

Abstract

The application of synthetic biology requires characterized tools to precisely control gene expression. This toolbox of genetic parts previously did not exist for the industrially promising cyanobacterium, Synechococcus sp. strain PCC 7002. To address this gap, two orthogonal constitutive promoter libraries, one based on a cyanobacterial promoter and the other ported from Escherichia coli, were built and tested in PCC 7002. The libraries demonstrated 3 and 2.5 log dynamic ranges, respectively, but correlated poorly with E. coli expression levels. These promoter libraries were then combined to create and optimize a series of IPTG inducible cassettes. The resultant induction system had a 48-fold dynamic range and was shown to out-perform Ptrc constructs. Finally, a RBS library was designed and tested in PCC 7002. The presented synthetic biology toolbox will enable accelerated engineering of PCC 7002.
At the most basic level, synthetic biology relies on the ability to integrate a set of well-defined DNA sequences to obtain a desired phenotype. Protein levels are commonly controlled by the modulation of transcription and translation initiation rates. For example, transcription rates are changed by fusing a gene to promoters of varied strength or modulating the degree of repression by titrating inducers (e.g., the lac promoter family is modulated by IPTG addition)1 while translation rates are controlled by changing the strength and accessibility of ribosome binding sites.2 Libraries of constitutive promoters have been developed via mutagenesis of base promoters for several model organisms including Escherichia coli and Saccharomyces cerevisiae.3−6 Inducible promoter systems, such as T7/lac7 and Ptrc,8 have been created by fusing repressor sequences to promoter sites. Alternatively, biophysical models have been developed to predict and guide engineering of translation initiation signals in bacteria, thereby providing an additional point of control.2,9,10 While these approaches have allowed for the development of complex genetic circuits in E. coli, many of these tools have not been developed or validated for use in nonmodel organisms.
Cyanobacteria are an attractive host for metabolic engineering due to their innate ability to convert CO2 and sunlight directly into a chemical product of interest, thereby bypassing the need for expensive feedstocks. Model strains of cyanobacteria have been modified to overproduce a wide variety of products,11 including sucrose,12 2,3-butanediol,13 and ethylene;14 however, none of these photosynthetically produced products have been at a rate that is economically viable. Attempts to increase productivity and titer have been hampered by a limited set of genetic tools available for metabolic engineering in cyanobacteria. Few promoters have been developed for applied projects in cyanobacteria; well-characterized endogenous promoters such as PpsbA or PcpcB are typically used in synthetic biology strategies. These promoters control transcription of photosystem genes making them often too strong and undesirably sensitive to light.15 Endogenous metal responsive promoters have been developed for use as induction systems with better than 100 fold dynamic range; however, the low sensitivity and complex off target regulatory effects make these systems difficult to use in practice.16 Finally, several orthogonal genetic tools originally developed in E. coli, such as Ptrc, PlacI, and Ptet, have been tested in cyanobacteria with poor or inconsistent results across model cyanobacterial systems.17
Recently, improvements have been made to small molecule induction systems in model cyanobacteria. In particular, new IPTG induction systems have been described for Synechocystis sp. strain PCC 6803 (PCC 6803) and Synechococcus sp. strain PCC 7942 (PCC 7942).18,19 While these works are improvements of previously available systems, the dynamic ranges were limited: 13-fold for PCC 6803 and 18-fold for PCC 7942. As an alternative to IPTG induction, an anhydrotetracyline induction system was developed for PCC 6803.20 This system demonstrated a 239-fold dynamic range under dark heterotrophic growth conditions but only 49-fold dynamic range under photoautotrophic growth conditions when induced with an exceptionally high concentration of anhydrotetracycline (10 ug/mL).20 While there has been some development of synthetic biology tools for PCC 6803 and PCC 7942, other cyanobacteria used in metabolic engineering remain limited to a few native promoters. In particular, Synechococcus sp. strain PCC 7002 (PCC 7002), an attractive strain for metabolic engineering due to its rapid growth rate, high CO2 fixation rate, and halotolerance, has very few promoters of known strength and only one reported inducible promoter.16,21
The objective of this work was to develop a set of defined synthetic biology tools that would enable precise control of gene expression in PCC 7002. Herein, we present two constitutive promoter libraries in PCC 7002 containing promoters with a range of strengths spanning 3 orders of magnitude. These promoter libraries were used to construct an IPTG induction system based on a promoter of cyanobacterial origin. Iterative modifications to this system resulted in stronger repression and higher intrinsic expression levels. The resulting IPTG induction system was capable of a 48 ± 7 fold increase of YFP expression. This induction ratio represents over an order of magnitude improvement over the next best small molecule inducible system in PCC 700216 but is an order of magnitude lower than Ptrc in E. coli.(22) Finally, a cyanobacterial ribosome binding site (RBS) library was designed with in silico modeling tools and validated in PCC 7002. The models were sufficient to predict gross changes in expression, but were not capable of predicting subtle differences. We predict that these tools will enable construction of more complex genetic circuits in PCC 7002 and increase the number of successful metabolic engineering projects in this promising cyanobacterium.
