

Abstract
Bacterial resistance to antibiotics continues to grow and pose serious challenges, while the discovery rate for new antibiotics declines. Kibdelomycin is a recently discovered natural-product antibiotic that inhibits bacterial growth by inhibiting the bacterial DNA replication enzymes DNA gyrase and topoisomerase IV. It was reported to be a broad-spectrum aerobic Gram-positive agent with selective inhibition of the anaerobic bacterium Clostridium difficile. We have extended the profiling of kibdelomycin by using over 196 strains of Gram-positive and Gram-negative aerobic pathogens recovered from worldwide patient populations. We report the MIC50s, MIC90s, and bactericidal activities of kibdelomycin. We confirm the Gram-positive spectrum and report for the first time that kibdelomycin shows strong activity (MIC90, 0.125 μg/ml) against clinical strains of the Gram-negative nonfermenter Acinetobacter baumannii but only weak activity against Pseudomonas aeruginosa. We confirm that well-characterized resistant strains of Staphylococcus aureus and Streptococcus pneumoniae show no cross-resistance to kibdelomycin and quinolones and coumarin antibiotics. We also show that kibdelomycin is not subject to efflux in Pseudomonas, though it is in Escherichia coli, and it is generally affected by the outer membrane permeability entry barrier in the nonfermenters P. aeruginosa and A. baumannii, which may be addressable by structure-based chemical modification.
INTRODUCTION
Bacterial resistance to antibiotics continues to emerge in clinical practice with frightening frequency, posing a serious health threat. Klevens et al. (1) recently reported that methicillin-resistant Staphylococcus aureus (MRSA) infections alone are responsible for about 18,000 deaths per year in the United States. The dearth of new antibiotics is due to a lack of discovery of novel chemical scaffolds that can be developed into clinically useful antibiotics, making this problem dire particularly for the ESKAPE (Enterococcus faecium, S. aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) pathogens (2). Most of the structural leads that have been used to develop antibiotic drugs were discovered over 5 decades ago, with the exception of linezolid and daptomycin, which were discovered in the 1980s. Most of these chemical leads originated from nature. Chemical modifications of the antibiotic lead structures discovered decades ago have led to incrementally improved antibiotics that continue to serve well and provide the current reservoir of clinical antibiotics (3, 4). However, the capacity of such modifications is not limitless. As a result, additional improvements of existing chemical scaffolds are proving increasingly challenging. The dearth of new antibiotics can be overcome by the discovery of new antibiotic scaffolds with either known or novel modes of action that can be developed as effective treatment options against drug-resistant bacteria.
We recently reported the discovery of a series of novel natural-product antibiotics with novel modes of action by the application of antisense-based screening technology exemplified by platensimycin, (5, 6) platencin (7, 8), and most recently kibdelomycin (9) and kibdelomycin A (Fig. 1) (10). The broad-spectrum Gram-positive antibiotic kibdelomycin was reported in 2011 and was isolated from a Kibdelosporangium sp. Kibdelomycin exerts its activity by inhibiting bacterial DNA synthesis through specific inhibition of the β subunits of DNA gyrase (GyrB) and topoisomerase IV (ParE). Kibdelomycin has been shown to be a potent inhibitor of Escherichia coli (50% inhibitory concentration [IC50], 60 nM) and S. aureus (IC50, 9 nM) gyrase supercoiling activity and a less potent inhibitor of the corresponding topoisomerase IV decatenating activity (E. coli IC50, 29,000 nM; S. aureus IC50, 500 nM). Kibdelomycin potently inhibited the catalytic E. coli ATPase activity of gyrase B (IC50, 11 nM) and topoisomerase IV (ParE) (IC50, 900 nM) (9). Kibdelomycin A is a less potent inhibitor of S. aureus gyrase supercoiling (IC50, 400 nM) and topoisomerase IV catenation (ParE IC50, 5,000 nM) but has been shown to be a potent inhibitor of E. coli gyrase B ATPase activity (IC50, 9 nM) though a poor inhibitor of the E. coli ParE ATPase (IC50, 6,400 nM) (10). We reported that kibdelomycin is a selective and potent inhibitor of Clostridium difficile growth without significantly affecting anaerobic Gram-negative bacteria, including Bacteroides species (11). It showed potent in vivo activity against C. difficile infection without systemic exposure (11). We recently reported an X-ray crystal structure of kibdelomycin bound to S. aureus and E. coli GyrB and ParE (12). The crystal structure showed that kibdelomycin binds uniquely in a U-shaped multicontact binding mode, occupying the ATP binding site with extension to another part of the pocket (12). Kibdelomycin exhibits a low frequency of resistance and shows no cross-resistance in S. aureus strains resistant to other known gyrase inhibitors, such as novobiocin, coumermycin, and quinolones, which is consistent with the novel dual-arm U-shaped binding mode described above. We describe here the time-kill kinetics of kibdelomycin against S. aureus and the activity of kibdelomycin against an expanded panel of wild-type and resistant strains of Gram-positive and Gram-negative bacteria. We also studied the effects of efflux pumps and the permeability barrier on the susceptibility of key Gram-negative pathogens to kibdelomycin. Interestingly, kibdelomycin demonstrates strong activity against geographically diverse clinical strains of A. baumannii and weak activity against P. aeruginosa.
