

Key Points
IL8-CXCR2 is overexpressed in purified stem cells from AML and MDS, and CXCR2 expression is associated with worse prognosis.
Inhibition of CXCR2 by genetic and pharmacologic means leads to decreased viability in AML/MDS stem cells and in vitro and in vivo models.
Abstract
Acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) are associated with disease-initiating stem cells that are not eliminated by conventional therapies. Novel therapeutic targets against preleukemic stem cells need to be identified for potentially curative strategies. We conducted parallel transcriptional analysis of highly fractionated stem and progenitor populations in MDS, AML, and control samples and found interleukin 8 (IL8) to be consistently overexpressed in patient samples. The receptor for IL8, CXCR2, was also significantly increased in MDS CD34+ cells from a large clinical cohort and was predictive of increased transfusion dependence. High CXCR2 expression was also an adverse prognostic factor in The Cancer Genome Atlas AML cohort, further pointing to the critical role of the IL8-CXCR2 axis in AML/MDS. Functionally, CXCR2 inhibition by knockdown and pharmacologic approaches led to a significant reduction in proliferation in several leukemic cell lines and primary MDS/AML samples via induction of G0/G1 cell cycle arrest. Importantly, inhibition of CXCR2 selectively inhibited immature hematopoietic stem cells from MDS/AML samples without an effect on healthy controls. CXCR2 knockdown also impaired leukemic growth in vivo. Together, these studies demonstrate that the IL8 receptor CXCR2 is an adverse prognostic factor in MDS/AML and is a potential therapeutic target against immature leukemic stem cell–enriched cell fractions in MDS and AML.
Introduction
Acute myeloid leukemia (AML) and myelodysplastic syndromes (MDSs) are heterogeneous malignancies that are thought to arise from a small pool of cancer-initiating cells residing within hematopoietic stem (HSC) and progenitor (HSPC) compartments.1-9 Despite the use of poly-chemotherapy and the development of newer agents, clinical outcome remains poor. The disease course is frequently characterized by relapse or failure to achieve durable remission, indicating that current treatment regimens do not target the cancer-initiating cells. Recently, there have been increasing efforts to identify molecular aberrations that could serve as pharmacologic targets within AML and MDS stem cell compartments.10-16 Resulting therapies have shown promising effects in preclinical studies,17,18 but further work will be necessary to identify therapeutic targets against preleukemic stem cells that would lead to long-term remission and prevention of relapse in AML and MDS.19,20 Previously, we identified quantitative and qualitative alterations in AML and MDS HSCs and progenitors and showed that genetically aberrant HSCs survive during morphologic remissions and expand before clinical relapse.2 In an effort to identify aberrant targets in MDS/AML HSPCs, we conducted transcriptomic profiling of stem cells (long-term HSCs [LT-HSCs], short-term HSCs [ST-HSCs]) and progenitors (granulocyte-monocyte progenitors [GMPs]) and compared them to healthy aged-matched control counterparts. Interleukin 8 (IL8) was one of the most significantly upregulated genes in both HSCs and GMPs, suggesting that it might play a crucial role in the carcinogenesis of AML and MDS. IL8 is a known potent proinflammatory cytokine that exerts its effects through binding to its G protein-coupled receptors CXCR1 and CXCR2. Extensive work in solid tumors has shown that IL8 is critical in survival, invasion, and proliferation of cancer cells21-24 and might be an important regulator of cancer stem cell activity.25-27 Activation of multiple pathways by IL8, including phosphatidylinositol 3-kinase/protein kinase B (AKT), phospholipase C/protein kinase C, and mitogen-activated protein kinase (MAPK) signaling, leads to increased expression of various transcription factors such as hypoxia inducible factor 1, nuclear factor-κB, activator protein-1, signal transducer and activator of transcription 3, and β-catenin, which promote tumor growth and survival.21 Blocking the IL8-CXCR1/CXCR2 axis has shown to be of therapeutic potential in a variety of solid tumors27-30; however, there is little known of its role in hematologic malignancies. Our study reveals that IL8 is significantly overexpressed in AML and MDS LT-HSCs, ST-HSCs, and GMPs compared with healthy controls. The IL8 receptor CXCR2 was highly expressed in a variety of leukemic cell lines, as well as in AML and MDS patient cohorts, and higher expression levels correlated with worse clinical outcomes. Functional studies showed that inhibiting and/or downregulating CXCR2 leads to decreased viability and clonogenic capacity of primary AML/MDS patients’ cells, but had no effect on healthy controls. Interestingly, CXCR2 inhibition decreased viability in the CD34+/CD38− HSC fraction from AML/MDS patients but had no impact on healthy control HSCs. Our studies suggest that the IL8-CXCR2 pathway is frequently dysregulated in AML and MDS stem cells and can serve as a novel therapeutic target in these diseases.
Methods
Patient samples, cell lines, and reagents
Specimens were obtained from patients diagnosed with MDS and AML after institutional review board approval by the Albert Einstein College of Medicine (supplemental Table 1, available on the Blood Web site), in accordance with the Declaration of Helsinki. The AML cell lines KG1, HL-60, Molm 13, U937, and THP-1 were grown in RPMI supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. CXCR2 inhibitor SB-332235 was obtained from GlaxoSmithKline, dissolved in dimethylsulfoxide, and stored at −20°C at a concentration of 100 mM.
Cell viability assay
Cell lines and primary samples were incubated at varying concentrations of the CXCR2 inhibitor SB-332235. Viability was assessed by addition of Cell Titer Blue (Promega) and measured via Fluostar Omega Microplate reader (BMG Labtech).