Results and Discussion
Promoter Library Based on the PCC 6803 cpcB Promoter
Until recently, the cpcB promoter from PCC 6803 (PPCC6803cpcB) was the best characterized promoter for use in PCC 7002. It was initially designed to drive gene overexpression as part of the high-copy plasmid pAQ1 homologous recombination system.21 Recently, a chromosomal recombination system based on acrylic acid counter selection was developed that also used PPCC6803cpcB for gene expression.23 In preliminary studies, we found that a single strong constitutive promoter was not applicable for most metabolic engineering applications. Therefore, we mutagenized PPCC6803cpcB to create a library of constitutive promoters capable of generating more modest levels of gene expression. To simplify library construction, PPCC6803cpcB derived from plasmid pAQ1_Exp21 was truncated from its original 500 bp length to a more easily modifiable 89 bp core promoter region. This region was chosen based on homology of cpcB promoters in closely related cyanobacteria. The resulting promoter was called Pcpt for cyanobacterial phycocyanin truncated promoter. PPCC6803cpcB strength was compared to Pcpt using a homozygous chromosomal YFP system in PCC 7002. The truncation reduced YFP fluorescence by 50%; however, Pcpt expression was not significantly affected by high light intensity; in contrast, a ∼30% reduction was seen in PPCC6803cpcB controlled YFP expression under high light (Figure 1). Previous transcription profiling in PCC 7002 showed a similar decrease in full length PcpcB expression under high light conditions.24 These data suggest that additional transcription factors beyond core RNA polymerase may be involved in generating maximum transcription rates from the PcpcB core promoter.
Figure 1.

Expression of full length and truncated PCC 6803 cpcB promoters under high and low light conditions. Light intensity is shown as PAR (photosynthetically available radiation) as measured in μE/m2·s. The full length PPCC6803cpcB exhibited reduced expression under high light (p-value <0.005). No significant difference was observed between high and low light conditions for the truncated promoter. Error bars represent standard deviations of biological replicates (n = 3).
To generate a promoter library, Pcpt was mutagenized with error prone PCR and fused with a YFP expression cassette. The promoter sequences from 107 strains were determined and aligned based on YFP expression. Several of these promoters had duplications or mutations outside of the core promoter region and were eliminated from further analysis, leaving 29 unique promoters (see Supporting Information Table 4 for sequences). YFP expression levels were screened in these segregated strains of PCC 7002 (Figure 2A). Of these promoters, 11 representative samples were chosen for further testing, covering the full range of YFP expression levels (Figure 2B). Expression from individual members of the Pcpt library spanned 3 orders of magnitude. Several strains displayed higher fluorescence than the basal Pcpt construct. One such strain, c223, displayed two times the YFP fluorescence as Pcpt, approaching the levels observed with the full length PPCC6803cpcB promoter. The sequence of c223 contained only two point mutations relative to the wild type promoter sequence (Figure 2C). These two mutations, A30T and A67T, were tested individually. The latter mutation was sufficient to double YFP expression by itself. Interestingly, this mutation changed the Pcpt −10 σ70 recognition site from TATAAA to TATAAT, the E. coli consensus sequence.25
Figure 2.

Normalized expression levels and sequences of members of the Pcpt promoter library. (A) Fluorescence values of library members normalized to expression level of Pcpt (yellow). (B) Fluorescence values of a representative sample of library members normalized to Pcpt (yellow). PPCC6803cpcB is shown in orange and wild type PCC 7002 without a YFP expression construct is shown in solid green. Error bars represent the standard deviation of biological replicates (n = 3). (C) Sequence alignment of the representative library members. The predicted −35 and −10 regions are highlighted in purple and blue, respectively.
Development of a Synthetic Promoter Library in PCC 7002
The latter finding raised the question of whether promoter libraries would be consistent between E. coli and PCC 7002. We therefore cloned and tested a series of well-characterized E. coli promoters to determine if they could serve as a second library in PCC 7002. A subset of the BioBrick promoters derived from part BBa_J23119, a σ70 consensus promoter, were combined with a synthetic 5′ UTR containing a RBS designed using a ribosome binding site calculator2 and YFP (Figure 3A).
Figure 3.

Schematic drawing and expression levels of the synthetic promoter library. (A) Drawing of synthetic promoter and 5′ UTR. Sequences for promoters J23119, pMB1, pMB2, and pMB3 are presented. The labeled transcription start site was determined for J23119 and pMB2. The promoter region is outlined in light green with the −35 and −10 regions in purple and light blue, respectively. The 5′ UTR is shown in blue with the RBS in red. (B) Expression levels of the synthetic promoter library relative to J23119. Error bars represent standard deviations of biological replicates (n = 3).
