10.1111/j.1471-4159.2009.06534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Au C, Benedetto A, Anderson J, Labrousse A, Erikson K, Ewbank JJ, Aschner M. PloS one. 2009;4:e7792. doi: 10.1371/journal.pone.0007792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson CP, Leibold EA. Frontiers in pharmacology. 2014;5:113. doi: 10.3389/fphar.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Domenico I, Lo E, Yang B, Korolnek T, Hamza I, Ward DM, Kaplan J. Cell metabolism. 2011;14:635–646. doi: 10.1016/j.cmet.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Higashi Y, Asanuma M, Miyazaki I, Hattori N, Mizuno Y, Ogawa N. Journal of neurochemistry. 2004;89:1490–1497. doi: 10.1111/j.1471-4159.2004.02445.x. [DOI] [PubMed] [Google Scholar]
- 23.Sriram K, Lin GX, Jefferson AM, Roberts JR, Wirth O, Hayashi Y, Krajnak KM, Soukup JM, Ghio AJ, Reynolds SH, Castranova V, Munson AE, Antonini JM. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:4989–5002. doi: 10.1096/fj.10-163964. [DOI] [PubMed] [Google Scholar]
- 24.Bornhorst J, Chakraborty S, Meyer S, Lohren H, Brinkhaus SG, Knight AL, Caldwell KA, Caldwell GA, Karst U, Schwerdtle T, Bowman A, Aschner M. Metallomics: integrated biometal science. 2014;6:476–490. doi: 10.1039/c3mt00325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. The Journal of Neuroscience. 2014;34:14079–14095. doi: 10.1523/JNEUROSCI.2329-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Hunter SE, Jung D, Di Giulio RT, Meyer JN. Methods. 2010;51:444–451. doi: 10.1016/j.ymeth.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahman I, Kode A, Biswas SK. Nature protocols. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 30.Lee HC, Yin PH, Lu CY, Chi CW, Wei YH. The Biochemical journal. 2000;348(Pt 2):425–432. [PMC free article] [PubMed] [Google Scholar]
- 31.Sawin ER, Ranganathan R, Horvitz HR. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 32.Roth JA, Singleton S, Feng J, Garrick M, Paradkar PN. Journal of neurochemistry. 2010;113:454–464. doi: 10.1111/j.1471-4159.2010.06607.x. [DOI] [PubMed] [Google Scholar]
- 33.Saini N, Georgiev O, Schaffner W. Molecular and cellular biology. 2011;31:2151–2161. doi: 10.1128/MCB.05207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saini N, Oelhafen S, Hua H, Georgiev O, Schaffner W, Bueler H. Neurobiology of disease. 2010;40:82–92. doi: 10.1016/j.nbd.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 35.Springer W, Hoppe T, Schmidt E, Baumeister R. Human molecular genetics. 2005;14:3407–3423. doi: 10.1093/hmg/ddi371. [DOI] [PubMed] [Google Scholar]
- 36.Papaevgeniou N, Chondrogianni N. Redox biology. 2014;2:333–347. doi: 10.1016/j.redox.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Finley EJ, Chakraborty S, Slaughter JC, Aschner M. Neurochemical research. 2013;38:1543–1552. doi: 10.1007/s11064-013-1054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aboud AA, Tidball AM, Kumar KK, Neely MD, Ess KC, Erikson KM, Bowman AB. Neurotoxicology. 2012;33:1443–1449. doi: 10.1016/j.neuro.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA. Human molecular genetics. 2014;23:2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- 40.Chesi A, Kilaru A, Fang X, Cooper AA, Gitler AD. PloS one. 2012;7:e34178. doi: 10.1371/journal.pone.0034178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:14079–14095. doi: 10.1523/JNEUROSCI.2329-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nunez MT, Garrick MD, Raisman-Vozari R, Hirsch EC. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:18578–18583. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sriram K, Lin GX, Jefferson AM, Roberts JR, Chapman RS, Chen BT, Soukup JM, Ghio AJ, Antonini JM. Archives of toxicology. 2010;84:521–540. doi: 10.1007/s00204-010-0525-9. [DOI] [PubMed] [Google Scholar]