Cell lysis and immunoblotting
Cells were lysed in phosphorylation lysis buffer as previously described.31 Immunoblotting was performed as previously described.31
Multiparameter high-speed fluorescence-activated cell sorter of stem and progenitor cells
Processing and sorting of hematopoietic stem and progenitor cells was performed as previously described.10 Briefly, mononuclear cells were isolated from bone marrow aspirates and peripheral blood samples by density gradient centrifugation and then subjected to immunomagnetic enrichment of CD34+ cells (MiltenyiBiotec). CD34+ cells were stained with PE-Cy5 (Tricolor)-conjugated monoclonal antibodies against lineage antigens (CD2 [RPA-2.10], CD3 [UCHT1], CD4 [S3.5], CD7 [6B7], CD8 [3B5], CD10 [CB-CALLA], CD11b [VIM12], CD14 [TueK4], CD19 [HIB19], CD20 [2H7], CD56 [MEM-188], Glycophorin A [CLB-ery-1(AME-1)]), as well as fluorochrome-conjugated antibodies against CD34 [581/CD34 (class III epitope)], CD38 (HIT2), CD90 (5E10), CD45RA (MEM-56), and CD123 (6H6). Cells were sorted with a 5-laser FACSAria II Special Order System flow cytometer (Becton Dickinson). Based on established surface marker characterization, we distinguished and sorted LT-HSCs (Lin−, CD34+, CD38−, CD90+), ST-HSCs (Lin−, CD34+, CD38−, CD90−), and GMPs (Lin−, CD34+, CD38+, CD123+, CD45RA+).32 Flow cytometry data were analyzed with BD FACSDiva (Becton Dickinson) and FlowJo (TreeStar) software. After sorting, RNA was extracted with the RNeasy Micro kit (Qiagen).
RNA amplification and real-time polymerase chain reaction
RNA was extracted with the RNeasy Micro kit (Qiagen), and quantity and quality were assessed with a Nanodrop 3000 instrument (Thermo Scientific). RNA extracted from sorted primary LT-HSCs, ST-HSCs, and GMP cells was amplified with the Ovation One Direct System (Nugen) and converted to single-strand cDNA. RNA extracted from cell lines and primary CD34+ cells was reverse-transcribed with Superscript III reverse transcriptase (Invitrogen). IL8 expression was measured by quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) with SYBR green on a CFX 96 Real Time System (BioRad).
Lentiviral short hairpin RNA vectors and transduction
For knockdown studies, 2 short hairpin RNAs (shRNAs) against CXCR2 were used (GIPZ system, catalog nos. RHS4430-99883569 and RHS4430-99891604; Open Biosystems). For production of lentiviral particles, lentiviral shRNA expression constructs were transfected together with packaging vectors into 293T producer cells using Lipofectamine LTX transfection reagent (Invitrogen), and supernatants were harvested after 48 and 72 hours and concentrated by ultracentrifugation. AML cell lines were transduced with the shRNA-containing lentiviruses (multiplicity of infection = 6-10). After culture in fresh medium, green fluorescent protein (GFP)-positive and 4,6 diamidino-2-phenylindole (DAPI)-negative cells were sorted 48 to 72 hours after infection using a FACSAria II sorter (BD Biosciences) and used for experiments. CXCR2 knockdown efficiency was measured by real-time PCR and flow cytometry.
Clonogenic assays
For clonogenic assays, primary patient samples and healthy controls were plated in methylcellulose (Stem Cell Technologies H4435) in 35-mm dishes and incubated with the CXCR2 inhibitor at 10 μM. Colonies were counted after 14 to 17 days in culture. Clonogenic assays with shRNA-mediated CXCR2 knockdown were performed 48 to 72 hours after lentiviral transduction of the respective cell line (U937, THP-1, and MOLM13). DAPI-negative and GFP-positive cells were sorted and plated in methylcellulose (StemCell Technologies H4435) in 35-mm dishes. Colonies were counted after 7 to 10 days in culture.
Cell cycle analysis
Cell cycle analysis was performed as previously described.33 In brief, 1 × 106 AML cells were incubated at varying doses of the CXCR2 inhibitor. After 24 hours, the cells were rinsed with ice-cold phosphate-buffered saline (PBS) and incubated for 45 minutes at 37°C in the dark with 0.5 mL Hoechst buffer (20 μg/mL Hoechst 33342 in Hanks balanced salt solution containing 10% fetal bovine serum, 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.2, 1 g/L glucose, and 50 μg/mL verapamil; Sigma-Aldrich). Pyronin Y (Sigma-Aldrich) was added at 1 μg/mL, and cells were incubated for 15 minutes at 37°C in the dark, washed with PBS, and analyzed by flow cytometry using a FACSAria II Special Order System (BD Biosciences).
