

Abstract
Over the past 4 decades, basic research has provided crucial information regarding the cellular and molecular biology of cancer. In particular, the relevance of cancer microenvironment (including both cellular and noncellular elements) and the concept of clonal evolution and heterogeneity have emerged as important in cancer pathogenesis, immunologic escape, and resistance to therapy. Multiple myeloma (MM), a cancer of terminally differentiated plasma cells, is emblematic of the impact of cancer microenvironment and the role of clonal evolution. Although genetic and epigenetic aberrations occur in MM and evolve over time under the pressure of exogenous stimuli, they are also largely present in premalignant plasma cell dyscrasia such as monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM), suggesting that genetic mutations alone are necessary, but not sufficient, for myeloma transformation. The role of bone marrow microenvironment in mediating survival, proliferation, and resistance to therapy in myeloma is well established; and although an appealing speculation, its role in fostering the evolution of MGUS or SMM into MM is yet to be proven. In this review, we discuss MM pathogenesis with a particular emphasis on the role of bone marrow microenvironment.
Characteristics of the myeloma cancer clone
Ontogenesis of myeloma
Multiple myeloma (MM) represents the far end of the spectrum of B cell–derived neoplasms. It is the neoplastic counterpart of terminally differentiated, immunoglobulin-producing, long-lived plasma cells (PCs). Long-lived PCs are a subset of PCs characterized by long-term (months to years) survival within the bone marrow (BM) and thought to be key for immunologic memory.1,2 Based on the sequencing of the immunoglobulin heavy chain (IgH) variable region of MM cells (MMCs), the first oncogenic events in MM appear to occur in the germinal center, likely during the processes of isotype class switching and somatic hypermutation, which are, by nature, mutation prone.3 The observation that patients with premalignant PC dyscrasia monoclonal gammopathy of undetermined significance (MGUS) and/or smoldering MM (SMM) also carry these initial mutations suggests that they are necessary, but not sufficient, in MM pathogenesis. Late oncogenic events are thought to occur in the BM, after the founder cancer clone is completely differentiated into a long-lived PC (Figure 1).4
Figure 1.

Pathogenesis of MM. The orange round cell represents a normal B cell, whereas the yellow round cell is a mutated, post–germinal center (GC) B lymphocyte that later differentiates into a long-lived PC (yellow oval). In MM pathogenesis, the initial genetic event (red square) is thought to occur in the GC, facilitated by the processes of somatic hypermutation and isotype switching, and characterizes the founder clone (F). Later genetic mutations occur at the time of transformation to MM (red circle), with de novo mutations (red geometric shapes) acquired during disease evolution and heterogeneously present in different subclones (S1 and S2). The genetic, epigenetic, and biological events occurring in the cancer clones and BM microenvironment during the evolution of premalignant dyscrasia to MM are outlined in the pink, green, and blue boxes, respectively. ECM, extracellular matrix.
There is ongoing debate regarding the identity of the MM stem cells. Different groups have shown that both CD138+ and CD19+/CD27+/CD38−/CD138− cells are capable of tumorigenesis in mouse models. However, CD138+ tends to lose self-renewing potential after a few cycles of serial transplantation, whereas the putative B-cell stem clone was never proved to be clonally related to its putative CD138+ progeny. Overall, modification in the cytokine composition of the media used to maintain CD138+ ex vivo successfully overcame the first issue, suggesting that the MM stem cell may be CD138+.5
Evolution of MM from precursor dyscrasia
In 2 independent retrospective studies, MGUS was proved almost universally to precede the development of MM with a lifelong rate of malignant transformation of 1% per year.6-8 MGUS is a common condition, being present in about 3% of white individuals aged >70 years and having an incidence increasing with age.9 Although several laboratory and clinical predictive factors of neoplastic evolution of MGUS have been identified, the molecular basis of this transformation remains unclear. The neoplastic BM microenvironment has been hypothesized as a major determinant of such evolution.9 Certain intrinsic characteristics of the MGUS clone (in particular, presence of cytogenetic abnormalities and/or DNA aneuploidy, non-IgG isotype of immunoglobulin production, and monoclonal protein quantification over 1.5 g/L) are predictors of MM progression. Free light chain–only MGUS has a lower rate of evolution to MM compared to full immunoglobulin-producing MGUS.10 Infiltration of BM by malignant PCs exceeding 5%, presence of circulating PCs, and suppression of polyclonal normal BM PCs as assessed by multiparametric flow cytometry and/or serum immunoglobulin level are also predictive factors of malignant transformation. Detectable Bence Jones proteinuria and radiologically occult bone lesions on magnetic resonance imaging and/or positron emission tomography with computed tomography scans represent early signs of PC dyscrasia–related organ dysfunction.11,12 Finally, dynamic changes in the level of the monoclonal (M) component with a progressive increase in the size of the M spike are also predictive of disease progression.13 Similar risk factors exist for evolution of SMM to active MM. Whereas MGUS progresses to MM and related dyscrasia with an unremitting rate of 1% per year, SMM patients have a 10%-per-year rate of progression within the first 5 years of diagnosis, which then progressively reduces but never disappears, thus prompting more intense monitoring as compared to MGUS.14 Recently, diagnostic criteria for PC dyscrasia have been revised in light of the natural history of a subset of SMM patients whose disease is characterized by rapid progression to MM, thus biologically behaving as active disease.14 This is the case for SMM patients with BM infiltration by malignant PCs exceeding 60% and a free light-chain ratio favoring the malignant light chain by >100-fold or with >1 bone lesion detected via magnetic resonance imaging.14
Molecular and signaling mechanisms in myeloma pathogenesis
Based on karyotype, MM is classified as nonhyperdiploid and hyperdiploid, with the latter accounting for 50% to 60% of cases and characterized by trisomies in odd chromosomes (3, 5, 7, 9, 11, 15, 19, and 21). In contrast, nonhyperdiploid MMCs harbor translocations between 14q32, the locus of IgH, and one of several partner oncogenes like MAF (v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog), MAFB, MM SET domain (MMSET), fibroblast growth factor receptor 3 (FGFR3), and cyclins D1 and D3.
Later oncogenic events in MM pathogenesis include mutation- or expression-mediated activation of oncogenes such as Kirsten rat sarcoma RAS viral (v-ras) oncogene homolog (K-RAS) and neuroblastoma RAS viral (v-ras) oncogene homolog (N-RAS), v-myc avian myelocytomatosis viral oncogene homolog (MYC), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), v-akt murine thymoma viral oncogene homolog (AKT), and B-Raf proto-oncogene (BRAF); and/or loss of function of oncosuppressor genes such as tumor protein 53 (TP53) and retinoblastoma 1 (RB1) (Figure 1).