FIG 1.
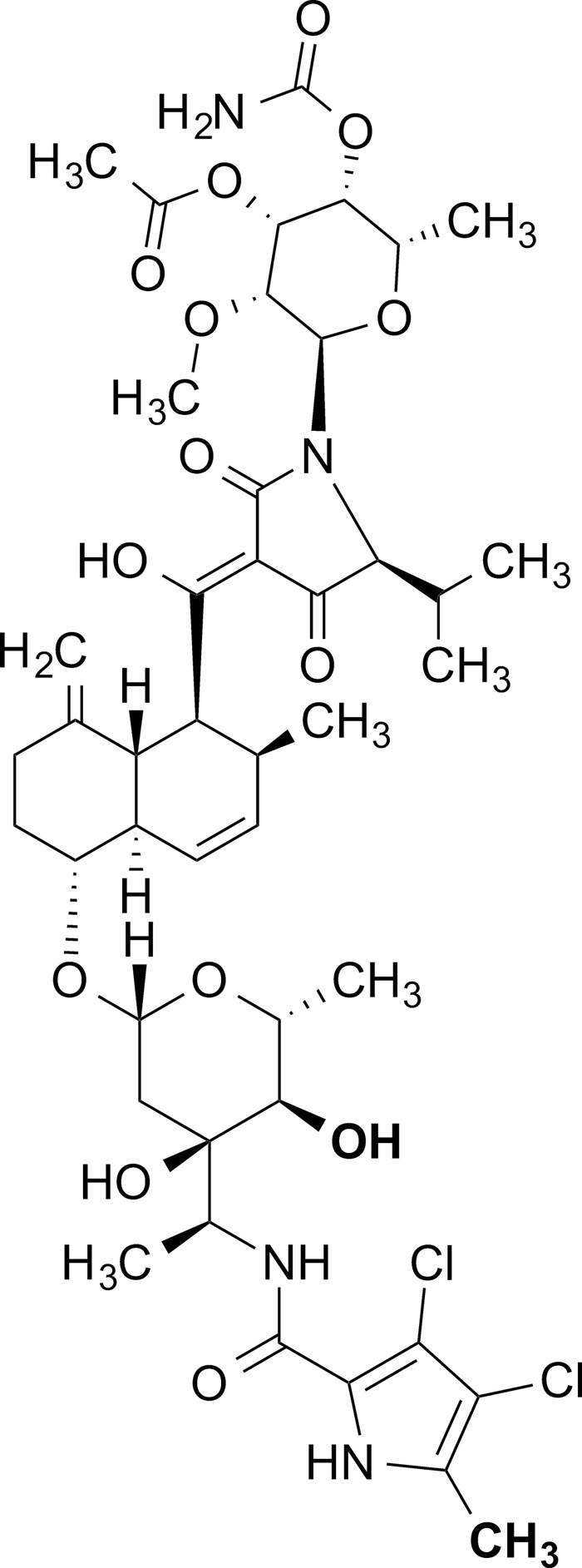
Chemical structure of kibdelomycin.
