

Key Points
Selinexor exhibits synergy with ibrutinib in CLL.
Selinexor is effective in vitro in ibrutinib-resistant CLL.
Abstract
Despite the therapeutic efficacy of ibrutinib in chronic lymphocytic leukemia (CLL), complete responses are infrequent, and acquired resistance to Bruton agammaglobulinemia tyrosine kinase (BTK) inhibition is being observed in an increasing number of patients. Combination regimens that increase frequency of complete remissions, accelerate time to remission, and overcome single agent resistance are of considerable interest. We previously showed that the XPO1 inhibitor selinexor is proapoptotic in CLL cells and disrupts B-cell receptor signaling via BTK depletion. Herein we show the combination of selinexor and ibrutinib elicits a synergistic cytotoxic effect in primary CLL cells and increases overall survival compared with ibrutinib alone in a mouse model of CLL. Selinexor is effective in cells isolated from patients with prolonged lymphocytosis following ibrutinib therapy. Finally, selinexor is effective in ibrutinib-refractory mice and in a cell line harboring the BTK C481S mutation. This is the first report describing the combined activity of ibrutinib and selinexor in CLL, which represents a new treatment paradigm and warrants further evaluation in clinical trials of CLL patients including those with acquired ibrutinib resistance.
Introduction
Chronic lymphocytic leukemia (CLL) is a lymphoid malignancy of clonal B cells that exhibit aberrant activation of the B-cell receptor (BCR) signaling pathway. A critical component of this pathway is Bruton agammaglobulinemia tyrosine kinase (BTK), a nonreceptor tyrosine kinase expressed predominantly in B lymphocytes.3 Ibrutinib, which irreversibly binds and inhibits BTK activity, has shown promising results in CLL, mantle cell lymphoma, and a subset of diffuse large B-cell lymphoma driven by BCR signaling.4-6 Despite encouraging results, complete responses are infrequent.7 Additionally, acquired resistance to ibrutinib represents an important clinical challenge wherein no standard treatment approach currently exists. Mechanisms of ibrutinib resistance were elucidated by our group and others and involve mutations at the C481S site of BTK or in the immediate downstream target, PLCγ2.1,2,8
Exportin-1 (CRM1/XPO1) is the sole nuclear exporter of tumor suppressor proteins such as p53, inhibitory nuclear factor-κB, and FOXO3a.9,10 Selective inhibitors of nuclear export (SINEs) inhibit XPO1 and restore subcellular localization of dysregulated molecules. Our previous published work showed XPO1 is a therapeutic target for CLL11 and has facilitated translation of selinexor, a SINE, to a phase 1 clinical trial (#NCT01607892), where antitumor activity has been observed in lymphoma,12 CLL,12 multiple myeloma,13 and acute myeloid leukemia.14 We recently showed that selinexor inhibits activation of downstream BCR targets such as extracellular signal-regulated kinase and protein kinase B and suppresses BTK gene expression.15 Based on these observations, we hypothesized that (1) targeting XPO1 via selinexor might be effective in patients with acquired resistance to ibrutinib and (2) dual targeting of XPO1 alongside BTK function might produce synergistic activity in CLL and prevent onset of ibrutinib-resistant clones.