The expression level of these constructs were quantified under standard growth conditions and compared to constructs using PPCC6803cpcB.21 The expression cassette containing the promoter BBa_J23119, the strongest E. coli promoter, generated 35 ± 3% of the fluorescence of the strain using promoter PPCC6803cpcB, one of the strongest promoters in PCC 7002. The closely related PCC 7002 homolog of PPCC6803cpcB, PPCC7002cpcB, drives the expression of SYNPCC7002_A2209, the third most abundant transcript in PCC 7002.24 Fluorescence from the other library members were all lower than BBa_J23119 spanning 2.5 orders of magnitude (Figure 3B). To increase the range of expression levels, additional permutations of BBa_J23119 were made, resulting in promoters pMB1, pMB2, and pMB3 (Figure 3A). When the relative fluorescence generated by these promoters in PCC 7002 were compared with the corresponding E. coli strengths published on the BioBrick Web site (http://parts.igem.org/Part:BBa_J23119), a weak correlation was observed (R2 = 0.434) (Figure 4A). The mechanism behind this weak correlation is unclear but could be due to differences in the transcription machinery of these two organisms. Interestingly, the σ70 transcription factors of E. coli and PCC 7002 share strong homology, particularly at regions involved in DNA binding. In response to this weak interspecies correlation, the Pcpt promoter library was evaluated in E. coli. YFP expression cassettes from the 11 representative library members were inserted into an E. coli plasmid in order to test relative expression. As was seen with the synthetic promoter library, there was only weak correlation between the relative expression of the Pcpt library in PCC 7002 and E. coli (R2 = 0.366) (Figure 4B).
Figure 4.
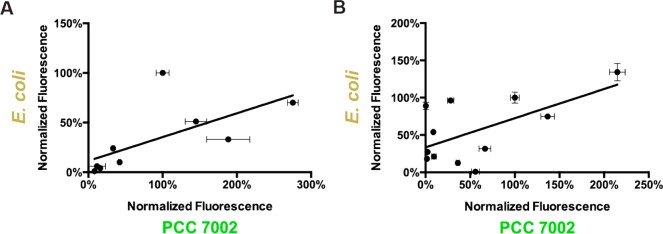
Comparison of promoter libraries in PCC 7002 and E. coli. (A) Comparison of BBa_J23119 derived promoters in E. coli and PCC 7002 relative to the expression level of J23100. R2 = 0.434. (B) Comparison of the cpcB derived promoter library in E. coli and PCC 7002 relative to the truncated cpcB promoter. R2 = 0.366. Error bars represent the standard deviation of biological replicates of biological replicates (n = 3).
Redesigning the Pcpt Promoter for IPTG Induction
While the constitutive promoter libraries offered a range of expression levels, many metabolic engineering projects require promoters that can be controlled in response to environmental signals. To make Pcpt responsive to the common laboratory inducer IPTG, the core promoter sequence was modified by replacing native sequence with lac operators to allow for binding of the lac repressor, LacI. Specifically, two lac operator sequence variants Oid and O17,17 were individually inserted 42 base pairs upstream of the predicted −35 promoter region and directly downstream of the predicted −10 promoter region of Pcpt, respectively, resulting in a promoter called PcptOO (Figure 5A). Insertion of these two operators caused a 76 ± 5% decrease in its intrinsic promoter strength relative to Pcpt (Figure 5B). Next, the lacI gene under the E. coli PlacIQ promoter was inserted downstream of the YFP terminator. Addition of the lacI cassette resulted in decreased fluorescence relative to PcptOO. Fluorescence was restored upon addition of 1 mM IPTG (Figure 5B). The resulting construct demonstrated only 4-fold induction but provided a starting point for promoter engineering.
Figure 5.

Initial development of an IPTG induction system based on the Pcpt promoter. (A) Fluorescence of PCC 7002 strains harboring promoters constructs normalized to Pcpt expression. Insertion of lac operators resulted in a ∼80% decrease in expression relative to Pcpt. Introduction of lacI resulted in repression that was removed by the addition of IPTG. (B) Expression levels of uninduced lac induction strains with various lacI promoters relative to expression of Pcpt. (C) Optimization of lacI expression and activity using different promoters driving lacI and the W220F mutation improving lacI binding. Error bars represent the standard deviation of biological replicates (n = 3).
Since this construct was fully inducible, an increased dynamic range would require stronger repression in the absence of the inducer and/or stronger expression in the absence of LacI. Poor repression could be attributed to suboptimal spacing of the operators or poor LacI expression. Attempts to move the operators closer together resulted in dramatic decreases in the intrinsic expression level of the promoter, resulting in constructs with limited utility and little increase in repression (data not shown). As an alternative, the promoter driving lacI, PlacI was replaced with members of the synthetic promoter library (Figure 5C). Several of these promoters, in particular PMB2, resulted in reduced induction system leakiness relative to the E. coli lacIQ promoter. Replacing the PlacIQ promoter in the original induction system with PMB2 resulted in a strain with 12-fold induction. This system was further improved by making a single amino acid substitution, W220F, in lacI which had previously been shown to strengthen LacI dimerization and provide tighter repression than wild type LacI.22 This construct, cLac94, with PMB2 driving the expression of a modified lacI, resulted in a system with 38 ± 6 fold induction using 5 mM IPTG (Figure 6A). A higher IPTG concentration was needed to achieve maximum induction, possibly due to the tighter DNA binding exhibited by lacI W220F.