The balance between proapoptotic and antiapoptotic members of the B-cell CLL/lymphoma 2 (BCL-2) family of proteins is key to determine the fate of a cell. Cancer cells (in particular, hematologic cells) frequently show upregulation of antiapoptotic members such as BCL-2, BCL2-like 1 (BCL2L1, also known as BCL-XL), and myeloid cell leukemia 1 (MCL-1). Translocation t(14;18) involving the BCL-2 locus was reported in 2% to 3% of MM patients, but protein level is elevated more frequently in primary MMCs and MM cell lines (MMCLs), suggesting a different mechanism of upregulation.15 BCL2L1 and MCL-1 are similarly upregulated in MM via the interleukin (IL)-6–signal transducer and activator of transcription 3 (STAT3) axis.16 Overexpression of the antiapoptotic BCL-2 proteins correlates with resistance to apoptosis in vitro and genomic instability, and it was predictive of treatment failure with interferon.17
The transcription repressor B-cell CLL/lymphoma 6 (BCL-6) is a protooncogene in MM whose expression is significantly upregulated in the context of the BM milieu through the IL-6/STAT3, Janus kinase (JAK), and tumor necrosis factor (TNF)-α/canonical nuclear factor (NF)-κB pathways.18
Constitutive signaling via the transcription factor X-box binding protein 1 (XBP-1), a key molecule in guiding commitment and differentiation to PCs and a downstream effector of the endoplasmic reticulum to nucleus signaling 1 (ERN1, also known as IRE1) branch of the unfolded protein response, was shown to be pathogenic in a MM murine model.19 In line with this hypothesis, elevated messenger RNA level of the active, spliced form of XBP-1 (sXBP-1) correlated with poor prognosis in MM.20 However, most recently, lack of sXBP-1 was proved to lead to decommitment from PC fate, decreased immunoglobulin production, escape from proteasome inhibitor–induced death, and eventually therapy failure and disease relapse, suggesting a dual role of this protein.21
Overactivation of the transcription factors paired box 5 (PAX5), involved in the early process of B lymphomagenesis, and interferon-regulating factor 4 (IRF-4), a modulator of innate and adaptive immunity, have been reported as critical for MM pathogenesis.22,23
The pathogenic significance of other observed mutations in primary MMCs such as family with sequence similarity 46, member C (FAM46C), a putative oncosuppressor gene encoding for a protein of unclear function; SP140 nuclear body protein (SP140), a homolog of SP100 nuclear antigen (SP100) involved in B-cell antigenic response; the transmembrane receptor roundabout 1 (ROBO1), previously described as a key element of axonal guidance during neuronal development in embryos; and the transcription factor early growth response 1 (EGR1) remains largely unknown, and validation of these candidate genes is currently ongoing.24,25
Epigenetics
Modulation of gene expression by means other than from alteration of DNA sequence has emerged as an important pathogenic mechanism in cancer, with epigenetic studies revealing novel molecular mechanisms of tumorigenesis.26,27 Large-scale epigenomic analysis of MMCs has been undertaken in an effort to further understand the pathogenesis of the disease and, in particular, the mechanisms underlying malignant transformation of MGUS and SMM.
Methylation of cytosines embedded in cytosine-phosphate guanine (CpG) islands in the promoter region of target genes is a well-defined epigenetic mechanism of transcriptional silencing.28 A number of tumor suppressor genes are hypermethylated early in the course of MM, with evidence that methylation increases during the process of MM evolution, reaching its acme at the PC leukemia stage.29,30 Histone methylation and acetylation are also altered in MM.31 Histone deacetylases are generally hyperactive in MM, resulting in globally increased gene transcription; and alteration of the MMSET gene via the t(4;14) results in aberrant histone methylation, with the net effect of increasing transcription of several oncogenes.32 Bromodomains exert an epigenetic function via their direct interaction with acetylated lysine residues. The bromo and extra-terminal (BET) family of bromodomains was recently proved to induce Myc expression in MM and proved to be a potential drug target in anti-MM therapy.33
MicroRNA (miR) microarray analysis identified altered expression of several miRs in primary MMCs derived from MGUS and MM patients.34 miR-21, miR-32, miR-17-92, miR-106b, miR-181a and b, miR-221, miR-222, and miR-382 are overexpressed in MMCs compared to normal PCs, whereas miR-15a and miR-16 are downregulated. Interestingly, although certain miRs are overexpressed also in MGUS patients, others such as miR-32 and miR-17-92 are only upregulated in MM, suggesting a potential pathogenic role of the latter in malignant evolution from MGUS. Although the pool of genes targeted by the miRs deregulated in MM is different, the overall result of their dysregulation in MM is increased proliferation and resistance to apoptosis.35
Genomic instability: a pathogenic mechanism for progressive disease
In a 1976 seminal paper, Dr Nowell first introduced the concept of clonal heterogeneity and evolution in cancer by adapting the Darwinian theory of random genetic mutations and environment-based selection of the fittest clones to cancer pathogenesis.36,37 Array comparative genomic hybridization, whole-exome sequencing, and whole-genome sequencing proved that clonal heterogeneity increased during MM pathogenesis from MGUS via SMM to MM and eventually PC leukemia, consistent with worsening genomic instability during disease evolution.24,25,38 The potential impact of certain therapies (in particular, alkylating agents and lenalidomide) on increased genomic instability and clonal evolution has been reported.39,40
Deep sequencing of IgH and immunoglobulin light chain (IgL) loci proved oligoclonality in 12% of MMCs obtained from a cohort of 193 MM patients. The immunoglobulin sequence was related in two-thirds of these patients, suggestive of evolution from a common founder clone, with immunoglobulin isotype switch and somatic hypermutation responsible for the formation of clones in 73% and 27% of patients, respectively.41 These observations suggest that the processes of isotype switching and somatic hypermutation can persist after evolution of MGUS to MM and terminal differentiation in PCs.
Analysis of paired samples obtained from MM patients at different times during the course of their illness provided the opportunity to study the pattern of evolution of clones over time. Four patterns have been described most recently, underscoring the intricate and dazzling nature of biological variability.25
High-density single nucleotide polymorphism array analysis in newly diagnosed MM patients revealed structural abnormalities (deletions and amplifications) to be almost universally present in MM patients (98% of individuals in a cohort of 192).42
Recently, ongoing DNA damage response was found activated at baseline in MMCLs and primary cells in association with active nonhomologous end joining and homologous recombination.43,44 Eukaryotic cells are equipped with a number of stress response pathways, such as the unfolded protein response or the DNA damage response, whose function is the maintenance of homeostasis in the face of its perturbation. Physiologically, if cell stress is overwhelming in duration or intensity, or if the damage to the cell cannot be repaired, these pathways activate the apoptosis cascade. Most recently, loss of function of the transcription cofactor Yes-associated protein 1 (YAP1), a downstream molecule in the Hippo pathway, was reported as relevant in MM escape from DNA damage, providing some insight into the molecular mechanisms behind resistance to stress-induced apoptosis.44
Beyond the cancer clone(s): the importance of the BM milieu
Tumor cells do not grow isolated from their surroundings, but they rather establish close ties with the microenvironment important for tumor survival and progression (Figure 2).45 Unlike solid malignancies, where the sites of primary disease and metastases are typically distinct, MM is characterized by widespread cancer involvement of multiple sites within the same microenvironment: the BM. The BM niche, therefore, acquires primary interest as a pathogenic factor in MM. Although the BM milieu has been shown to induce tumor proliferation, resistance to apoptosis, and cancer cell trafficking and homing, a definite pathogenic role for the BM niche in the progression of MGUS and SMM to active MM remains under investigation.46 It is well established that a bidirectional signaling loop exists between MMCs and BM microenvironment cells and that BM microenvironment of MM patients differs in its cellular and noncellular composition from that of healthy individuals (Figure 2).47
Figure 2.