Evaluation of viability in primary CD34+CD38− HSCs
Primary AML, MDS, and healthy control samples were incubated with 1 and 10 μM of the CXCR2 inhibitor SB-332235 in Iscove's modified Dulbecco's medium with 10% fetal bovine serum at 37°C. After 24 hours, cells were rinsed with PBS and stained with antibodies against CD34 (8G12) and CD38 (HIT2). After washing with PBS, cells were mixed with prediluted fluorescein isothiocyanate-conjugated annexin V (Invitrogen), incubated at room temperature for 15 minutes, and resuspended in 0.5 mL of annexin binding buffer (Invitrogen). Just before flow cytometric analysis, DAPI (Acros Organics) was added to the cells at a final concentration of 1 μg/mL. Viability, apoptosis, and necrosis of CD34+/CD38− and CD34+/CD38+ cells were analyzed by flow cytometry using a FACSAria II Special Order System (BD Biosciences) as performed previously.34-36
Patient database and survival data
Gene expression data from 183 MDS CD34+ samples and 17 controls were obtained from Gene Expression Omnibus (GEO) (GSE19429).37 Data on 200 AML samples were obtained from The Cancer Genome Atlas (TCGA).38 Gene expression data on sorted LT-HSCs, ST-HSCs, and GMPs from AML/MDS patients and healthy controls are deposited in the GEO database (accession nos. GSE35008 and GSE35010). Gene Set Enrichment Analysis was performed by comparing the CXCR2 high signature to 2 published preleukemic HSC signatures (GSE3500810 and GSE303774).
Leukemia xenografts
NOD/SCID IL2Rγ knockout mice were injected with 105 U937 cells that had been transduced with nonsilencing shRNA and CXCR2 knockdown shRNAs. Overall survival was measured and analyzed by Kaplan-Meier plotting and the log-rank test.
Results
IL8 is overexpressed in MDS and AML HSCs and progenitors
Leukemia- and MDS-initiating cells including preleukemic stem cells reside in the lineage-negative phenotypic stem and progenitor compartments and need to be targeted for curative strategies. To determine aberrantly expressed genes in MDS and AML stem and progenitor cells, we previously conducted a parallel transcriptional study comparing LT-HSCs, ST-HSCs, and GMPs from AML samples with deletion of chromosome 7 (−7) to healthy control counterparts10 (Figure 1A). A small set of 7 genes was found to be consistently overexpressed in all compartments (LT-HSCs, ST-HSCs, and GMPs) and included IL8. We expanded our initial study to include sorted samples from normal karyotype and complex karyotype AML/MDS samples (Figure 1A). Our sorting strategy uses rigorous lineage depletion to avoid analysis of the leukemic bulk (blast) population and to focus on the earliest known stem cell and committed myeloid progenitor populations in humans. Sorting of 12 AML/MDS samples and 4 controls followed by gene expression analysis revealed that the IL8 gene was significantly overexpressed by several log-fold in all samples in both HSCs and GMP populations, across normal karyotype, complex karyotype, and −7 cases (Figure 1B). These results were validated in an independent cohort of samples by qRT-PCR. Two AML, 2 MDS, and 2 control samples were sorted (Figure 1E-F shows representative samples), analyzed, and confirmed significant upregulation of IL8 in the MDS/AML samples (Figure 1G).
Figure 1.
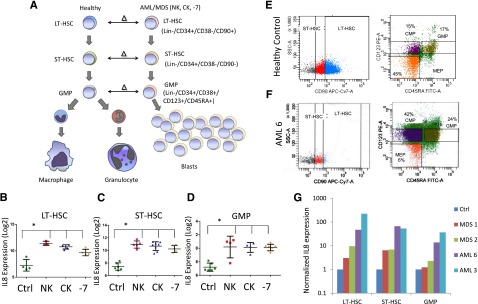
IL8 is overexpressed in stem and progenitor cells in MDS and AML. (A) Gene expression data from sorted AML/MDS bone marrow samples were compared with healthy controls and (B-D) revealed significantly increased IL8 expression in LT-HSCs (Lin-ve, CD34+, CD38−, CD90+, N = 12 AML/MDS, HC = 4), ST-HSCs (Lin-ve, CD34+, CD38−, CD90−), and GMP (Lin-ve, CD34+, CD38+, CD123+, CD45RA+) (P < .001, FDR < 5%). Cytogenetic abnormalities are depicted as follows: NK, normal karyotype; CK, complex karyotype; −7 = deletion of chr7. Representative flow plots show sorting strategy for (E) healthy control and (F) AML sample. (G) qRT-PCR on sorted cells reveals increased expression of IL8 in all AML/MDS HSCs (LT/ST) and GMP.
CXCR2 is expressed in primary AML and MDS samples and is associated with worse clinical outcomes
IL8 and other CXCL ligands bind to the CXCR1 and CXCR2 receptors to trigger many different biological processes. We evaluated the expression of these ligands and receptors in a large cohort of AML samples analyzed by RNA-seq (TCGA, n = 200).38 We observed that among the CXCR1 and 2 ligands, IL8 was most significantly expressed in AML (mean reads per kilobase per million = 36 [IL8] vs 12 [CXCL2], 3 [CXCL3], 2 [CXCL1], and 0.2 [CXCL5]; P < .001), and CXCR2 expression was significantly higher than CXCR1 expression (mean reads per kilobase per million = 4 [CXCR2] vs 2 [CXCR1]; P < .001) in leukemic cells (Figure 2A-B). We evaluated CXCR2 overexpression for prognostic impact in this cohort and observed that samples with higher expression of the receptor had a significantly worse prognosis compared with CXCR2 lower-expressing patients (median overall survival of 245 days in high CXCR2 cases vs 854 in low CXCR2 cases; log rank, P = .003; Figure 2C). Next, we evaluated the expression of CXCR2 in MDS HSPCs in another large cohort of 183 samples.37 CXCR2 expression was found to be higher in MDS samples compared with healthy controls (P = .001, false discovery rate [FDR] < 10%; Figure 2D). Cases with high CXCR2 were found to present with worse disease phenotype manifesting with lower hemoglobin levels and a higher percentage of transfusion dependence (53% for CXCR high vs 35% for CXCR2 low; P < .001; Figure 2E-F). Correlation with mutations obtained from targeted sequencing of a subset of MDS cases39 revealed significant enrichment of TET2 mutations in CXCR2 low expressers, with no other significant correlations with other mutations (supplemental Table 2). Taken together, these data demonstrate that high CXCR2 expression in HSPCs is an adverse prognostic factor in both AML and MDS.