Figure 6.

Diagram and induction levels for IPTG inducible promoters. (A) Expression of induced and uninduced IPTG inducible promoters relative to Pcpt. With the exception of the Pcpt control, all strains contain lacI expressed by PMB2. All cLac### strains have the additional W220F mutation in lacI while cLacMB2 contains wildtype lacI. Error bars represent standard deviation of biological replicates (n = 3) (B) List of IPTG inducible promoters with fold induction potential (fold change between 0 and 5 mM IPTG) and sequence comparison. The lac operators are highlighted in yellow, the −35 sequence is highlighted in purple, and the −10 sequence is highlighted in green.
Increasing the Intrinsic Strength of PcpcB-Based Induction System
Optimizing lacI expression resulted in tight repression and a larger dynamic range upon induction of the PcptOO promoter. However, with these modifications, the maximum expression reached only 15 ± 2% relative to Pcpt (Figure 6A). To address this limitation, the PcptOO region of cLac94 was mutated to incorporate advantageous base-pair changes discovered during the construction of the Pcpt promoter library (Figure 6B). The first PcptOO mutation tested was A67T, which was identified by testing the mutations present in the strong Pcpt library member c223. The A67T mutation in the PcptOO construct, cLac109 increased YFP expression by 255 ± 21% upon induction with IPTG; however, it also significantly increased the leakiness of the promoter (defined as expression in the absence of IPTG), resulting in only a 11 ± 1 fold induction range (Figure 6B). Deletion of A67 (cLac124) provides a shorter overlap between the −10 and the downstream operator, analogous to the strong PtrcE. coli promoter. This deletion resulted in 96 ± 10% derepressed expression but only an 8-fold dynamic range. Finally, the spacing between the PcptOO −35 and −10 binding site was reduced from 18 to the more common 17 base pairs, resulting in construct cLac142 (See Figure 6B). This resulted in a maximum derepressed expression of 30 ± 10% and a 34-fold induction. See Supporting Information Table 5 for all lac induction promoter sequences.
The increased expression seen when mutating the −10 promoter sequence to the E. coli σ70 consensus sequence led us to try mutating the −35 PcptOO from CAGACA to the E. coli consensus sequence TTGACA, creating the construct cLac143. This mutation increased the derepressed promoter strength of PcptOO to 235 ± 32% relative to constitutive Pcpt. In the presence of LacI and absence of IPTG, YFP was expressed at 4.9 ± 0.2% of Pcpt resulting in a potential 48 ± 7 fold range of expression upon addition of IPTG (Figure 6A). Titration of IPTG resulted in qualitatively similar fluorescence curves (basal expression up to 10 μM and maximum expression at 1 mM) for the constructs with the highest dynamic range, cLac94 and cLac143 (Figure 7).
Figure 7.

Titration of inducible promoter constructs cLac94 and cLac143 with varying concentrations of IPTG. Promoter cLac94 is shown in blue and promoter cLac143 is shown in red. Both strains contain lacI W220F expressed with pMB2. Error bars represent standard deviation of biological replicates (n = 3).
Comparison with trc Promoters
The performance of the PcptOO induction system increased as mutations moved its sequence toward the E. coli consensus. This observation led us to reconsider Ptrc as a viable inducible promoter in PCC 7002. Using our optimized lacI repression cassette, we replaced the Pcpt core promoter with Ptrc using either one or two lac operators. The maximum derepressed YFP expression was similar for the one and two operator constructs, 188 ± 9% and 179 ± 10% relative to constitutive Pcpt, respectively. However, the noninduced Ptrc promoter expression with one operator was significantly more leaky than the version with two operators resulting in a 6 (cLac145) and 23 fold induction range (cLac146), respectively (Figure 6A). The cLac146 induction system has the advantage of using a shorter promoter region; however, it has a lower dynamic range than both cLac94 and cLac143 making it less useful for genetic control in PCC 7002. PtrcOO could prove be a useful compliment to PcptOO, avoiding unwanted chromosomal recombination events due to regions of strong homology in strains with multiple gene insertions.