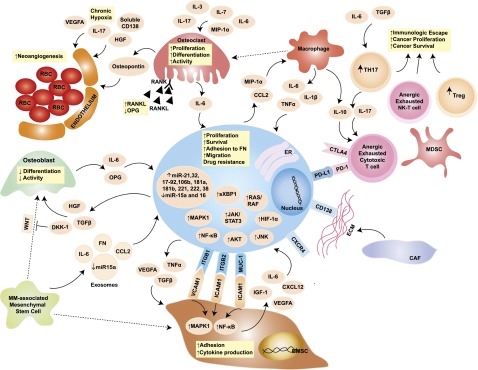
Role of the BM niche in MM pathogenesis. The blue oval in the center is the MMC, with its close interplay with cellular and acellular components of the BM. The pale orange ovals represent relevant cytokines/chemokines in the BM milieu. Dotted arrows indicate differentiation, whereas solid arrows indicate secretion and/or effect on a target cell. Yellow squares contain a synopsis of the overall effect of cytokines and cell-to-cell contact on the target cell. Key signaling cascades, transmembrane proteins, and intracellular organelles, which are of interest for molecularly targeted therapies, are represented. BMSC, BM stromal cell; CAF, cancer-associated fibroblast; CCL2, chemokine (C-C motif) ligand 2; CTLA4, cytotoxic T-lymphocyte-associated protein 4; CXCL12, chemokine (C-X-C motif) ligand 12; CXCR4, chemokine (C-X-C motif) receptor 4; DKK-1, dickkopf WNT signaling pathway inhibitor 1; ER, endoplasmic reticulum; FN, fibronectin; HGF, hepatocyte growth factor; HIF-1α, hypoxia-inducible factor 1α; ICAM1, intercellular adhesion molecule 1; IGF-1, insulinlike growth factor 1; ITGB1, integrin β1; ITGB2, integrin β2; JNK, c-JUN N-terminal kinase; MAPK1, mitogen-activated protein kinase 1; MDSC, myeloid-derived suppressor cell; MIP-1α, macrophage inflammatory protein 1α; MUC-1, mucin 1; NK-T cells, natural killer T cells; OPG, osteoprotegerin; PD-1, programmed cell death 1; PD-L1, programmed ligand death 1; RANK, receptor activator of NF-κB; RANKL, RANK ligand; RBC, red blood cell; TGF-β, transforming growth factor β; TH17, T helper 17 cell; Treg, regulatory T cell; VCAM1, vascular cell adhesion molecule 1; VEGFA, vascular endothelial growth factor A; WNT, wingless-type. Adapted from Bianchi and Anderson47 with permission.
BM microenvironment cells
Similar to other cancers with bone-metastatic potential, such as breast and prostate adenocarcinoma, MMCs tend to localize in proximity to osteoblasts (OBs) and vascular endothelium, hijacking the BM niche, which in healthy physiological conditions, supports hematopoietic stem cells.48,49 OBs and endothelial cells (ECs) are only 2 of the cellular components of the heterogeneous BM microenvironment. Stromal cells, fibroblasts, adipocytes, osteoclasts (OCs), macrophages, dendritic cells (DCs), and lymphocytes are other important players in MM pathogenesis and are discussed separately in the following paragraphs.
BMSCs.
BMSCs are a major constituent of the BM space, where they support and nurture the hematopoietic cells.50 Similarly to other cellular components of the BM, malignant and normal BMSCs are functionally different, and BMSCs derived from MM patients support MM proliferation, resistance to apoptosis, drug resistance, migration, and invasion via both direct cell-to-cell contact and through secretion of cytokines and chemokines.46 Several molecular mechanisms mediating such functions have been identified. The interaction of BMSCs with MMCs via the vascular cell adhesion molecule 1/integrin β1 (VCAM1/ITGB1), intercellular adhesion molecule 1/integrin β2 (ICAM1/ITGB2), and mucin 1 cell surface–associated (MUC1) axes triggers a bidirectional signaling cascade, leading to activation of NF-κB and MAPK1 pathways in BMSCs and of MAPK1 in MMCs.46 In turn, NF-κB and MAPK1 signaling causes the secretion of proproliferative, antiapoptotic, and chemotactic cytokines such as IL-6, CXCL12, IGF-1, and VEGFA, which further support myelomagenesis. Furthermore, engagement of the Notch1 receptor on MMCs by BMSCs results in protection from alkylating and intercalating agents by a p21-dependent and NF-κB–independent mechanism.51
Mesenchymal stem cells.
Mesenchymal stem cells (MSCs) are precursor cells retaining the capacity to self-renew and differentiate into a variety of cell types, including fibroblasts, adipocytes, chondrocytes, and OBs/osteocytes.52 In vitro, they are the precursors of BMSCs. MM-associated MSCs (MM-MSCs) and healthy donor–derived (HD)-MSCs differ at baseline in their genomic and cytokine production profile. MM-MSCs produce more VEGFA, IL-6, IL-1β, and TNF compared to their normal counterparts, thus favoring MMC proliferation and inhibiting OB function and differentiation.53 MM-MSCs further favor tumorigenesis by transferring oncogenic factors, including IL-6, chemokine (C-C motif) ligand 2 (CCL2), and fibronectin, to MMCs via exosomes.54 Exosomes released by MM-MSCs were also noted to contain a different pattern of miRs; in particular, a lower level of the oncosuppressor miR-15a. Overall, in vitro exposure of MMCs to MM-MSC-derived exosomes induced MMC proliferation, secretion of IL-6, and adhesion to fibronectin, which reflected tumor growth and increased BM homing in animal models. However, HD exosomes displayed an antitumorigenic effect on MMCs in vitro and in vivo.54 Recently, MM-MSCs were also found to have upregulated miR-135b, resulting in decreased SMAD family member 5 (SMAD5) expression and impaired capacity of OB differentiation.55 Whether these functional differences between MM-MSCs and HD-MSCs are acquired or intrinsic is still a matter of debate. Indeed, MM-MSCs carry genomic abnormalities that are not present in HD-MSCs, and their normal regenerative function appears to remain impaired long-term, even in disease-free patients.56,57 However, the contribution of such genetic mutations to the phenotype is unclear because HD-MSCs cocultured with MMCs in vitro acquire a phenotype similar to MM-MSCs in a matter of hours.58
Despite these differences, in vitro coculture of an MMCL with either type of MSC resulted in similar upregulation of genes involved in MM chemotaxis, neoangiogenesis, OC induction, and OB inhibition. However, genes associated with ribosome function, the ubiquitin-proteasome pathway, and the noncanonical WNT pathway were modulated only in patient-derived MSCs.59 These results suggest that myeloma cells exert a major role in influencing the function of MSCs, but that healthy and cancer-derived MSCs also respond differently to this interaction.
The osteoblastic niche.
OBs are the bone cellular component responsible for apposition of new bone, thus counterbalancing the function of OCs, the bone-resorbing cells. Various hormones, bone paracrine molecules, nutrients, drugs, and disease states influence the function of either cell type, modulating bone remodeling. In a physiological healthy state, equilibrium between OB and OC function exists, thus guaranteeing adequate bone mass.60 OBs differentiate from BM MSCs under the effect of both hormonal and paracrine stimuli, with parathyroid hormone, glucocorticoid hormones, and estrogens being the major hormonal regulators, whereas TGF-β and bone morphogenetic proteins are important paracrine factors. Murine models unequivocally showed that the transcription factor Runt-related transcription factor 2 (RUNX2) is the master regulator of OB differentiation and that the canonical WNT pathway plays a key role in osteoblastogenesis.61-65
In the long bones, OBs localize in the endosteum at the junction between trabecular bone and red marrow, where they play an important role in the support of hematopoietic stem cells.66 In MM patients, the osteoblastic niche is depleted in favor of an overabundance of OCs, which support cancer cell proliferation and resistance to apoptosis and whose exuberant activity is responsible for MM-related bone disease and lytic lesions.67 In vitro, MMCs favor osteoclastogenesis while inhibiting osteoblastogenesis. Molecular mechanisms of the antiosteoblastic effect of MMCs include downregulation of Runx2 in MSCs and differentiated OB progenitors; increased production of WNT pathway inhibitors, including dickkopf WNT signaling pathway inhibitor 1 (DKK1); secretion of antiosteoblastic factors such as TGF-β and hepatocyte growth factor; and constitutive activation of the Notch pathway.53
On the other hand, in coculture with MMCs, OBs increase the secretion of IL-6, thus supporting MM proliferation.68 Furthermore, the paracrine release of osteoprotegerin seems to be protective against TNF-related apoptosis-inducing ligand/Apo2 ligand (TRAIL), thus functioning as an antiapoptotic stimulus.69
OCs.