MATERIALS AND METHODS
Reagents.
All reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. Kibdelomycin and kibdelomycin A were isolated from a Kibdelosporangium sp. now named Kibdelosporangium banguiesis, as described earlier (9, 10).
Bacterial strains.
All of the strains in Tables 1 and 2 were collected from clinical samples in Japan or as otherwise stated. The ATCC and IID bacterial strains in Table 1 were obtained from the American Type Culture Collection and the Japanese Society for Bacteriology, respectively. The clinical strains in this study were isolated in Japan. Efflux-deficient E. coli was kindly provided by Okayama University (13). The quinolone-resistant S. aureus strains were selected sequentially from four clinical isolates (14). The quinolone-resistant S. pneumoniae strain was selected from IID553 (15). The P. aeruginosa nfxB and nfxC mutant strains were made from PAO4009, and the nalB mutant was made from PAO6006 (16).
TABLE 1.
In vitro MICsa of kibdelomycin and the quinolone levofloxacin
| Organism | Phenotype | MIC (μg/ml) |
|
|---|---|---|---|
| Kibdelomycin | Levofloxacin | ||
| Gram-positive bacteria | |||
| S. aureus Smith | Wild type | 0.25 | 0.125 |
| S. aureus OITI 1-971 | MRSA clinical isolate | 0.25 | 32 |
| S. pneumoniae IID553 | Wild type | 0.25 | 1 |
| E. faecium A2373 | Vancomycin resistant | 1 | 4 |
| Gram-negative bacteria | |||
| E. coli ATCC 25922 | Wild type | >16 | 0.031 |
| E. coli TG1 | Wild type | >16 | 0.031 |
| P. aeruginosa PAO1 | Wild type | >16 | 0.5 |
| P. aeruginosa PAO4009 | Wild type | >16 | 0.5 |
| P. aeruginosa KH4013E (nfxB) | MexCD-OprJ overexpressed | >16 | 2 |
| P. aeruginosa KH4014a (nfxC) | MexEF-OprN overexpressed | >16 | 8 |
| P. aeruginosa PAO969 | Wild type | >16 | 0.5 |
| P. aeruginosa PAO6006 (nalB) | MexAB-OprM overexpressed | 16 | 4 |
| P. aeruginosa TOHOKU1 | Multidrug resistant | 16 | 64 |
| A. baumannii IID876 | Wild type | 2 | 0.125 |
MICs were determined by the CLSI agar diffusion method. All strains were obtained from the ATCC or the Japanese Society of Bacteriology. P. aeruginosa strains KH4013E (nfxB) and KH4014a (nfxC) were prepared from PAO4009, and PAO6006 (nalB) was prepared from PAO6006 (16).
TABLE 2.
In vitro MICsa of kibdelomycin and levofloxacin for quinolone-resistant Gram-positive strains
| Organism | Phenotype | Mutation(s) | MIC (μg/ml) |
|
|---|---|---|---|---|
| Kibdelomycin | Levofloxacin | |||
| S. aureus | Wild-type parent | None | 0.25 | 0.25 |
| S. aureus MS5935A | 1st-step mutant | grlA (S80F) | 0.5 | 1 |
| S. aureus MS5935B | 2nd-step mutant | grlA (S80F), gyrA (S84L) | 0.5 | 16 |
| S. aureus MS5935C | 3rd-step mutant | grlA (S80F), gyrA (S84L), grlA (E84K) | 0.5 | 64 |
| S. aureus MS5935D | 4th-step mutant | grlA (S80F), gyrA (S84L), grlA (E84K), gyrA (E88V) | 0.5 | >128 |
| S. pneumoniae IID553 | Wild-type parent | None | 0.5 | 1 |
| S. pneumoniae NC9971 | Quinolone resistant | parC (S79Y), gyrA (S81F) | 0.5 | 32 |
MICs were determined by the CLSI agar diffusion method. Similar results were obtained with quinolone-resistant strains generated from S. aureus parent strains MS5952, MS5867, and MR6009. The quinolone-resistant S. aureus mutants were selected sequentially from clinical isolate MS5935 (14). The quinolone-resistant S. pneumoniae strain was selected from strain IID553 (15).