Figure 2.
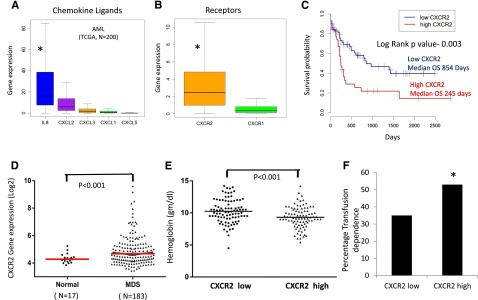
CXCR2 is expressed in primary AML and MDS samples and is associated with worse clinical outcomes. Expression of (A) chemokine ligands and (B) receptors was evaluated in the TCGA RNA-seq dataset from 200 AML samples (data shown as boxplots). (C) Median overall survival of AML patients with high CXCR2 was significantly worse than in patients with lower expression (log rank, P = .003). (D) Gene expression data of CD34+ cells from 183 MDS patients and 17 healthy controls shows significantly higher expression of CXCR2 in MDS CD34+ cells (t test, P < .001, FDR < 5%). MDS patients with (E) high CXCR2 expression (>median) have lower hemoglobin levels (t test, P < .001) and (F) higher RBC transfusion dependence (test of proportions, P < .05).
Gene expression signature of MDS HSPCs with high CXCR2 is similar to known preleukemic stem cell profiles and includes many important functional pathways
To determine the genetic pathways that were differentially expressed in MDS HSPCs with high expression of CXCR2, we identified differentially expressed transcripts between the CXCR2 high and low patient subsets (using median CXCR2 expression as cutoff in a cohort of 183 MDS CD34+ samples; FDR < 0.1; Figure 3A). Ingenuity pathway analysis revealed significant dysregulation of pathways involved in chemokine signaling, cytokine-cytokine receptor interaction, innate immunity signaling, hematopoietic cell lineage regulation, and others in CXCR2 high patients and also included many genes that play important roles in molecular leukemogenesis (Figure 3B; supplemental Table 3). Next, we tested whether the high CXCR2 expression signature had any overlap with known preleukemic stem cell gene expression profiles. Gene set enrichment analysis with 2 recently published preleukemic stem cell signatures (GSE3500810 and GSE303774), including 1 from our own group, revealed highly significant enrichment, demonstrating that HSPCs from CXCR2 high MDS patients have a similar transcriptomic profile than known preleukemic and leukemia-initiating cell populations (Figure 3C).
Figure 3.
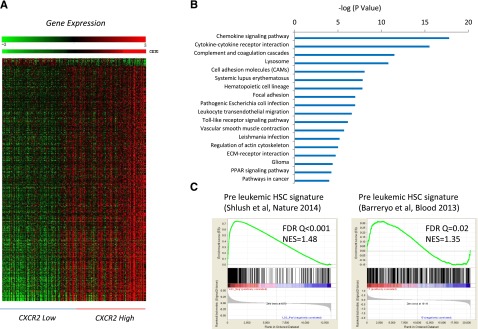
Important functional pathways are dysregulated in MDS cases with high expression of cxcr2. (A) Gene expression profiles from samples with low and high CXCR2 were compared, and differentially expressed transcripts were identified (FDR < 0.1). (B) Significantly dysregulated pathways are shown. The gene signature of high CXCR2 MDS cases is similar to previously published preleukemic stem cell signatures. (C) Gene Set Enrichment Analysis plots show significant enrichment of 2 recent preleukemic gene expression signatures.
Inhibition of CXCR2 leads to decreased proliferation and cell cycle arrest in leukemic cells
To determine the functional role of the IL8-CXCR2 pathway in leukemia biology, we first evaluated the expression of the receptor in AML cell lines. We determined that AML cell lines significantly overexpressed CXCR2, ranging from 3-fold (KG1) to 300-fold overexpression (THP1) compared with healthy CD34+ cells (Figure 4A).To determine the functional role of CXCR2 in these cells, we used a specific inhibitor of CXCR2, SB332235, that is a clinically available compound that has 100-fold selectivity for CXCR2 over CXCR1.40 CXCR2 inhibition led to a dose-dependent decrease in proliferation in all cell lines (Figure 4B). CXCR2 inhibition also led to a significant inhibition of proliferation in primary samples from AML and high-risk MDS cases (Figure 4C). Next, we designed shRNAs against CXCR2 (Figure 4D; supplemental Figure 1) and found that decreased expression of this receptor also led to significantly reduced leukemic colony formation capacity of AML cell lines (P < .001; Figure 4E). The reduction in colony numbers was also accompanied by decreased size of individual leukemic colonies (Figure 4F).
Figure 4.