A maladaptive prosurvival and bidirectional loop also exists among OCs (the bone-resorbing cells) and MMCs.70 This interaction is clinically relevant because it results in MM-related bone disease, manifesting as osteopenia, lytic lesions, and eventually pathological fractures.71 Increased osteoclastogenesis in MM is largely determined by the aberrant composition of the soluble BM milieu. The normal balance between the proosteoclastogenic TNF ligand superfamily member 11a (TNFSF11A, also known as RANKL) and the antiosteoclastogenic RANK-decoy receptor osteoprotegerin is lost in MM in favor of the former. Other pro-OC cytokines such as MIP-1α, IL-6, IL-7, and IL-3 are also detected at higher concentration in the serum of MM patients compared with healthy donors and contribute to osteoclastogenesis and MM-related bone disease.71 Moreover, as discussed later, a number of hematopoietic cells can differentiate into OCs in the context of the abnormal MM BM milieu, thus further fueling the maladaptive MMC-OC axis.
OCs contribute to MM pathogenesis, not only via their bone-resorbing properties but also by secreting IL-6 and osteopontin, thus stimulating MM proliferation and angiogenesis, respectively.72
Vascular endothelial niche and angiogenesis.
Neoangiogenesis and increased vessel density, hallmarks of MM evolution from MGUS and SMM, portend an adverse prognosis after high-dose therapy and hematopoietic stem cell transplant.73,74 Although constitutive activation of HIF-1α; aberrant expression of HIF-2 by MMCs; and elevated levels of VEGFA, hepatocyte growth factor, and syndecan-1 have all been shown to promote angiogenesis in MM, increased tumor burden and loss of an inhibitor of angiogenesis during evolution from MGUS to MM have proved to be major determinants of increased microvessel density in MM patients.75,76
Similarly to MSCs, endothelial cells derived from MM patients display a gene expression profile that is characteristically different from ECs obtained from MGUS patients and, overall, is characterized by the upregulation of chemotactic, neoangiogenetic, and proproliferative factors, including CXCL12 and elements of the ECM.77
CAFs and adipocytes.
Fibroblasts differentiate from MSCs and generally play a structural, supportive role in tissues. Fibroblasts infiltrating the cancer microenvironment have been shown to functionally differ from their normal counterparts. In particular, CAFs play a pivotal role in inducing epithelial-mesenchymal transition and promoting metastasis in solid malignancies.78 In mouse models, a bidirectional loop between MMCs and CAFs was demonstrated, with the former inducing CAF proliferation, which in turn promoted MM progression and angiogenesis.79
Furthermore, BM fibroblasts from patients with PC dyscrasia produce ECM components that differ from those of healthy individuals.80 In particular, during the evolution from MGUS to MM, a pool of ECM proteins is progressively upregulated; most prominently, integrin α5β5, periostin, matrix metalloproteinase 2 (MMP2), platelet-derived growth factor receptor β, laminin α4, plasminogen activator inhibitor-1, lysyl-hydroxylase 2, prolyl 4-hydroxylase 1, nidogen-2, c-type mannose receptor 2, and basigin.
Adipocytes were also shown to support MMC proliferation, survival, and migration in vitro partially through the effect of the soluble factor leptin, suggesting that these cells, abundant in the BM of elderly individuals, might have a role in MM pathogenesis.81
T, natural killer, and NK-T lymphocytes.
Similar to other cancers, MMCs are capable of escaping immunologic surveillance by inducing immune tolerance and T-cell anergy. Indirect evidence of the importance of an immune response in limiting MMC growth and survival is provided by the prolonged disease-free survival experienced by a number of MM patients post–allogeneic hematopoietic stem cell transplant, where the graft-versus-myeloma effect is thought to be the main determinant of therapeutic success. The composition of lymphocytes present in the MM microenvironment substantially differs from that in a healthy subject.82,83 Under the priming of elevated concentration of IL-6 and TGF-β, TH17 cells are abundant in the BM of MM patients.84,85 These are a distinct subset of CD4+ T helper lymphocytes characterized by a peculiar pattern of cytokine production: IL-17, IL-17F, IL-21, and IL-22.86 In the context of the BM cytokine milieu of MM patients, TH17 cells suppress cancer immune surveillance by secreting IL-17 and IL-10. IL-17 also serves as a proosteoclastogenesis factor, providing an indirect mechanism of MMC support and contributing to MM-related bone disease.85 Moreover, in solid tumors, IL-17 has been shown to be a proangiogenic factor and a chemoattractant for MDSCs, providing 2 other mechanisms of tumor pathogenicity.87,88 Such functions have not been described in MM to date but raise the possibility of a broader procancer activity of TH17 cells. Tregs have a similar immunosuppressive function on cytotoxic T cells and antigen-presenting cells via both cell-to-cell contact and release of IL-10 and TGF-β.89 MM-derived Tregs are characterized by a higher secretion of these cytokines, consistent with a functional aberrancy, and increased functional Tregs in the peripheral blood of MM patients directly correlate with worse prognosis.90
Cytotoxic CD8+ T cells in the BM microenvironment of MM patients differ from their counterparts in healthy subjects in the repertoire of T-cell receptor (TCR) co-receptor molecules. Increased expression of PD-1, a TCR co-receptor with inhibitory function, was shown on BM-resident cytotoxic T cells of MM patients. Coupled with overexpression of PD-L1 on the surface of MMCs, the increased expression of PD-1 on cytotoxic T cells account as one of the major mechanisms of immunologic tolerance in MM.91,92 Clinical trials implementing pharmacologic blockade of PD-1 or PD-L1 are currently ongoing, predicated on these observations and the positive results obtained with these molecules in solid cancer, particularly melanoma.93-95
Beyond their role in the immunologic escape of MMCs, T cells also contribute to MM-related bone disease by supporting osteoclastogenesis. In an IL-6– and IL-7–mediated process, activated T cells modify their pattern of cytokine production by increasing RANKL and by decreasing interferon γ secretion, thus favoring OC proliferation and activity.96
Natural killer (NK) cells are also functionally impaired in MM patients with downregulation of the NK group 2D (NKG2D) activating receptor and upregulation of the inhibitory co-receptor PD-1.90 During the evolution of MGUS to MM, cells acquire the capacity of shedding major histocompatibility complex class I polypeptide-related sequence A (MICA), a ligand for NKG2D that triggers NK cell–mediated lysis when expressed on the cell surface. However, soluble MICA causes downregulation of NKG2D, contributing to MMC immune escape.97
NK-T cells are CD-1d restricted T cells that bind to lipidic antigens. Type I NK-T (iNKT) cells, expressing an invariant TCR, have been described to play an important role in anticancer surveillance, largely by stimulating DC and NK cell activity.98 In MM, progression of disease and relapse is associated with deficiency in NK-T cells.99 Stimulation of iNKT cells via treatment with α-galactosylceramide–loaded monocyte-derived DCs has been reported, with augmentation with lenalidomide in vitro.100 Therapy with such iNKT cells with low-dose lenalidomide resulted in clinical responses in a small cohort of SMM patients, associated with activation of iNKT cells, NK cells, and monocytes.101
MDSCs.
MDSCs represent the result of a short-circuit arrest in the normal differentiation process of myeloid precursors in the BM induced by the abnormal BM cytokine milieu in states of chronic inflammation such as cancer, infection, and trauma.102 These pathological states cause accumulation of a heterogeneous population of immature myeloid precursors with an immunosuppressive function, thus the name MDSCs. Several groups demonstrated the capacity of MMCs to expand MDSCs in murine models, which is hypothesized to contribute to myeloma pathogenesis at least in 2 ways: first, by exerting an antiproliferative effect against lymphocytes via increased nitric oxide production, l-arginine depletion, and IL-10 secretion; and second, by differentiating into OCs in the context of a RANKL-rich milieu.103,104
Macrophages.