Study design
Human CLL and normal B cells were isolated and cultured as previously described.11 Blood was obtained from CLL patients under an institutional review board-approved protocol with informed consent according to the Declaration of Helsinki. Cell death was assessed using annexin-V/propidium iodide (PI) staining as previously described.11 Chicken DT40 BTK-null cell lines (RCB1468) were obtained from RIKEN Bioresource. Lentiviral constructs pReceiver-LV125 and A0534-Lv125 were obtained from GeneCopoeia and were used to stably transfect DT40 BTK-null cells with empty vector and BTK. The mutation was made using QuikChange site-directed mutagenesis (Stratagene) in the kinase domain at cysteine 481 to serine (see the primer sequence in supplemental Materials available on the Blood Web site). Confirmation of the DNA sequence and infection of the DT40 cell lines was performed as previously described.16 Cells were selected with puromycin. All animal experiments were carried out under protocols approved by the Ohio State University Institutional Animal Care and Use Committee. C57BL/6 cells were engrafted with CD19+CD5+ leukemia cells from an Eμ-TCL1 mouse with active CLL-like leukemia. Leukemia onset was defined as ≥10% CD45+CD5+CD19+ B cells in peripheral blood by flow cytometry. At leukemia onset, engrafted mice were randomly assigned to treatment groups. Overall survival was the primary end point. An in vivo model of ibrutinib resistance was developed using C57BL/6 mice engrafted with splenocytes derived from ibrutinib-resistant Eµ-TCL1 mice that were passaged through 2 C57BL/6 animals. Ibrutinib-resistant Eµ-TCL1 mice were generated by continuous dosing of animals with ibrutinib in drinking water from the time of weaning. Ibrutinib-resistant Eμ-TCL1 mice with active leukemia were injected intraperitoneally with 100 μg EdU (5-ethynyl-2′-deoxyuridine), single-cell suspensions were prepared from spleen and bone marrow, and EdU incorporation was detected by flow cytometry according to the manufacturer’s protocol (Life Technologies). All statistical analyses were performed by the Ohio State University Center for Biostatistics using previously described models.11 Selinexor was provided by Karyopharm. Ibrutinib for in vivo studies was provided by Pharmacyclics and for in vitro studies was purchased from Selleck.
Results and discussion
We previously showed that selinexor exhibits proapoptotic activity against CLL cells via inhibition of nuclear export of tumor suppressor proteins.11 Additionally we showed that selinexor counteracts BCR signaling partially through the downmodulation of BTK protein expression.15 We therefore hypothesized that selinexor would synergize with ibrutinib as it targets BTK through a completely different mechanism. We examined this hypothesis in primary CLL patient samples and found that ibrutinib and selinexor in combination exhibit significant synergistic cytotoxicity (Figure 1A). We repeated this assay in patient samples stimulated via TLR9 using synthetic CpG oligodeoxynucleotides and in patient samples cocultured with the human bone marrow-derived fibroblast cell line HS-5 that induces survival of normal B cells and CLL cells ex vivo.17,18 Synergistic cytotoxicity of ibrutinib and selinexor was maintained with CpG stimulation (Figure 1B). The combination showed a significant increase in cytotoxicity compared with each agent alone during stromal cell coculture (Figure 1C). It is well established that CLL cells rely on prosurvival signals from the microenvironment to resist cytotoxic agents. This suggests dual inhibition of BTK kinase function by ibrutinib and BTK protein expression by selinexor may be an effective strategy to target CLL cells localized to many different compartments including peripheral blood, bone marrow, and other secondary lymphoid tissues.
Figure 1.
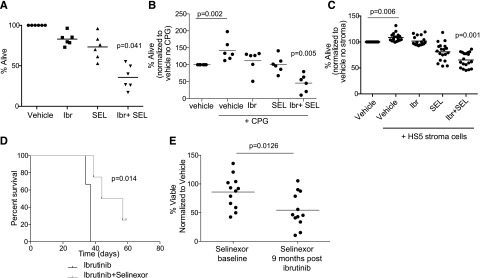
Selinexor synergizes in vitro and in vivo with ibrutinib. (A) CD19+ cells from CLL patients (n = 6) were isolated from peripheral blood and incubated with vehicle, 0.5 μM selinexor (SEL), 1 μM ibrutinib (Ibr) or selinexor + ibrutinib. Ibr was given as a 1-hour pulse exposure followed by washout, and SEL was given continuously for 24 hours. Viability was determined by annexin-V/PI flow cytometry and is shown relative to time-matched dimethylsulfoxide controls for each group. Horizontal bars represent averages. Each agent alone (SEL or Ibr) significantly decreased cell viability compared with vehicle (P < .03). The combination produced a synergistic effect on viability (P = .041). (B) CD19+ cells from CLL patients (n = 6) were unstimulated or 3.2 µM CpG-stimulated in the presence of vehicle, 0.5 μM SEL (24-hour continuous exposure), 1 μM Ibr (1-hour pulse exposure with washout), or SEL + Ibr. Cytotoxicity was measured by annexin/PI. Horizontal bars represent averages. Each agent alone (SEL or Ibr) significantly decreased cell viability compared with vehicle (P = .001). The