Design and Construction of a PCC 7002 RBS Library
To test whether an RBS library could be predictably constructed for PCC 7002, the Shine–Delgarno region of the previously described pACSA_J23119_YFP (AGGGAT) was modified using the RBS Library Calculator26 to create an 8-member library (degenerate base sequence ASGMGR) specific to PCC 7002 with a predicted 143 fold range of translation initiation rates.2 Two additional Shine–Delgarno sequences that were predicted to induce strong translation initiation rates were added (AGGGGG, AGGGAG), which increased the predicted library range to 213-fold. YFP fluorescence data in PCC 7002 showed that, in practice, this library provided a 30-fold range of protein expression with a weak correlation to their predicted strengths (Figure 8A). qPCR analysis of these strains showed that one library member, AGGAGA, had a significantly higher YFP transcript level than any of the other library members (Figure 8B). The YFP expression cassettes were sequenced and no mutations were present (data not shown). Excluding AGGAGA removed an outlier and improved the fit of the model from R2 = 0.3760 to R2 = 0.7443 (Figure 9A and B). The RBS library sequences were also back calculated with two alternative translation rate prediction programs: the UTR Designer9 and the RBSDesigner.10 In this case, the correlation of the experimental data to the predicted expression level provided by the UTR Designer (Figure 9C) was similar to the RBS Calculator prediction; however, the analogous correlation to the RBSDesigner prediction was weaker (Figure 9D). These data suggest that the RBS Calculator is useful for designing RBS parts in PCC 7002; however, as shown in a recent paper by Li et al.,27 fine control of RBS strength may not be possible with this prediction program.
Figure 8.
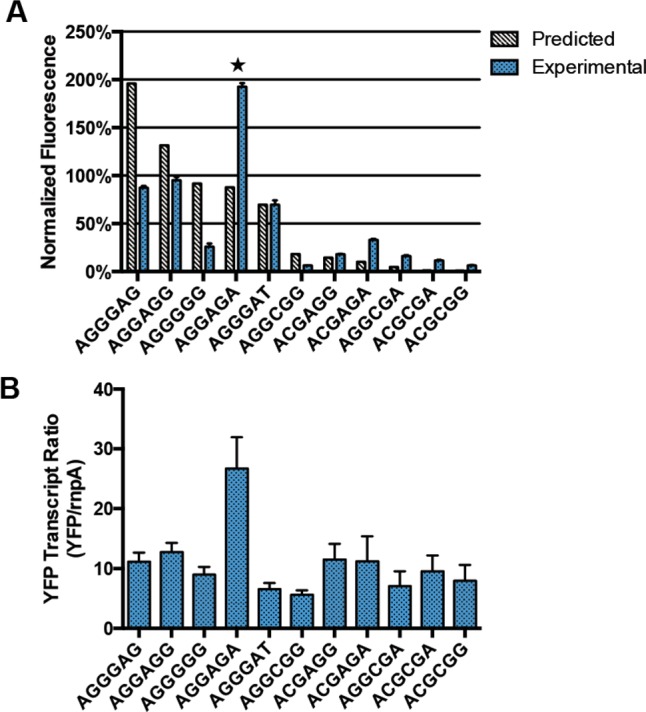
Evaluation of the RBS library versus predicted transcription initiation rates. (A) RBS Calculator v1.1 transcription initiation rate data was compared with YFP fluorescence by anchoring both data sets to AGGGAT, which is the RBS sequence of the original pJ23119_YFP construct. YFP fluorescence is represented as a percentage of constitutive Pcpt YFP fluorescence. Error bars on experimental data represent standard deviation of biological replicates (n = 3). Starred library member AGGAGA had significantly higher YFP transcript than every other library member (Figure 8B), which likely explains its observed deviation from the prediction. (B) Relative YFP transcript levels quantified by qPCR normalized to rnpA. Transcript levels for AGGAGA was significantly higher (95% CI) than every other strain in the RBS library. All other library members showed no significant difference between them (95% CI).
Figure 9.

Correlation of the predicted translation initiation rates from the RBS Calculator to experimentally determined YFP fluorescence. (A) Correlation with AGGAGA included (R2 = 0.3760). (B) Correlation with AGGAGA removed (R2 = 0.7443). (C) Correlation of the experimental data set, excluding AGGAGA, to the UTR Designer (R2 = 0.7843). (D) Correlation of the experimental data set, excluding AGGAGA, to the RBS Designer (R2 = 0.04448). Error bars on experimental data represent standard deviation of biological replicates (n = 3).
Conclusions
PCC 7002 has the potential to become an important photosynthetic chemical production platform. To date, few products have been successfully made with this strain. This may, in part, be due to the dearth of genetic tools available for the strain. In this study, we developed two orthogonal constitutive promoter libraries with a 3 and 2.5-fold dynamic range respectively and used these libraries to optimize the most tightly controlled IPTG induction system available for any cyanobacterial species. Finally, we performed the first evaluation of the RBS Calculator in PCC 7002. The resultant RBS library showed a moderate correlation to the predicted values suggesting that the RBS library calculator can be used in PCC 7002 to predict gross differences in expression. A representative subset of the promoter library, RBS library, and IPTG inducible strains were also tested for relative expression at high cell densities (OD730 ∼ 5) with high correlation to the data collected during log phase (OD730 0.5, data not shown). This suggests that strains produced using these tools can be reliably used in higher density chemical production environments. The gene expression tools presented here, along with our previously published acrylic acid counter selection system23 enable rapid construction of finely controlled genetic circuits within PCC 7002 and should accelerate the field of synthetic biology within cyanobacteria.