Within the tumor microenvironment, MDSCs can also differentiate into tumor-associated macrophages (TAMs), which are phenotypically and functionally distinct from their precursor cells. TAMs can also be the product of differentiation of circulating monocytes, which are attracted to cancer cells via chemoattractants.105 MMCs secrete 2 potent macrophage attractants: CCL2 and MIP-1α.106 TAMs contribute to MM pathogenesis in 3 different ways. First, they are a major source of IL-6 as well as other pro-MM cytokines such as TNF-α and IL-1β. Second, they produce IL-10, a major mediator of cancer immune tolerance by suppressing the function of T cells. Lastly, macrophages contribute substantially to angiogenesis by releasing VEGFA and nitric oxide and acquiring the capability of differentiating into EC-like cells under the autocrine/paracrine effect of VEGFA.107,108
The role of hypoxia in MM pathogenesis
The BM microenvironment is hypoxic, resulting in elevated expression of HIF-1α and VEGFA and increased neoangiogenesis in MM patients.109 In vitro, HIF-1α was shown to be necessary for BMSC- and IL-6–induced MM proliferation and survival.110 Mouse models harboring MMCs lentivirally transduced with short hairpin RNA against HIF-1α demonstrated decreased tumor burden, neoangiogenesis, and MM-related bone destruction, thus supporting the development of molecularly targeted HIF-1α inhibitors for MM treatment.111 Moreover, hypoxia drives epithelial-to-mesenchymal transition in MM, thus promoting tumor trafficking and metastasis, and appears to induce a stemlike phenotype in vitro.112-114 Recently, recapitulation of chronic hypoxia in vitro caused increased exosome production in MMCs, with upregulation of miR-135b content.115 miR-135b directly downregulated factor-inhibiting HIF-1 in endothelial cells, resulting in increased angiogenesis. Finally, hypoxia has been exploited to selectively deliver cytotoxic drugs to MMCs via the hypoxia-activated prodrug TH-302, now in clinical development.116,117
Soluble factors
MMCs and BMSCs profoundly influence each other’s behavior via a variety of molecular mechanisms. Among these, changes in the secretion of cytokines, growth factors, and chemokines play a prominent role in supporting cancer cell growth, survival, trafficking, and resistance to therapies.
IL-6.
BMSCs and macrophages are the main source of IL-6 production.118 In a bidirectional loop, binding of BMSCs to MMCs triggers increased IL-6 production by BMSCs, which in turn acts in a paracrine fashion on MMCs.119 Other BM milieu cytokines such as TNF-α, IL-1β, and IL-17 also induce increased secretion of IL-6 by BMSCs.120,121 The binding of IL-6 to its receptor on the MMC membrane triggers signaling via the MAPK, JAK2/STAT3, and PI3K/AKT pathways, resulting in MMC proliferation and survival. Importantly, IL-6 is one of the mediators of dexamethasone resistance in MMCs via protein tyrosine phosphatase, nonreceptor type 11 (PTPN11, also known as SHP2).122,123
IGF-1.
Similar to IL-6, IGF-1 is an important mediator of MMC growth, survival, migration, and dexamethasone resistance.124 In vitro, IGF-1 signaling via the PI3K/AKT, MAPK, and NF-κB pathways results in increased telomerase activity and upregulation of the antiapoptotic molecules, such as surviving cellular FADD-like IL-1β–converting enzyme (FLICE)-inhibitory protein (c-FLIP), X-linked inhibitor of apoptosis protein (XIAP), cellular inhibitor of apoptosis 2 (cIAP-2), and BCL-2–related protein A1 (BFL1).125,126
CXCL12/CXCR4 and the relevance of BM homing in MM.
CXCL12 (also known as stromal cell–derived factor 1α, SDF-1α) is the ligand of CXCR4. The former is produced by BMSCs, whereas MMCs express the latter.127 CXCL12 is highly expressed at BM sites of metastatic disease from solid tumor or MM and plays a major role in directing homing and trafficking of MMCs.128,129 Signaling through CXCL12-CXCR4 results in modest direct effect on MM proliferation and resistance to dexamethasone. However, CXCL12 induces IL-6 and VEGF secretion by BMSCs, thus indirectly promoting MMC proliferation, antiapoptosis, neoangiogenesis, and resistance to therapy. Importantly, blockade of the CXCL12/CXCR4 pathway in murine MM models results in decreased MMC homing and growth, thus halting disease progression.129
Exosomes
Most recently, cancer cells and microenvironment cells have been shown to exchange macromolecules, including nucleic acid, via exosomes and microvesicles.130-132 MMCs have the capability of uptaking miRs contained in BM mesenchymal cell/BMSC–derived exosomes.54 The exogenously acquired miRs modulate gene expression similarly to endogenous miRs, providing a further layer of complexity in the epigenetic modulation of genes in MM. Of note, Roccaro and colleagues63 reported that the content of exosomes differs significantly between normal and MM-derived BM mesenchymal cells/BMSCs. For instance, the oncosuppressor miR-15a is downregulated in exosomes derived from BM mesenchymal cells/BMSCs derived from MM patients. Furthermore, exosomes have been shown to mediate drug resistance in MM.133 This observation further supports the hypothesis that the cancer microenvironment is not just an innocent bystander in the oncogenic process but contributes actively to cancer pathogenesis. Although there is no scientific evidence at this time, it has been postulated that exosomes may transfer genetic material (in particular, oncogenes) into tumor cells, suggesting a completely novel mechanism for oncogenesis.133
Extracellular matrix
There is growing evidence that proteins in the ECM play an active role in supporting MMC growth and survival. Proteomic analysis of BM-resident fibroblasts obtained from healthy individuals vs patients with MGUS and MM demonstrated a clearly different spectrum of ECM protein production, suggesting that a bidirectional signaling loop exists between fibroblasts and MMCs, resulting in the production of an ECM component more suitable for cancer cell growth.80 Indeed, in vitro studies demonstrated that hyaluronan and fibronectin, 2 components of the ECM, mediate drug resistance in MM.134
Research efforts are now directed at understanding the molecular mechanisms by which MMCs modify the pattern of ECM protein production in BM-resident fibroblasts and investigating whether MMCs themselves are responsible for the secretion of ECM components that are predominant during disease evolution from MGUS.
Syndecan-1 (also known as CD138), a type I transmembrane heparan sulfate proteoglycan, is highly expressed by MMCs and can undergo proteolytic cleavage and shedding in the extracellular environment. Although the transmembrane form binds to collagen type I in the ECM and is relevant for MMC adhesion, the shed form promotes MMC growth, invasion, and migration in a murine myeloma model. Consistent with this model, high levels of soluble syndecan-1 correlate with tumor burden and worse prognosis in MM patients.135
Conclusions and remarks
High-throughput technologies, most prominently deep sequencing, have identified various genomic aberrations that provide MMCs with the ability to proliferate in an uncontrolled fashion. A comprehensive understanding of the genomic landscape of MM and of the pivotal role of the BM microenvironment in providing the right milieu for further growth and survival explains the overall MM biology, particularly during the evolution from premalignant PC dyscrasia to active disease. The repercussions for patient care are immediate because an improved knowledge of the molecular mechanisms of MM pathogenesis continues to be the foundation for the development of more efficacious therapies targeting the cancer cells in the context of the malicious microenvironment.
Acknowledgments
This work was partly supported by National Institutes of Health National Cancer Institute grants PO1-155258, RO1-124929, P50-100707, and PO1-78378 and US Department of Veterans Affairs Merit Review Award I01 BX001584-01 (N.C.M.).
Authorship
Contribution: G.B. and N.C.M. wrote the paper.
Conflict-of-interest disclosure: N.C.M. is a consultant for Celgene, Onyx, Janssen, and Oncopep, and has an ownership interest in Oncopep. G.B. declares no competing financial interests.
Correspondence: Nikhil C. Munshi, 450 Brookline Ave, Boston, MA 02215; e-mail: nikhil_munshi@dfci.harvard.edu.