- 44.He Q, Du T, Yu X, Xie A, Song N, Kang Q, Yu J, Tan L, Xie J, Jiang H. Neuroscience letters. 2011;501:128–131. doi: 10.1016/j.neulet.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Prohaska JR, Broderius M. Biometals: an international journal on the role of metal ions in biology, biochemistry, and medicine. 2012;25:633–642. doi: 10.1007/s10534-012-9521-2. [DOI] [PubMed] [Google Scholar]
- 46.Prohaska JR. Annals of the New York Academy of Sciences. 2014;1314:1–5. doi: 10.1111/nyas.12354. [DOI] [PubMed] [Google Scholar]
- 47.Ayton S, Lei P, Adlard PA, Volitakis I, Cherny RA, Bush AI, Finkelstein DI. Molecular neurodegeneration. 2014;9:27. doi: 10.1186/1750-1326-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mounsey RB, Teismann P. International journal of cell biology. 2012;2012:983245. doi: 10.1155/2012/983245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-Shachar D, Eshel G, Riederer P, Youdim MB. Annals of neurology. 1992;32(Suppl):S105–110. doi: 10.1002/ana.410320718. [DOI] [PubMed] [Google Scholar]
- 50.Angeli S, Barhydt T, Jacobs R, Killilea DW, Lithgow GJ, Andersen JK. Metallomics: integrated biometal science. 2014;6:1816–1823. doi: 10.1039/c4mt00168k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aboud AA, Tidball AM, Kumar KK, Neely MD, Han B, Ess KC, Hong CC, Erikson KM, Hedera P, Bowman AB. Neurobiology of disease. 2014;73C:204–212. doi: 10.1016/j.nbd.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Vries RL, Przedborski S. Molecular and cellular neurosciences. 2013;55:37–43. doi: 10.1016/j.mcn.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 53.Podlesniy P, Figueiro-Silva J, Llado A, Antonell A, Sanchez-Valle R, Alcolea D, Lleo A, Molinuevo JL, Serra N, Trullas R. Annals of neurology. 2013;74:655–668. doi: 10.1002/ana.23955. [DOI] [PubMed] [Google Scholar]
- 54.Gu F, Chauhan V, Kaur K, Brown WT, LaFauci G, Wegiel J, Chauhan A. Translational psychiatry. 2013;3:e299. doi: 10.1038/tp.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gavin CE, Gunter KK, Gunter TE. Neurotoxicology. 1999;20:445–453. [PubMed] [Google Scholar]
- 56.Aracena P, Aguirre P, Munoz P, Nunez MT. Biological research. 2006;39:157–165. doi: 10.4067/s0716-97602006000100017. [DOI] [PubMed] [Google Scholar]
- 57.Benedetto A, Au C, Avila DS, Milatovic D, Aschner M. PLoS genetics. 2010;6 doi: 10.1371/journal.pgen.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nass R, Hall DH, Miller DM, 3rd, Blakely RD. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Graumann R, Paris I, Martinez-Alvarado P, Rumanque P, Perez-Pastene C, Cardenas SP, Marin P, Diaz-Grez F, Caviedes R, Caviedes P, Segura-Aguilar J. Polish journal of pharmacology. 2002;54:573–579. [PubMed] [Google Scholar]
- 60.Garner CD, Nachtman JP. Chemico-biological interactions. 1989;69:345–351. doi: 10.1016/0009-2797(89)90120-8. [DOI] [PubMed] [Google Scholar]
- 61.Bridelli MG, Tampellini D, Zecca L. FEBS letters. 1999;457:18–22. doi: 10.1016/s0014-5793(99)01001-7. [DOI] [PubMed] [Google Scholar]