Methods
Chemicals, Reagents, and Media
Acrylic acid was obtained from Fisher Scientific (AC16425). Restriction enzymes, Phusion DNA polymerase, and T4 DNA ligase were purchased from New England Biolabs (Ipswich, MA). All other chemicals and reagents were purchased from either Fisher Scientific or Sigma-Aldrich. Unless otherwise noted, PCC 7002 was grown in Media A+ and maintained on 1.5% agar plates.28 Unless otherwise noted, cultures were grown in 10 mL volumes bubbled with air at 35 °C with a light intensity of 200 μE/m2·s. E. coli were grown in lysogeny broth (LB).
Strain Construction
All strains used in this study are listed in Supporting Information Table 1. E. coli DH5α was used for cloning of plasmids. Wild Type PCC 7002 was obtained from the Pasteur Culture Collection. Unless otherwise indicated, all genetic tools (i.e., promoters, ribosome binding sites) were constructed as fusions to the EYFP reporter gene29 and cloned into pACSA_pcpcB_YFP.23 The resulting plasmids contained the YFP expression cassette flanked with DNA sequences that would enable homologous recombination at the acsA loci of the PCC 7002 chromosome. Plasmids were amplified and stored in E. coli DH5α prior to transformation into PCC 7002 according to published protocols.21 Homozygous recombinants were isolated using an acrylic acid counter selection as previously described.23 Segregation was confirmed by colony PCR. See Supporting Information Table 3 for the oligonucleotide sequences used in this study. The standard deviations of the correlated values were propagated whenever average YFP fluorescence levels were compared.
Measurement of Fluorescence
All experiments began with isolation of single colonies from freezer stocks of segregated mutants. Cultures of PCC 7002 expressing YFP were grown in triplicate to an OD730 0.5–1. Cultures were normalized to 1.5–3 OD730·mL and centrifuged at 3000g for 12 min. The resulting pellets were aspirated, resuspended in 300 μL BugBuster Protein Extraction Reagent (Novagen), rocked at room temperature for 30 min, and centrifuged at 16 000g for 25 min at 4 °C. The florescence of the resulting supernatant was measured (excitation 514 nm, emission 527 nm) using a Tecan M1000 plate reader.
YFP expression in E. coli was similarly tested by growing DH5α containing pACSA plasmids with promoter or RBS library members in 5 mL of LB overnight at 37 °C with shaking. After 16 h the cultures were normalized to OD600 and split into triplicate 5 mL tubes and grown overnight again. The cultures were normalized to OD600 and 200 μL of each culture was analyzed for YFP fluorescence by plate reader.
Construction of a Promoter Library Based on the PCC 6803 PcpcB Promoter
Pcpt, a truncated version of the PPCC6803cpcB promoter, was constructed by annealing two complementary, phosphorylated oligonucleotides. The resulting double stranded DNA cassette was cloned by restriction digestion and ligation to yield pACSA_Pcpt_YFP. The library of Pcpt mutants was created using the GeneMorph II Random Mutagenesis Kit (Agilent). Error prone PCR was conduced using the kit’s instructions with primers cpt error prone F and cpt error prone R (Supporting Information Table 3). pACSA_Pcpt_YFP plasmid DNA (0.1 ng) was used as a template to maximize mutation frequency. The resulting PCR products were serially used as a template for additional rounds (four total) of error-prone PCR, each time using 0.1 ng template per reaction. Once amplified (using Phusion polymerase), the promoter libraries were cloned into pACSA_Pcpt_YFP cassette using EcoR I/Sac I restriction sites. The resulting plasmids were replicated in DH5α, pooled, and transformed into PCC 7002 for double homologous recombination into the acsA locus as previously described.23,30 Approximately 100 mutants were segregated and confirmed to be homozygous. The truncated Pcpt promoter region was amplified from each isolate using GoTaq DNA polymerase (Promega, Madison WI) and sequenced (Functional Biosciences, Madison WI). Promoters from 11 of the library members were moved back into the pACSA_Pcpt_YFP construct using in vitro recombination assembly with overlapping primers (Gibson 2011) using the primer pairs indicated in Supporting Information Table 3. The resulting assemblies were transformed into E. coli DH5α and confirmed by sequencing.
Construction of a Synthetic Promoter Library
The J23119 promoter sequence was obtained from the BioBrick part BBa_J23119 (iGEM Registry of Standard Biological Parts, www.partsregistry.org). The RBS was designed to have 40 000 arbitrary translation initiation units as predicted by the RBS calculator.2 These two parts were combined with the plasmid pACSA_Pcpt_YFP23 to create pACSA_J23119_YFP in a two part Gibson Assembly using primer pairs indicated in Supporting Information Table 3. Derivatives of this construct were made by replacing promoter Bba_J23119 with Bba_J23100, Bba_J23101, Bba_J23105, Bba_J23108, Bba_J23109, Bba_J23110, Bba_J23114, and Bba_J23117, along with three novel promoter sequences pMB1, pMB2, and pMB3. These constructs were made using a two piece Gibson Assembly using primer pairs indicated in Supporting Information Table 3. Transcription start sites were mapped using the 5′ RACE System for Rapid Amplification of cDNA Ends, Version 2.0 (Invitrogen).