References
- 1.Anderson KC, Carrasco RD. Pathogenesis of myeloma. Annu Rev Pathol. 2011;6:249–274. doi: 10.1146/annurev-pathol-011110-130249. [DOI] [PubMed] [Google Scholar]
- 2.Hauser AE, Muehlinghaus G, Manz RA, et al. Long-lived plasma cells in immunity and inflammation. Ann N Y Acad Sci. 2003;987:266–269. doi: 10.1111/j.1749-6632.2003.tb06059.x. [DOI] [PubMed] [Google Scholar]
- 3.Seifert M, Scholtysik R, Küppers R. Origin and pathogenesis of B cell lymphomas. Methods Mol Biol. 2013;971:1–25. doi: 10.1007/978-1-62703-269-8_1. [DOI] [PubMed] [Google Scholar]
- 4.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–348. doi: 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 5.Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012;122(10):3456–3463. doi: 10.1172/JCI61188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 7.Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–5417. doi: 10.1182/blood-2008-12-194241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418–5422. doi: 10.1182/blood-2008-12-195008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kyle RA, Durie BG, Rajkumar SV, et al. International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24(6):1121–1127. doi: 10.1038/leu.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet. 2010;375(9727):1721–1728. doi: 10.1016/S0140-6736(10)60482-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van de Donk NW, Palumbo A, Johnsen HE, et al. European Myeloma Network. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica. 2014;99(6):984–996. doi: 10.3324/haematol.2013.100552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548. doi: 10.1016/S1470-2045(14)70442-5. [DOI] [PubMed] [Google Scholar]
- 13.Rosiñol L, Cibeira MT, Montoto S, et al. Monoclonal gammopathy of undetermined significance: predictors of malignant transformation and recognition of an evolving type characterized by a progressive increase in M protein size. Mayo Clin Proc. 2007;82(4):428–434. doi: 10.4065/82.4.428. [DOI] [PubMed] [Google Scholar]
- 14.Kyle RA, Remstein ED, Therneau TM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–2590. doi: 10.1056/NEJMoa070389. [DOI] [PubMed] [Google Scholar]
- 15.Pettersson M, Jernberg-Wiklund H, Larsson LG, et al. Expression of the bcl-2 gene in human multiple myeloma cell lines and normal plasma cells. Blood. 1992;79(2):495–502. [PubMed] [Google Scholar]
- 16.Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 17.Shaughnessy JD, Jr, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 18.Hideshima T, Mitsiades C, Ikeda H, et al. A proto-oncogene BCL6 is up-regulated in the bone marrow microenvironment in multiple myeloma cells. Blood. 2010;115(18):3772–3775. doi: 10.1182/blood-2010-02-270082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrasco DR, Sukhdeo K, Protopopova M, et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell. 2007;11(4):349–360. doi: 10.1016/j.ccr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagratuni T, Wu P, Gonzalez de Castro D, et al. XBP1s levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood. 2010;116(2):250–253. doi: 10.1182/blood-2010-01-263236. [DOI] [PubMed] [Google Scholar]
- 21.Leung-Hagesteijn C, Erdmann N, Cheung G, et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24(3):289–304. doi: 10.1016/j.ccr.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaffer AL, Emre NC, Lamy L, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226–231. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. 2007;8(5):463–470. doi: 10.1038/ni1454. [DOI] [PubMed] [Google Scholar]
- 24.Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236–1242. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 28.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 29.Heuck CJ, Mehta J, Bhagat T, et al. Myeloma is characterized by stage-specific alterations in DNA methylation that occur early during myelomagenesis. J Immunol. 2013;190(6):2966–2975. doi: 10.4049/jimmunol.1202493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaiser MF, Johnson DC, Wu P, et al. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood. 2013;122(2):219–226. doi: 10.1182/blood-2013-03-487884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimopoulos K, Gimsing P, Grønbæk K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014;4:e207. doi: 10.1038/bcj.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Garcia E, Popovic R, Min DJ, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117(1):211–220. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calvo KR, Landgren O, Roccaro AM, Ghobrial IM. Role of microRNAs from monoclonal gammopathy of undetermined significance to multiple myeloma. Semin Hematol. 2011;48(1):39–45. doi: 10.1053/j.seminhematol.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pichiorri F, Suh SS, Ladetto M, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci USA. 2008;105(35):12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 37.Darwin C. London, England: John Murray; 1859. On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. [PMC free article] [PubMed] [Google Scholar]
- 38.Walker BA, Wardell CP, Melchor L, et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia. 2014;28(2):384–390. doi: 10.1038/leu.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jelinek DF, Ahmann GJ, Greipp PR, et al. Coexistence of aneuploid subclones within a myeloma cell line that exhibits clonal immunoglobulin gene rearrangement: clinical implications. Cancer Res. 1993;53(21):5320–5327. [PubMed] [Google Scholar]
- 40.Palumbo A, Bringhen S, Kumar SK, et al. Second primary malignancies with lenalidomide therapy for newly diagnosed myeloma: a meta-analysis of individual patient data. Lancet Oncol. 2014;15(3):333–342. doi: 10.1016/S1470-2045(13)70609-0. [DOI] [PubMed] [Google Scholar]
- 41.Martinez-Lopez J, Fulciniti M, Barrio S, et al. Deep sequencing reveals oligoclonality at the immunoglobulin locus in multiple myeloma patients [abstract]. Blood. 2013;122(21). Abstract 401. [Google Scholar]
- 42.Avet-Loiseau H, Li C, Magrangeas F, et al. Prognostic significance of copy-number alterations in multiple myeloma. J Clin Oncol. 2009;27(27):4585–4590. doi: 10.1200/JCO.2008.20.6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood. 2009;113(10):2290–2297. doi: 10.1182/blood-2007-05-089193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cottini F, Hideshima T, Xu C, et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat Med. 2014;20(6):599–606. doi: 10.1038/nm.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 47.Bianchi G, Anderson KC. Understanding biology to tackle the disease: multiple myeloma from bench to bedside, and back. CA Cancer J Clin. 2014;64(6):422-444. [DOI] [PubMed]
- 48.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 49.Xie Y, Yin T, Wiegraebe W, et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature. 2009;457(7225):97–101. doi: 10.1038/nature07639. [DOI] [PubMed] [Google Scholar]
- 50.Bianco P, Gehron Robey P. Marrow stromal stem cells. J Clin Invest. 2000;105(12):1663–1668. doi: 10.1172/JCI10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nefedova Y, Cheng P, Alsina M, Dalton WS, Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103(9):3503–3510. doi: 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- 52.Olechnowicz SW, Edwards CM. Contributions of the host microenvironment to cancer-induced bone disease. Cancer Res. 2014;74(6):1625–1631. doi: 10.1158/0008-5472.CAN-13-2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toscani D, Bolzoni M, Accardi F, Aversa F, Giuliani N. The osteoblastic niche in the context of multiple myeloma. Ann N Y Acad Sci. 2014;1335:45–62. doi: 10.1111/nyas.12578. [DOI] [PubMed] [Google Scholar]