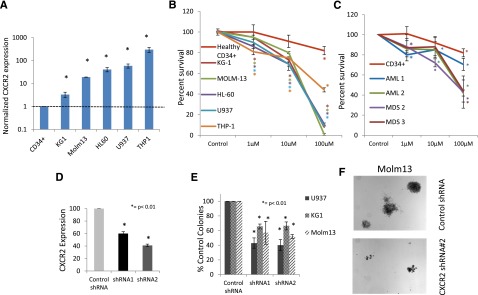
CXCR2 is highly expressed in human leukemic cell lines and regulates viability in cell lines and primary samples. (A) qRT-PCR for CXCR2 demonstrates significantly increased expression in AML cell lines (t test, P < .05). (B) Viability of AML cell lines is inhibited by the CXCR2 inhibitor SB332235 compared with healthy CD34+ cells at 48 hours (t test, *P < .05, **P < .01). (C) Viability of primary MNCs from 2 AML and 2 high-risk MDS samples inhibited by the CXCR2 inhibitor. (D) shRNA against CXCR2 leads to significant knockdown by qRT-PCR in U937 cells (t test, P < .05). Colony growth shows significantly (E) decreased number and (F) size of leukemic colonies AML cells on CXCR2 knockdown compared with scrambled controls (t test, P < .01).
To determine the mechanism of growth arrest, we performed cell cycle analysis and observed a significant arrest of AML cells in the G0 stage (P < .05) after CXCR2 inhibition (Figure 5A-B). This was seen consistently in both cell lines examined (t test, P < .05; Figure 5B-C). At the molecular level, immunoblotting demonstrated that IL8 stimulation led to activation of proliferative extracellular signal-regulated kinase (ERK) MAPK and AKT pathways in leukemia cells, whereas no stimulation of the apoptotic p38 MAPK pathway was observed (Figure 5D-F). Pharmacologic inhibition of CXCR2 led to abrogation of IL8-stimulated signaling in these cells.
Figure 5.
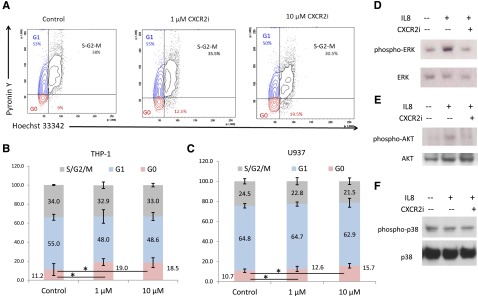
CXCR2 inhibition leads to cell cycle arrest in AML cells and inhibits IL8-stimulated signaling cascades. (A) Representative flow plots of THP-1 cells treated with the CXCR2 inhibitor demonstrate a significantly increased percentage of cells in G0 stage. (B-C) Data from 3 independent experiments demonstrate significantly increased G0 cell cycle arrest in THP-1 and U937 cell lines after CXCR2 inhibition (t test, P < .05). (D-F) THP-1 cells were stimulated with IL8 in the presence and absence of CXCR2 inhibitor (10 μm) and immunoblotted for phospho/activated ERK MAP kinase, p38 MAPK, and AKT with appropriate loading controls.
CXCR2 inhibition leads to selective targeting of AML/MDS HSCs
Next, we determined the functional role of the IL8-CXCR2 pathway in primary HSCs from patients, and treated 5 AML, 1 MDS, and 3 healthy control bone marrow samples with the CXCR2 inhibitor. Fluorescence-activated cell sorter analysis revealed that CXCR2 inhibition led to significantly decreased viability of CD34+/CD38− HSCs from AML/MDS patients (Figure 6A-B). In contrast, no significant decrease in cell viability was seen in healthy control HSCs (Figure 6C-D). No significant decrease in viability of the CD34+/CD38+ population in AML/MDS samples was observed (supplemental Figure 2), demonstrating that CXCR2 inhibition preferentially targets leukemic HSCs.
Figure 6.
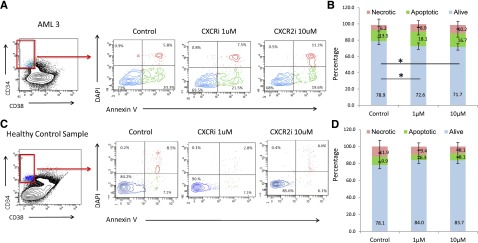
CXCR2 inhibition reduces viability in leukemic CD34+/CD38− HSCs. Four AML and 1 MDS samples were treated with CXCR2 inhibitor, and viability was assessed in the CD34+/CD38− HSC compartment. Viability in AML/MDS HSCs was significantly decreased after CXCR2 inhibition (t test, P < .05) with no change in the healthy controls (N = 3).
CXCR2 inhibition leads to inhibition in primary AML/MDS samples and increased survival in vivo
Next, we determined the efficacy of CXCR2 inhibition in inhibiting leukemic colony formation from primary MDS and AML samples. CXCR2 inhibitor treatment led to a significant decrease in leukemic colony size and numbers (Figure 7A-B). This effect was seen in both the AML and high-risk MDS cases, whereas no decrease in erythroid/myeloid colonies was seen in healthy CD34+ cells (Figure 7A). Finally, we examined the efficacy of CXCR2 knockdown in vivo using xenografts with U937 cells. U937 cells were infected with lentiviruses containing shRNA directed against CXCR2 or scrambled control shRNAs and a fluorescent reporter gene (GFP); cells were sorted for GFP and xenografted into NSG mice. CXCR2 knockdown led to significant improvement in overall survival (Figure 7C; P = .02, log rank), demonstrating CXCR2 as a therapeutic target in vivo.
Figure 7.