Construction of the IPTG Inducible PCC 7002 Strains
pACSA_Pcpt_YFP was modified to be IPTG inducible by sequentially inserting two operators and the PlacIQ_lacI repressor cassette proximal to YFP. The downstream operator was added by Quikchange site directed mutagenesis (Agilent) and then the upstream operator was inserted by Gibson assembly to produce the construct, pACSA_PcptOO_YFP. The PlacIQ_lacI −expression cassette was added to pACSA_PcptOO_YFP using the Xba I restriction site. Primers used to produce these constructs are described in Supporting Information Table 3. LacI mediated repression was improved by replacing the PlacIQ promoter in pACSA_PcptOO_YFP_PlacIQ_lacI with members of the synthetic promoter library using Gibson Assembly (see Supporting Information Table 3 for primers used). LacI repression was further improved by making a W220F point mutation via Quikchange site directed mutagenesis. Additional mutations within PcptOO were created using Quikchange (see Supporting Information Table 3 for primers used). PTrcO_YFP_PMB2_lacIWF and PTrcOO_YFP_PMB2_lacIWF were constructed using two part Gibson Assembly using the optimized pACSA_PcptOO_YFP_PMB2_lacIWF plasmid as a template and primers identified in Supporting Information Table 3. All lac constructs were stably inserted into the AcsA locus of PCC 7002 as previously described. Segregation was confirmed by colony PCR.
Design and Construction of the RBS Library
The RBS Library Calculator26 was used to design an RBS library based on the untranslated region and YFP coding sequence of pACSA_J23119_YFP. The library was constructed using Quikchange mutagenesis with primers shown in Supporting Information Table 3. A degenerate oligonucleotide pair was used to construct the majority of the library. All library members were stably inserted into the AcsA locus of PCC 7002 as previously described. Segregation was confirmed by colony PCR.
qPCR Analysis of the RBS Library in PCC 7002
RNA extraction methods and DNase treatment were adapted from prior studies31 by adding an additional glass bead lysis step during the lysozyme treatment. Samples were vortexed every 2 min during the 6 min incubation at 64 °C. cDNA was synthesized using random hexamers and 500 ng total RNA by the iScript cDNA Synthesis Kit (Bio-Rad) according to the given instructions. RT-qPCR was performed on 10 μL reactions using iQ SYBR Green Supermix on a CFX Connect Real-Time PCR Detection System (Bio-Rad). Then, 1 μL of the cDNA reaction was added to each reaction, and purified PCR products of genes of interest were used to create standard curves for each primer pair. Denaturation at 95 °C for 3 min was followed by 36 cycles of 95 °C for 15 s, 54 °C for 30 s, and 72 °C for 1 min. A melt curve with was generated with increments of 0.5 °C every 5 s. The statistical significance of transcript levels differences between the library members were calculated using Tukey’s multiple comparisons test.
Acknowledgments
This research was funded by the National Science Foundation (NSF) (EFRI-1240268, CBET-1149678, SEES 12-15871), the US Air Force Office of Scientific Research (FA9550-11-1-0038), and the Department of Energy (DE-SC0010329). G.C.G. is the recipient of a National Institutes of Health (NIH) Biotechnology Training Fellowship (NIGMS-5 T32 GM08349).
Supporting Information Available
Tables 1–5. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ A.L.M. and M.B.B. contributed equally to the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Balzer S.; Kucharova V.; Megerle J.; Lale R.; Brautaset T.; Valla S. (2013) A comparative analysis of the properties of regulated promoter systems commonly used for recombinant gene expression in Escherichia coli. Microb. Cell Fact. 12, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espah Borujeni A.; Channarasappa A. S.; Salis H. M. (2014) Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 42, 2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannig G.; Makrides S. C. (1998) Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 16, 54–60. [DOI] [PubMed] [Google Scholar]
- Alper H.; Fischer C.; Nevoigt E.; Stephanopoulos G. (2005) Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. U.S.A. 102, 12678–12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer K.; Mijakovic I.; Jensen P. R. (2006) Synthetic promoter libraries—Tuning of gene expression. Trends Biotechnol. 24, 53–55. [DOI] [PubMed] [Google Scholar]
- Curran K. A.; Crook N. C.; Karim A. S.; Gupta A.; Wagman A. M.; Alper H. S. (2014) Design of synthetic yeast promoters via tuning of nucleosome architecture. Nat. Commun. 5, 4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubendorf J. W.; Studier F. W. (1991) Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J. Mol. Biol. 219, 45–59. [DOI] [PubMed] [Google Scholar]
- Brosius J.; Erfle M.; Storella J. (1985) Spacing of the −10 and −35 regions in the tac promoter. Effect on its in vivo activity. J. Biol. Chem. 260, 3539–3541. [PubMed] [Google Scholar]