- 54.Roccaro AM, Sacco A, Maiso P, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123(4):1542–1555. doi: 10.1172/JCI66517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu S, Cecilia Santini G, De Veirman K, et al. Upregulation of miR-135b is involved in the impaired osteogenic differentiation of mesenchymal stem cells derived from multiple myeloma patients. PLoS ONE. 2013;8(11):e79752. doi: 10.1371/journal.pone.0079752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garayoa M, Garcia JL, Santamaria C, et al. Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia. 2009;23(8):1515–1527. doi: 10.1038/leu.2009.65. [DOI] [PubMed] [Google Scholar]
- 57.Reagan MR, Ghobrial IM. Multiple myeloma mesenchymal stem cells: characterization, origin, and tumor-promoting effects. Clin Cancer Res. 2012;18(2):342–349. doi: 10.1158/1078-0432.CCR-11-2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yaccoby S, Wezeman MJ, Zangari M, et al. Inhibitory effects of osteoblasts and increased bone formation on myeloma in novel culture systems and a myelomatous mouse model. Haematologica. 2006;91(2):192–199. [PMC free article] [PubMed] [Google Scholar]
- 59.Garcia-Gomez A, De Las Rivas J, Ocio EM, et al. Transcriptomic profile induced in bone marrow mesenchymal stromal cells after interaction with multiple myeloma cells: implications in myeloma progression and myeloma bone disease. Oncotarget. 2014;5(18):8284–8305. doi: 10.18632/oncotarget.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423(6937):349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 61.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 62.Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 63.Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 64.Babij P, Zhao W, Small C, et al. High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res. 2003;18(6):960–974. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- 65.Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 66.Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425(6960):836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 67.Giuliani N, Rizzoli V. Myeloma cells and bone marrow osteoblast interactions: role in the development of osteolytic lesions in multiple myeloma. Leuk Lymphoma. 2007;48(12):2323–2329. doi: 10.1080/10428190701648281. [DOI] [PubMed] [Google Scholar]
- 68.Karadag A, Oyajobi BO, Apperley JF, Russell RG, Croucher PI. Human myeloma cells promote the production of interleukin 6 by primary human osteoblasts. Br J Haematol. 2000;108(2):383–390. doi: 10.1046/j.1365-2141.2000.01845.x. [DOI] [PubMed] [Google Scholar]
- 69.Shipman CM, Croucher PI. Osteoprotegerin is a soluble decoy receptor for tumor necrosis factor-related apoptosis-inducing ligand/Apo2 ligand and can function as a paracrine survival factor for human myeloma cells. Cancer Res. 2003;63(5):912–916. [PubMed] [Google Scholar]
- 70.Abe M, Hiura K, Wilde J, et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood. 2004;104(8):2484–2491. doi: 10.1182/blood-2003-11-3839. [DOI] [PubMed] [Google Scholar]
- 71.Raje N, Roodman GD. Advances in the biology and treatment of bone disease in multiple myeloma. Clin Cancer Res. 2011;17(6):1278–1286. doi: 10.1158/1078-0432.CCR-10-1804. [DOI] [PubMed] [Google Scholar]
- 72.Roodman GD. Pathogenesis of myeloma bone disease. Leukemia. 2009;23(3):435–441. doi: 10.1038/leu.2008.336. [DOI] [PubMed] [Google Scholar]
- 73.Kumar S, Witzig TE, Timm M, et al. Bone marrow angiogenic ability and expression of angiogenic cytokines in myeloma: evidence favoring loss of marrow angiogenesis inhibitory activity with disease progression. Blood. 2004;104(4):1159–1165. doi: 10.1182/blood-2003-11-3811. [DOI] [PubMed] [Google Scholar]
- 74.Kumar S, Gertz MA, Dispenzieri A, et al. Prognostic value of bone marrow angiogenesis in patients with multiple myeloma undergoing high-dose therapy. Bone Marrow Transplant. 2004;34(3):235–239. doi: 10.1038/sj.bmt.1704555. [DOI] [PubMed] [Google Scholar]
- 75.Zhang J, Sattler M, Tonon G, et al. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res. 2009;69(12):5082–5090. doi: 10.1158/0008-5472.CAN-08-4603. [DOI] [PubMed] [Google Scholar]
- 76.Andersen NF, Standal T, Nielsen JL, et al. Syndecan-1 and angiogenic cytokines in multiple myeloma: correlation with bone marrow angiogenesis and survival. Br J Haematol. 2005;128(2):210–217. doi: 10.1111/j.1365-2141.2004.05299.x. [DOI] [PubMed] [Google Scholar]
- 77.Ria R, Todoerti K, Berardi S, et al. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin Cancer Res. 2009;15(17):5369–5378. doi: 10.1158/1078-0432.CCR-09-0040. [DOI] [PubMed] [Google Scholar]
- 78.Malanchi I, Santamaria-Martínez A, Susanto E, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481(7379):85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 79.Frassanito MA, Rao L, Moschetta M, et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia. 2014;28(4):904–916. doi: 10.1038/leu.2013.254. [DOI] [PubMed] [Google Scholar]
- 80.Slany A, Haudek-Prinz V, Meshcheryakova A, et al. Extracellular matrix remodeling by bone marrow fibroblast-like cells correlates with disease progression in multiple myeloma. J Proteome Res. 2014;13(2):844–854. doi: 10.1021/pr400881p. [DOI] [PubMed] [Google Scholar]
- 81.Caers J, Deleu S, Belaid Z, et al. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia. 2007;21(7):1580–1584. doi: 10.1038/sj.leu.2404658. [DOI] [PubMed] [Google Scholar]
- 82.Favaloro J, Brown R, Aklilu E, et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk Lymphoma. 2014;55(5):1090–1098. doi: 10.3109/10428194.2013.825905. [DOI] [PubMed] [Google Scholar]
- 83.Braga WM, Atanackovic D, Colleoni GW. The role of regulatory T cells and TH17 cells in multiple myeloma. Clin Dev Immunol. 2012;2012:293479. [DOI] [PMC free article] [PubMed]
- 84.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 85.Noonan K, Marchionni L, Anderson J, Pardoll D, Roodman GD, Borrello I. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood. 2010;116(18):3554–3563. doi: 10.1182/blood-2010-05-283895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8(5):337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 87.Chang SH, Mirabolfathinejad SG, Katta H, et al. T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci USA. 2014;111(15):5664–5669. doi: 10.1073/pnas.1319051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: the ultimate identity crisis. Front Immunol. 2014;5:276. doi: 10.3389/fimmu.2014.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 90.Kawano Y, Moschetta M, Manier S, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. 2015;263(1):160–172. doi: 10.1111/imr.12233. [DOI] [PubMed] [Google Scholar]
- 91.Song W, van der Vliet HJJ, Tai YT, et al. Generation of antitumor invariant natural killer T cell lines in multiple myeloma and promotion of their functions via lenalidomide: a strategy for immunotherapy. Clin Cancer Res. 2008;14(21):6955–6962. doi: 10.1158/1078-0432.CCR-07-5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baxter AG, Hodgkin PD. Activation rules: the two-signal theories of immune activation. Nat Rev Immunol. 2002;2(6):439–446. doi: 10.1038/nri823. [DOI] [PubMed] [Google Scholar]
- 93.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rosenblatt J, Vasir B, Uhl L, et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood. 2011;117(2):393–402. doi: 10.1182/blood-2010-04-277137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rosenblatt J, Avivi I, Vasir B, et al. Blockade of PD-1 in combination with dendritic cell/myeloma fusion cell vaccination following autologous stem cell transplantation [abstract]. Blood. 2012;120(21). Abstract 578. [Google Scholar]
- 96.DiCapua Siegel DS, Moreau P, Avigan D, et al. A phase 1 (Ph1) trial of MK-3475 combined with lenalidomide (Len) and low-dose dexamethasone (Dex) in patients (pts) with relapsed/refractory multiple myeloma (RRMM) [abstract]. J Clin Oncol. 2014;32(5s). Abstract TPS3117. [Google Scholar]