combination produced a synergistic decrease in viability (P = .005). (C) CD19+ cells from CLL patients were incubated with 0.5 μM SEL (24-hour continuous exposure), 1 μM Ibr (1-hour pulse exposure with washout), or SEL + Ibr on an HS5 human bone marrow stromal cell layer for 24 hours. Cytotoxicity was measured by annexin-V/PI flow-based assay. Horizontal bars represent averages. SEL and Ibr together resulted in significantly more cytotoxicity than either agent alone (P < .001). (D) Overall survival (OS) curves for C57BL/6 mice engrafted with spleen lymphocytes derived from the Eμ-TCL1 transgenic mouse. Mice with active leukemia (defined as ≥10% CD5+/CD19+ cells in the leukocyte population) were randomized to treatment with ibrutinib (∼30 mg/kg/day via drinking water) or ibrutinib + selinexor (15 mg/kg on 2 consecutive days each week via oral gavage; n = 6 per group). (E) Persistent lymphocytes collected at baseline and 9 months after beginning ibrutinib from the same patients were treated in vitro with selinexor at 0.5 μM (n = 13). Cytotoxicity was measured by annexin-V/PI flow cytometry after 72 hours.
Our prior studies with ibrutinib19 and selinexor15 in the Eµ-TCL1 engraftment mouse model of CLL showed that each drug alone can inhibit the expansion phase of CLL in this model. To see whether selinexor has the potential to improve on ibrutinib therapy in vivo, because this agent is the current standard of care, we monitored overall survival in a cohort of engrafted mice randomized to receive either ibrutinib alone or selinexor and ibrutinib. As shown in Figure 1D, mice treated with the combination had significantly better survival compared with mice given ibrutinib alone. Similar to reports of other active agents in the Eµ-TCL1 model, disease eradication was not achieved for any treatment group due to the aggressive nature of this model. We next examined the efficacy of selinexor in the common clinical scenario of prolonged lymphocytosis following ibrutinib treatment in patients with CLL. Our previous data indicate that, although BTK is inhibited, downstream mediators of BCR signaling are activated in the persistent lymphocytes,20 and treatment with targeted kinase inhibitors shows that these cells are not dependent on a single survival pathway.20 Lymphocytes collected at baseline and 9 months after beginning of ibrutinib therapy from the same patients were treated with targeted kinase inhibitors20 or selinexor. Although all the other inhibitors remain equally active at both time points,20 selinexor was significantly more effective in persistent lymphocytosis (after ibrutinib) samples (Figure 1E), providing additional evidence for therapeutic combination of these 2 agents.
Selinexor targets multiple BCR signaling nodes, including BTK, in a manner independent of BTK kinase activity, suggesting that selinexor may possess the ability to overcome or prevent ibrutinib-mediated resistance in CLL by blocking adaptive signaling responses in resistant subclones. Our group previously identified a major mechanism of acquired ibrutinib resistance in CLL patients involving mutation of the BTK cysteine residue where ibrutinib binding occurs (C481S), changing the binding of ibrutinib from irreversible to reversible.1,2 To focus on this important resistance mechanism, we cloned human wild-type or C481S BTK into DT40 cells lacking endogenous BTK (supplemental Materials). Viability was assessed after treating DT40 cells with selinexor for 24 hours. Selinexor remains active in the presence of the BTK C481S mutation (Figure 2A). To test our hypothesis in vivo, C57BL/6 mice were engrafted with CD19+CD5+ leukemia derived from ibrutinib-resistant Eµ-TCL1 mice. Although these mice are not known to possess the C481S mutation, they maintain functionally resistant disease as a result of selective pressure from ibrutinib exposure, mimicking acquired resistance in patients. At leukemia onset, mice were randomized to receive vehicle, ibrutinib alone, or selinexor alone. As expected, mice retained their resistance to ibrutinib. However, treatment with selinexor induced a significant improvement in survival (Figure 2B). We further demonstrated that selinexor effectively inhibited the fraction of proliferating leukemic cells, based on a significant decrease in the percentage of EdU-positive leukemic cells of ibrutinib-resistant mice treated with selinexor (Figure 2C). The ability of selinexor to overcome acquired resistance to ibrutinib was confirmed in vitro in primary CLL cells derived from patients on ibrutinib that have relapsed with BTK C481S mutations (n = 3), as confirmed by Ion Torrent deep sequencing performed at the time of ibrutinib relapse (Figure 2D). These data show that selinexor has single-agent activity in ibrutinib-resistant CLL in vitro, suggesting it may be effective in ibrutinib-resistant CLL patients and may have the potential to prevent expansion of ibrutinib-resistant subclones when used in combination with ibrutinib.