- Seo S. W.; Yang J.-S.; Kim I.; Yang J.; Min B. E.; Kim S.; Jung G. Y. (2013) Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab Eng. 15, 67–74. [DOI] [PubMed] [Google Scholar]
- Na D.; Lee D. (2010) RBSDesigner: Software for designing synthetic ribosome binding sites that yields a desired level of protein expression. Bioinformatics 26, 2633–2634. [DOI] [PubMed] [Google Scholar]
- Wang B.; Wang J.; Zhang W.; Meldrum D. R. (2012) Application of synthetic biology in cyanobacteria and algae. Front. Microbiol. 3, 344. 10.3389/fmicb.2012.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducat D. C.; Avelar-Rivas J. A.; Way J. C.; Silver P. A. (2012) Rerouting carbon flux to enhance photosynthetic productivity. Appl. Environ. Microbiol. 78, 2660–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver J. W. K.; Machado I. M. P.; Yoneda H.; Atsumi S. (2013) Cyanobacterial conversion of carbon dioxide to 2,3-butanediol. Proc. Natl. Acad. Sci. U.S.A. 110, 1249–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerer J.; Tao L.; Davis M.; Ghirardi M.; Maness P.-C.; Yu J. (2012) Sustained photosynthetic conversion of CO2 to ethylene in recombinant cyanobacterium Synechocystis 6803. Energy Environ. Sci. 5, 8998–9006. [Google Scholar]
- Heidorn T.; Camsund D.; Huang H.-H.; Lindberg P.; Oliveira P.; Stensjö K.; Lindblad P. (2011) Synthetic biology in cyanobacteria engineering and analyzing novel functions. Methods Enzymol. 497, 539–579 10.1016/B978-0-12-385075-1.00024-X. [DOI] [PubMed] [Google Scholar]
- Berla B. M.; Saha R.; Immethun C. M.; Maranas C. D.; Moon T. S.; Pakrasi H. B. (2013) Synthetic biology of cyanobacteria: Unique challenges and opportunities. Front. Microbiol. 4, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-H.; Camsund D.; Lindblad P.; Heidorn T. (2010) Design and characterization of molecular tools for a Synthetic Biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res. 38, 2577–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederholtmeyer H.; Wolfstadter B. T.; Savage D. F.; Silver P. A.; Way J. C. (2010) Engineering cyanobacteria to synthesize and export hydrophilic products. Appl. Environ. Microbiol. 76, 3462–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camsund D.; Heidorn T.; Lindblad P. (2014) Design and analysis of LacI-repressed promoters and DNA-looping in a cyanobacterium. J. Biol. Eng. 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-H.; Lindblad P. (2013) Wide-dynamic-range promoters engineered for cyanobacteria. J. Biol. Eng. 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Alvey R. M.; Byrne P. O.; Graham J. E.; Shen G.; Bryant D. A. (2011) Expression of genes in cyanobacteria: Adaptation of endogenous plasmids as platforms for high-level gene expression in Synechococcus sp. PCC 7002. Methods Mol. Biol. 684, 273–293. [DOI] [PubMed] [Google Scholar]
- Gatti-Lafranconi P.; Dijkman W. P.; Devenish S. R. A.; Hollfelder F. (2013) A single mutation in the core domain of the lac repressor reduces leakiness. Microb. Cell Fact. 12, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann M. B.; Zess E. K.; Walters E. M.; Schmitt E. F.; Markley A. L.; Pfleger B. F. (2013) An organic acid based counter selection system for cyanobacteria. PLoS One 8, e76594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus Ludwig D. A. B. (2011) Transcription profiling of the model cyanobacterium Synechococcus sp. strain PCC 7002 by next-gen (SOLiD) sequencing of cDNA. Front. Microbiol. 2, 41. 10.3389/fmicb.2011.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisser S.; Margalit H. (1994) Determination of common structural features in Escherichia coli promoters by computer analysis. Eur. J. Biochem. 223, 823–830. [DOI] [PubMed] [Google Scholar]
- Salis H. M.; Mirsky E. A.; Voigt C. A. (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 27, 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.-W.; Burkhardt D.; Gross C.; Weissman J. S. (2014) Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens S. E.; Patterson C. O.; Myers J. (1973) The production of hydrogen peroxide by blue–green algae: A survey. J. Phycol. 9, 427–430. [Google Scholar]
- Miyawaki A.; Griesbeck O.; Heim R.; Tsien R. Y. (1999) Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc. Natl. Acad. Sci. U.S.A. 96, 2135–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigaard N.-U., Sakuragi Y., and Bryant D. A. (2004) Gene inactivation in the cyanobacterium Synechococcus sp. PCC 7002 and the green sulfur bacterium Chlorobium tepidum using in vitro-made DNA constructs and natural transformation, in Photosynthesis Research Protocols, pp 325–340, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Khodursky A., Bernstein J., Peter B., Rhodius V., Wendisch V., and Zimmer D. (2003) Escherichia coli spotted double-strand DNA microarrays, in Methods Mol. Biol. (Brownstein M., and Khodursky A., Eds.), pp 61–78–78, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.