- 97.Giuliani N, Colla S, Sala R, et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: a potential role in multiple myeloma bone disease. Blood. 2002;100(13):4615–4621. doi: 10.1182/blood-2002-04-1121. [DOI] [PubMed] [Google Scholar]
- 98.Jinushi M, Vanneman M, Munshi NC, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci USA. 2008;105(4):1285–1290. doi: 10.1073/pnas.0711293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12(4):239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chan AC, Neeson P, Leeansyah E, et al. Natural killer T cell defects in multiple myeloma and the impact of lenalidomide therapy. Clin Exp Immunol. 2014;175(1):49–58. doi: 10.1111/cei.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Richter J, Neparidze N, Zhang L, et al. Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood. 2013;121(3):423–430. doi: 10.1182/blood-2012-06-435503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Van Valckenborgh E, Schouppe E, Movahedi K, et al. Multiple myeloma induces the immunosuppressive capacity of distinct myeloid-derived suppressor cell subpopulations in the bone marrow. Leukemia. 2012;26(11):2424–2428. doi: 10.1038/leu.2012.113. [DOI] [PubMed] [Google Scholar]
- 104.Zhuang J, Zhang J, Lwin ST, et al. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS ONE. 2012;7(11):e48871. doi: 10.1371/journal.pone.0048871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mantovani A, Germano G, Marchesi F, Locatelli M, Biswas SK. Cancer-promoting tumor-associated macrophages: new vistas and open questions. Eur J Immunol. 2011;41(9):2522–2525. doi: 10.1002/eji.201141894. [DOI] [PubMed] [Google Scholar]
- 106.Roussou M, Tasidou A, Dimopoulos MA, et al. Increased expression of macrophage inflammatory protein-1alpha on trephine biopsies correlates with extensive bone disease, increased angiogenesis and advanced stage in newly diagnosed patients with multiple myeloma. Leukemia. 2009;23(11):2177–2181. doi: 10.1038/leu.2009.130. [DOI] [PubMed] [Google Scholar]
- 107.Scavelli C, Nico B, Cirulli T, et al. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene. 2008;27(5):663–674. doi: 10.1038/sj.onc.1210691. [DOI] [PubMed] [Google Scholar]
- 108.Hope C, Ollar SJ, Heninger E, et al. TPL2 kinase regulates the inflammatory milieu of the myeloma niche. Blood. 2014;123(21):3305–3315. doi: 10.1182/blood-2014-02-554071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Colla S, Storti P, Donofrio G, et al. Low bone marrow oxygen tension and hypoxia-inducible factor-1α overexpression characterize patients with multiple myeloma: role on the transcriptional and proangiogenic profiles of CD138(+) cells. Leukemia. 2010;24(11):1967–1970. doi: 10.1038/leu.2010.193. [DOI] [PubMed] [Google Scholar]
- 110.Borsi E, Perrone G, Terragna C, et al. HIF-1α inhibition blocks the cross talk between multiple myeloma plasma cells and tumor microenvironment. Exp Cell Res. 2014;328(2):444–455. doi: 10.1016/j.yexcr.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 111.Storti P, Bolzoni M, Donofrio G, et al. Hypoxia-inducible factor (HIF)-1α suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia. 2013;27(8):1697–1706. doi: 10.1038/leu.2013.24. [DOI] [PubMed] [Google Scholar]
- 112.Azab AK, Hu J, Quang P, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of endothelial to mesenchymal transition-like features. Blood. 2012;119(24):5782–5794. doi: 10.1182/blood-2011-09-380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muz B, de la Puente P, Azab F, Luderer M, Azab AK. Hypoxia promotes stem cell-like phenotype in multiple myeloma cells. Blood Cancer J. 2014;4:e262. doi: 10.1038/bcj.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kawano Y, Kikukawa Y, Fujiwara S, et al. Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int J Oncol. 2013;43(6):1809–1816. doi: 10.3892/ijo.2013.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014;124(25):3748–3757. doi: 10.1182/blood-2014-05-576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hu J, Handisides DR, Van Valckenborgh E, et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood. 2010;116(9):1524–1527. doi: 10.1182/blood-2010-02-269126. [DOI] [PubMed] [Google Scholar]
- 117.Laubach JP, Raje NS, Yee AJ, et al. A Phase 1/2 trial of TH-302 and dexamethasone without or with bortezomib (TBorD) in patients with relapsed/refractory multiple myeloma [abstract]. Blood. 2014;124(21). Abstract 2142. [Google Scholar]
- 118.Chauhan D, Uchiyama H, Urashima M, Yamamoto K, Anderson KC. Regulation of interleukin 6 in multiple myeloma and bone marrow stromal cells. Stem Cells. 1995;13(suppl 2):35–39. [PubMed] [Google Scholar]
- 119.Dankbar B, Padró T, Leo R, et al. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000;95(8):2630–2636. [PubMed] [Google Scholar]
- 120.Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene. 2001;20(33):4519–4527. doi: 10.1038/sj.onc.1204623. [DOI] [PubMed] [Google Scholar]
- 121.Prabhala RH, Pelluru D, Fulciniti M, et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood. 2010;115(26):5385–5392. doi: 10.1182/blood-2009-10-246660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hardin J, MacLeod S, Grigorieva I, et al. Interleukin-6 prevents dexamethasone-induced myeloma cell death. Blood. 1994;84(9):3063–3070. [PubMed] [Google Scholar]
- 123.Chauhan D, Pandey P, Hideshima T, et al. SHP2 mediates the protective effect of interleukin-6 against dexamethasone-induced apoptosis in multiple myeloma cells. J Biol Chem. 2000;275(36):27845–27850. doi: 10.1074/jbc.M003428200. [DOI] [PubMed] [Google Scholar]
- 124.Qiang YW, Kopantzev E, Rudikoff S. Insulinlike growth factor-I signaling in multiple myeloma: downstream elements, functional correlates, and pathway cross-talk. Blood. 2002;99(11):4138–4146. doi: 10.1182/blood.v99.11.4138. [DOI] [PubMed] [Google Scholar]
- 125.Mitsiades CS, Mitsiades N, Poulaki V, et al. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene. 2002;21(37):5673–5683. doi: 10.1038/sj.onc.1205664. [DOI] [PubMed] [Google Scholar]
- 126.Akiyama M, Hideshima T, Hayashi T, et al. Cytokines modulate telomerase activity in a human multiple myeloma cell line. Cancer Res. 2002;62(13):3876–3882. [PubMed] [Google Scholar]
- 127.Hideshima T, Chauhan D, Hayashi T, et al. The biological sequelae of stromal cell-derived factor-1alpha in multiple myeloma. Mol Cancer Ther. 2002;1(7):539–544. [PubMed] [Google Scholar]
- 128.Alsayed Y, Ngo H, Runnels J, et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007;109(7):2708–2717. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roccaro AM, Sacco A, Purschke WG, et al. SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Reports. 2014;9(1):118–128. doi: 10.1016/j.celrep.2014.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Camussi G, Deregibus MC, Tetta C. Tumor-derived microvesicles and the cancer microenvironment. Curr Mol Med. 2013;13(1):58–67. [PubMed] [Google Scholar]
- 131.Martins VR, Dias MS, Hainaut P. Tumor-cell-derived microvesicles as carriers of molecular information in cancer. Curr Opin Oncol. 2013;25(1):66–75. doi: 10.1097/CCO.0b013e32835b7c81. [DOI] [PubMed] [Google Scholar]
- 132.Yang C, Robbins PD. The roles of tumor-derived exosomes in cancer pathogenesis. Clin Dev Immunol. 2011;2011:842849. [DOI] [PMC free article] [PubMed]
- 133.Wang J, Hendrix A, Hernot S, et al. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. 2014;124(4):555–566. doi: 10.1182/blood-2014-03-562439. [DOI] [PubMed] [Google Scholar]
- 134.Vincent T, Mechti N. Extracellular matrix in bone marrow can mediate drug resistance in myeloma. Leuk Lymphoma. 2005;46(6):803–811. doi: 10.1080/10428190500051448. [DOI] [PubMed] [Google Scholar]
- 135.Yang Y, Yaccoby S, Liu W, et al. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood. 2002;100(2):610–617. doi: 10.1182/blood.v100.2.610. [DOI] [PubMed] [Google Scholar]