Figure 2.

Selinexor is active in the setting of acquired resistance to ibrutinib. (A) DT40 BTK-null cells with WT or C481S BTK were exposed to 1 μM ibrutinib for 1 hour, 0.5 μM selinexor for 24 hours, or dimethylsulfoxide (vehicle) for 24 hours. Cytotoxicity after 24 hours was measured by annexinV/PI flow cytometry. Viable populations were calculated as a percent of viability of vehicle control. Three biological replicates were performed. Selinexor induced significantly more cell death compared with vehicle in cells expressing C481S (P = .042), WT (P = .027), or empty vector (P = .011). (B) C57BL/6 mice were engrafted with spleen lymphocytes derived from an Eμ-TCL1 transgenic mouse with acquired resistance to ibrutinib. Mice were followed for leukemia development (defined as ≥10% CD5+/CD19+ cells in the leukocyte population), and once leukemic, randomized to treatment with ibrutinib alone (∼30 mg/kg/day via drinking water), selinexor alone (15 mg/kg on 2 consecutive days each week via oral gavage), or vehicle. As expected, mice treated with ibrutinib did not show any survival advantage compared with vehicle control, whereas mice treated with selinexor showed improved survival (n = 12-14 per group). (C) In vivo EdU labeling was performed in a cohort of mice engrafted as described in B. Mice were treated for 2 days with vehicle, SEL, or Ibr (n = 5 for each group). EdU was injected on day 3. Spleens were analyzed by flow cytometry for percentage of Edu-positive cells within the leukemic population (CD45+/CD19+/CD5+ cells). (D) CLL cells derived from ibrutinib resistant patients (n = 3) were treated in vitro with selinexor at 0.5 μM. Cytotoxicity was measured by annexin-V/PI after 48 hours.
Together our data suggest the combination of selinexor and ibrutinib as a promising new therapeutic paradigm in CLL that may elicit more robust initial responses and provide activity in the setting of acquired resistance to ibrutinib.
Acknowledgments
The authors thank Pharmacyclics Inc. for providing the ibrutinib for the in vivo studies and Karyopharm Inc. for providing selinexor used in these studies. The authors thank the patients who provided blood for the abovementioned studies.
This work was supported by the Leukemia and Lymphoma Society in the form of translational grant SCORE LLS 7080-06/7004-11 and National Institutes of Health, National Cancer Institute grant R01 5R01CA177292.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Z.A.H., J.C.B., J.A.W., and R.L. designed the experiments, analyzed the data, wrote the paper, and reviewed and approved the final version; and R.M., K.A.B., D.G., E.W., L.L.S., K.W., A.J.J., and A.M.L. planned and contributed to components of the experimental work presented (chemistry, biologic, clinical, statistical or animal studies), reviewed and modified versions of the paper, and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rosa Lapalombella, Room 460 OSUCCC, 410 West 12th Ave, The Ohio State University, Columbus, OH 43210; e-mail: rosa.lapalombella@osumc.edu.
References
- 1.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woyach JA, Ruppert AS, Lozanski G, et al. Association of disease progression on ibrutinib therapy with the acquisition of resistance mutations: a single-center experience of 267 patients. J Clin Oncol. 2014;32:15s. Abstract 7010.