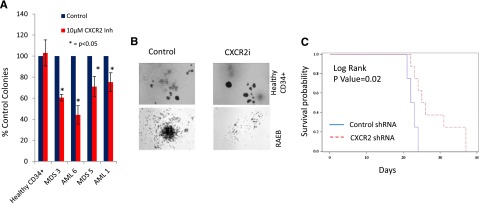
CXCR2 inhibition leads to inhibition in primary AML/MDS samples and increased survival in vivo. (A) Four primary MDS/AML samples were grown in the presence and absence of the CXCR2 inhibitor and demonstrated inhibition of leukemic colony growth after treatment. (B) The size of leukemic colonies was also reduced. NSG mice were xenografted with U937 cells containing control or CXCR2 shRNAs. (C) Mice with CXCR2 shRNA xenografts demonstrated significantly improved overall survival (log rank, P = .02).
Discussion
AML and MDS are diseases that are characterized by a high rate of relapse when treated with chemotherapy or non-chemotherapeutic approaches such as azacytidine and lenalidomide. Leukemia-initiating stem cells including preleukemic stem cells have been shown to persist even during morphologic remissions after these treatments and have to be targeted for future curative approaches.1,2,8,9,20,41 It has been shown that leukemia-initiating cells can reside in different phenotypic stem and progenitor compartments at relatively low frequencies and are associated with transcription factor alterations that result in pathogenic transcriptional changes.2-7,11-13,32,42-44 Thus, to uncover newer stem cell-directed targets, we conducted a transcriptomic analysis on rigorously defined stem and progenitor populations in AML and MDS in comparison with their respective healthy counterparts. With this approach, we were able to identify IL8 as one of the few genes most significantly overexpressed across different stem and progenitor subsets in AML and MDS patients, indicating it might play a crucial role in the pathogenesis of AML and MDS. We were also able to show that the IL8 receptor CXCR2 is overexpressed in AML and MDS patient samples and in several myeloid leukemia cell lines. We also found that high CXCR2 expression correlates with worse clinical outcome. These data implicate the IL8-CXCR2 pathway as a novel therapeutic target in in AML and MDS.
The IL8-CXCR2 pathway has been shown to be important in the pathogenesis of solid tumors, and preclinical studies have demonstrated a role for this pathway in the breast cancer microenvironment.22,27,28,30,45 Various small molecule CXCR2 inhibitors are being developed clinically, and one is being tested in a clinical trial in breast cancer. Our results demonstrate that this pathway is also functionally important for the viability of leukemic stem cell compartments in AML and MDS. Although healthy HSPCs also show a basal production of IL8, significant overexpression of IL8 within the malignant stem and progenitor compartment was detected in every case examined, with notably up to 100-fold upregulation seen in some cases compared with healthy controls. We also demonstrated that inhibition of the IL8 receptor, CXCR2, leads to a significant decrease in viability and colony formation in multiple leukemic cell lines, as well as patient samples. Most importantly, viability of the CD34+/CD38− stem cell-enriched fraction was significantly decreased in AML/MDS patient cells after CXCR2 inhibition, whereas the inhibitor had no impact on healthy control stem cells, indicating that inhibition of the IL8-CXCR2 axis might selectively target the malignant stem cell clones. The role of IL8 in healthy HSCs is not well understood. Although murine models with IL8 receptor-deficient mice show relatively normal hematopoiesis arguing against a crucial role for IL8 in the regulation of healthy HSCs,46 some reports suggest that IL8 might play a positive and protective role in mobilization of healthy HSCs and promotion of monocyte/macrophage growth and differentiation.47,48 Further studies report increased IL8 to exert an inhibitory effect on healthy hematopoiesis and myeloid proliferation,49-53 which could potentially explain the inhibition of growth and differentiation of healthy HSCs in an IL8-rich environment in AML and MDS. The IL8-CXCR2 signaling pathway has been shown to stimulate proliferative pathways such as the phosphatidylinositol 3-kinase/AKT, phospholipase C/protein kinase C, and ERK/MAPK pathways in various models. It has also been shown to activate oncogenic pathways such as hypoxia inducible factor 1, nuclear factor-κB, signal transducer and activator of transcription 3, and β-catenin, which help to promote tumor growth and survival.21 We and others have shown that these transcription factors are important for survival of cancer stem cells.2,34-36
IL8 is also an important regulator of innate immunity. Recent data have shown that innate immune signaling mediators such as IL receptor 1 receptor accessory protein (IL1RAP), Toll-like receptors, and myeloid-derived suppressor cells are activated in MDS.10,54-56 The signaling protein, IL-1 receptor-associated kinase (IRAK4), has also been shown to be a therapeutic target in MDS.57 Our findings further support the role of activated innate immune signaling pathways in MDS/AML and suggest that interaction of IL8-CXCR2 with these other pathways needs to be studied in future studies. Taken together, our results provide a rationale for further evaluating the targeting of the IL8-CXCR2 axis and its specificity for aberrant AML/MDS stem and progenitor cells in vitro, as well as in preclinical in vivo studies.
Acknowledgments
This work was supported by the Leukemia Lymphoma Society, Department of Defense, and National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK1039615 and National Heart, Lung, and Blood Institute grant R01HL116336.
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.S. designed research; C.S., O.G., W.L., A.S., S.G., L.B., T.B., S.B., N.R., and M.B. performed research; A.P. and J.B. contributed data; A.W. contributed reagents; Y.Y., B.W., and S.W. analyzed data; and U.S. and A.V. wrote the paper and analyzed the data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Amit Verma, Albert Einstein College of Medicine, 1300 Morris Park Ave, Chanin Building, Room 302B, Bronx, NY 10461; e-mail: amit.verma@einstein.yu.edu; or Ulrich Steidl, Albert Einstein College of Medicine, 1300 Morris Park Ave, Chanin Building, Room 606A, Bronx, NY 10461; e-mail: ulrich.steidl@einstein.yu.edu.