- 3.Mohamed AJ, Yu L, Bäckesjö C-M, et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228(1):58–73. doi: 10.1111/j.1600-065X.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 4.Cinar M, Hamedani F, Mo Z, Cinar B, Amin HM, Alkan S. Bruton tyrosine kinase is commonly overexpressed in mantle cell lymphoma and its attenuation by Ibrutinib induces apoptosis. Leuk Res. 2013;37(10):1271–1277. doi: 10.1016/j.leukres.2013.07.028. [DOI] [PubMed] [Google Scholar]
- 5.Wilson WH, Gerecitano JF, Goy A, et al. The Bruton's tyrosine kinase (BTK) inhibitor, ibrutinib (PCI-32765), has preferential activity in the ABC subtype of relapsed/refractory de novo diffuse large B-cell lymphoma (DLBCL): interim results of a multicenter, open-label, phase 2 Study [abstract]. Blood. 2012;120(21). Abstract 686. [Google Scholar]
- 6.Yang Y, Shaffer AL, III, Emre NC, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell. 2012;21(6):723–737. doi: 10.1016/j.ccr.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrd JC, Brown JR, O’Brien S, et al. RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–223. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furman RR, Cheng S, Lu P, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370(24):2352–2354. doi: 10.1056/NEJMc1402716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell. 2012;23(18):3673–3676. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennan CM, Gallouzi I-E, Steitz JA. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J Cell Biol. 2000;151(1):1–14. doi: 10.1083/jcb.151.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120(23):4621–4634. doi: 10.1182/blood-2012-05-429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuruvilla J, Gutierrez M, Shah BD, et al. Preliminary evidence of antitumor activity of Selinexor (KPT-330) in a phase I trial of a first-in-class oral selective inhibitor of nuclear export (SINE) in patients (pts) with relapsed/refractory non-Hodgkin’s lymphoma (NHL) and chronic lymphocytic leukemia (CLL) [abstract]. Blood. 2013;122(21). Abstract 90. [Google Scholar]
- 13.Chen CI, Gutierrez M, de Nully Brown P, et al. Antitumor activity of Selinexor (KPT-330), a first-in-class oral selective inhibitor of nuclear export (SINE) XPO1/CRM1 antagonist in patients (pts) with relapsed/refractory multiple myeloma (MM) or Waldenstrom’s macroglobulinemia (WM) [abstract]. Blood. 2013;122(21). Abstract 1942. [Google Scholar]
- 14.Savona M, Garzon R, de Nully Brown P, et al. Phase I trial of selinexor (KPT-330), a first-in-class oral selective inhibitor of nuclear export (SINE) in patients (pts) with advanced acute myelogenous leukemia (AML) [abstract]. Blood. 2013;122(21). Abstract 1440. [Google Scholar]
- 15.Zhong Y, El-Gamal D, Dubovsky JA, et al. Selinexor suppresses downstream effectors of B-cell activation, proliferation and migration in chronic lymphocytic leukemia cells. Leukemia. 2014;28(5):1158–1163. doi: 10.1038/leu.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lapalombella R, Yeh Y-Y, Wang L, et al. Tetraspanin CD37 directly mediates transduction of survival and apoptotic signals. Cancer Cell. 2012;21(5):694–708. doi: 10.1016/j.ccr.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roecklein BA, Torok-Storb B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood. 1995;85(4):997-1005. [PubMed] [Google Scholar]
- 18.Kay NE, Shanafelt TD, Strege AK, Lee YK, Bone ND, Raza A. Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an “angiogenic switch”. Leuk Res. 2007;31(7):899–906. doi: 10.1016/j.leukres.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woyach JA, Bojnik E, Ruppert AS, et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood. 2014;123(8):1207–1213. doi: 10.1182/blood-2013-07-515361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2014;123(12):1810–1817. doi: 10.1182/blood-2013-09-527853. [DOI] [PMC free article] [PubMed] [Google Scholar]