References
- 1.Elias HK, Schinke C, Bhattacharyya S, Will B, Verma A, Steidl U. Stem cell origin of myelodysplastic syndromes. Oncogene. 2014;33(44):5139–5150. doi: 10.1038/onc.2013.520. [DOI] [PubMed] [Google Scholar]
- 2.Will B, Zhou L, Vogler TO, et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood. 2012;120(10):2076–2086. doi: 10.1182/blood-2011-12-399683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. [DOI] [PMC free article] [PubMed]
- 4.Shlush LI, Zandi S, Mitchell A, et al. HALT Pan-Leukemia Gene Panel Consortium. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pang WW, Pluvinage JV, Price EA, et al. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc Natl Acad Sci USA. 2013;110(8):3011–3016. doi: 10.1073/pnas.1222861110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woll PS, Kjällquist U, Chowdhury O, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. 2014;25(6):794–808. doi: 10.1016/j.ccr.2014.03.036. [DOI] [PubMed] [Google Scholar]
- 7.Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 8.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29(5):591–599. doi: 10.1200/JCO.2010.31.0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jordan CT. Searching for leukemia stem cells—not yet the end of the road? Cancer Cell. 2006;10(4):253–254. doi: 10.1016/j.ccr.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Barreyro L, Will B, Bartholdy B, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120(6):1290–1298. doi: 10.1182/blood-2012-01-404699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarry JE, Murphy K, Perry R, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 2011;121(1):384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majeti R, Becker MW, Tian Q, et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci USA. 2009;106(9):3396–3401. doi: 10.1073/pnas.0900089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 14.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–1784. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 15.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 16.van Rhenen A, van Dongen GA, Kelder A, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110(7):2659–2666. doi: 10.1182/blood-2007-03-083048. [DOI] [PubMed] [Google Scholar]
- 17.Busfield SJ, Biondo M, Wong M, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. 2014;28(11):2213–2221. doi: 10.1038/leu.2014.128. [DOI] [PubMed] [Google Scholar]
- 18.Askmyr M, Ågerstam H, Hansen N, et al. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood. 2013;121(18):3709–3713. doi: 10.1182/blood-2012-09-458935. [DOI] [PubMed] [Google Scholar]
- 19.Pandolfi A, Barreyro L, Steidl U. Concise review: preleukemic stem cells: molecular biology and clinical implications of the precursors to leukemia stem cells. Stem Cells Transl Med. 2013;2(2):143–150. doi: 10.5966/sctm.2012-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corces-Zimmerman MR, Majeti R. Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia. 2014;28(12):2276–2282. doi: 10.1038/leu.2014.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14(21):6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 22.Sharma B, Nawandar DM, Nannuru KC, Varney ML, Singh RK. Targeting CXCR2 enhances chemotherapeutic response, inhibits mammary tumor growth, angiogenesis, and lung metastasis. Mol Cancer Ther. 2013;12(5):799–808. doi: 10.1158/1535-7163.MCT-12-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee YS, Choi I, Ning Y, et al. Interleukin-8 and its receptor CXCR2 in the tumour microenvironment promote colon cancer growth, progression and metastasis. Br J Cancer. 2012;106(11):1833–1841. doi: 10.1038/bjc.2012.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saintigny P, Massarelli E, Lin S, et al. CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res. 2013;73(2):571–582. doi: 10.1158/0008-5472.CAN-12-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Infanger DW, Cho Y, Lopez BS, et al. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013;73(23):7079–7089. doi: 10.1158/0008-5472.CAN-13-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh JK, Farnie G, Bundred NJ, et al. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin Cancer Res. 2013;19(3):643–656. doi: 10.1158/1078-0432.CCR-12-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh JK, Simões BM, Howell SJ, Farnie G, Clarke RB. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. 2013;15(4):210. doi: 10.1186/bcr3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Wu Y, Hou Y, et al. CXCR2 macromolecular complex in pancreatic cancer: a potential therapeutic target in tumor growth. Transl Oncol. 2013;6(2):216–225. doi: 10.1593/tlo.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ning Y, Labonte MJ, Zhang W, et al. The CXCR2 antagonist, SCH-527123, shows antitumor activity and sensitizes cells to oxaliplatin in preclinical colon cancer models. Mol Cancer Ther. 2012;11(6):1353–1364. doi: 10.1158/1535-7163.MCT-11-0915. [DOI] [PubMed] [Google Scholar]
- 30.Jamieson T, Clarke M, Steele CW, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122(9):3127–3144. doi: 10.1172/JCI61067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verma A, Deb DK, Sassano A, et al. Activation of the p38 mitogen-activated protein kinase mediates the suppressive effects of type I interferons and transforming growth factor-beta on normal hematopoiesis. J Biol Chem. 2002;277(10):7726–7735. doi: 10.1074/jbc.M106640200. [DOI] [PubMed] [Google Scholar]
- 32.Steidl U, Rosenbauer F, Verhaak RG, et al. Essential role of Jun family transcription factors in PU.1 knockdown-induced leukemic stem cells. Nat Genet. 2006;38(11):1269–1277. doi: 10.1038/ng1898. [DOI] [PubMed] [Google Scholar]
- 33.Will B, Kawahara M, Luciano JP, et al. Effect of the nonpeptide thrombopoietin receptor agonist Eltrombopag on bone marrow cells from patients with acute myeloid leukemia and myelodysplastic syndrome. Blood. 2009;114(18):3899–3908. doi: 10.1182/blood-2009-04-219493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassane DC, Guzman ML, Corbett C, et al. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood. 2008;111(12):5654–5662. doi: 10.1182/blood-2007-11-126003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guzman ML, Li X, Corbett CA, et al. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8). Blood. 2007;110(13):4436–4444. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guzman ML, Rossi RM, Neelakantan S, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. 2007;110(13):4427–4435. doi: 10.1182/blood-2007-05-090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellagatti A, Cazzola M, Giagounidis A, et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia. 2010;24(4):756–764. doi: 10.1038/leu.2010.31. [DOI] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerstung M, Pellagatti A, Malcovati L, et al. Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes. Nat Commun. 2015;6:5901. doi: 10.1038/ncomms6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braber S, Koelink PJ, Henricks PA, et al. Cigarette smoke-induced lung emphysema in mice is associated with prolyl endopeptidase, an enzyme involved in collagen breakdown. Am J Physiol Lung Cell Mol Physiol. 2011;300(2):L255–L265. doi: 10.1152/ajplung.00304.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Craddock C, Quek L, Goardon N, et al. Azacitidine fails to eradicate leukemic stem/progenitor cell populations in patients with acute myeloid leukemia and myelodysplasia. Leukemia. 2013;27(5):1028–1036. doi: 10.1038/leu.2012.312. [DOI] [PubMed] [Google Scholar]
- 42.Steidl U, Steidl C, Ebralidze A, et al. A distal single nucleotide polymorphism alters long-range regulation of the PU.1 gene in acute myeloid leukemia. J Clin Invest. 2007;117(9):2611–2620. doi: 10.1172/JCI30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bullinger L, Döhner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350(16):1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- 44.Valk PJ, Verhaak RG, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350(16):1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- 45.Du M, Qiu Q, Gruslin A, et al. SB225002 promotes mitotic catastrophe in chemo-sensitive and -resistant ovarian cancer cells independent of p53 status in vitro. PLoS ONE. 2013;8(1):e54572. doi: 10.1371/journal.pone.0054572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cacalano G, Lee J, Kikly K, et al. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265(5172):682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- 47.Corre I, Pineau D, Hermouet S. Interleukin-8: an autocrine/paracrine growth factor for human hematopoietic progenitors acting in synergy with colony stimulating factor-1 to promote monocyte-macrophage growth and differentiation. Exp Hematol. 1999;27(1):28–36. doi: 10.1016/s0301-472x(98)00032-0. [DOI] [PubMed] [Google Scholar]
- 48.Laterveer L, Lindley IJ, Hamilton MS, Willemze R, Fibbe WE. Interleukin-8 induces rapid mobilization of hematopoietic stem cells with radioprotective capacity and long-term myelolymphoid repopulating ability. Blood. 1995;85(8):2269–2275. [PubMed] [Google Scholar]
- 49.Broxmeyer HE, Cooper S, Cacalano G, Hague NL, Bailish E, Moore MW. Involvement of Interleukin (IL) 8 receptor in negative regulation of myeloid progenitor cells in vivo: evidence from mice lacking the murine IL-8 receptor homologue. J Exp Med. 1996;184(5):1825–1832. doi: 10.1084/jem.184.5.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broxmeyer HE, Sherry B, Cooper S, et al. Comparative analysis of the human macrophage inflammatory protein family of cytokines (chemokines) on proliferation of human myeloid progenitor cells. Interacting effects involving suppression, synergistic suppression, and blocking of suppression. J Immunol. 1993;150(8 Pt 1):3448–3458. [PubMed] [Google Scholar]
- 51.Emadi S, Clay D, Desterke C, et al. French INSERM Research Network on MMM. IL-8 and its CXCR1 and CXCR2 receptors participate in the control of megakaryocytic proliferation, differentiation, and ploidy in myeloid metaplasia with myelofibrosis. Blood. 2005;105(2):464–473. doi: 10.1182/blood-2003-12-4415. [DOI] [PubMed] [Google Scholar]
- 52.Daly TJ, LaRosa GJ, Dolich S, Maione TE, Cooper S, Broxmeyer HE. High activity suppression of myeloid progenitor proliferation by chimeric mutants of interleukin 8 and platelet factor 4. J Biol Chem. 1995;270(40):23282–23292. doi: 10.1074/jbc.270.40.23282. [DOI] [PubMed] [Google Scholar]
- 53.Gewirtz AM, Zhang J, Ratajczak J, et al. Chemokine regulation of human megakaryocytopoiesis. Blood. 1995;86(7):2559–2567. [PubMed] [Google Scholar]
- 54.Dimicoli S, Wei Y, Bueso-Ramos C, et al. Overexpression of the toll-like receptor (TLR) signaling adaptor MYD88, but lack of genetic mutation, in myelodysplastic syndromes. PLoS ONE. 2013;8(8):e71120. doi: 10.1371/journal.pone.0071120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei Y, Dimicoli S, Bueso-Ramos C, et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia. 2013;27(9):1832–1840. doi: 10.1038/leu.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X, Eksioglu EA, Zhou J, et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest. 2013;123(11):4595–4611. doi: 10.1172/JCI67580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rhyasen GW, Bolanos L, Fang J, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell. 2013;24(1):90–104. doi: 10.1016/j.ccr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]