The strains (n = 196) in Table 3 were collected as a part of the Merck SMART surveillance studies (17) throughout the world and represent Belgium (n = 4), China (n = 10), the Czech Republic (n = 1), France (n = 5), Germany (n = 6), Hong Kong (n = 10), Ireland (n = 1), Italy (n = 9), Japan (n = 8), Poland (n = 1), Portugal (n = 1), Singapore (n = 1), South Korea (n = 12), Spain (n = 6), Sweden (n = 2), Taiwan (n = 9), Thailand (n = 3), the United Kingdom (n = 12), and the United States (n = 96). These strains were recovered from abscess, blood, bronchial washing, ear, eye, nasopharynx/throat/nose, sinus, skin, sputum, tracheal aspirate, urine, wound, and soft tissue samples. The strains were selected randomly and include antibiotic-susceptible and -resistant strains. The sources of the membrane-permeating ability- and efflux-deficient strains presented in Table 4 are listed in the last column.
TABLE 3.
Comparative in vitro MICsa of kibdelomycin, linezolid, vancomycin, and levofloxacin for 196 clinical bacterial strains
| Organism (no. of isolates) and antimicrobial agent | MIC (μg/ml) |
||
|---|---|---|---|
| 50% of strains | 90% of strains | Range | |
| S. aureus (57) | |||
| Kibdelomycin | 1.00 | 2.00 | 0.25–4.00 |
| Linezolid | 2.00 | 16 | 1.00 to >16 |
| Vancomycin | 1.00 | 4.00 | 0.50 to >32 |
| Levofloxacin | 8 | >16 | 0.25 to >16 |
| S. epidermidis (7) | |||
| Kibdelomycin | ND | ND | 2.00–8.00 |
| Linezolid | ND | ND | 1.00–2.00 |
| Vancomycin | ND | ND | 1.00–2.00 |
| Levofloxacin | ND | ND | 0.50–8.00 |
| Coagulase-negative Staphylococcus (8) | |||
| Kibdelomycin | ND | ND | 0.50–8.00 |
| Linezolid | ND | ND | 1.00–2.00 |
| Vancomycin | ND | ND | 0.50–2.00 |
| Levofloxacin | ND | ND | 0.125–16 |
| S. pneumoniae (22) | |||
| Kibdelomycin | 2.00 | 2.00 | 0.25–4.00 |
| Linezolid | 1.00 | 1.00 | ≤0.25–1.00 |
| Vancomycin | 0.25 | 0.25 | 0.25–0.50 |
| Levofloxacin | 0.50 | 16 | 0.50–16 |
| S. pyogenes (15) | |||
| Kibdelomycin | 4.00 | 4.00 | 2.00–8.00 |
| Linezolid | 1.00 | 1.00 | 0.50–1.00 |
| Vancomycin | 0.25 | 0.25 | 0.25–0.50 |
| Levofloxacin | 0.50 | 0.50 | 0.25–2.00 |
| E. faecalis (21) | |||
| Kibdelomycin | 0.50 | 2.00 | 0.25–2.00 |
| Linezolid | 1.00 | 1.00 | 0.50–1.00 |
| Vancomycin | 2.00 | >32 | 1.00 to >32 |
| Levofloxacin | >16 | >16 | 1.00 to >16 |
| E. faecium (18) | |||
| Kibdelomycin | 4.00 | 4.00 | 2.00–4.00 |
| Linezolid | 2.00 | 2.00 | 1.00–2.00 |
| Vancomycin | >32 | >32 | 0.50 to >32 |
| Levofloxacin | >16 | >16 | 1.00 to >16 |
| M. catarrhalis (12) | |||
| Kibdelomycin | 0.25 | 0.50 | ≤0.015–1.00 |
| Linezolid | 4.00 | 4.00 | 2.00–4.00 |
| Vancomycin | >32 | >32 | 32 to >32 |
| Levofloxacin | 1 | 2 | 0.5–4 |
| H. influenzae (17) | |||
| Kibdelomycin | 2.00 | 4.00 | 0.50–8.00 |
| Linezolid | >16 | >16 | 8.00 to >16 |
| Vancomycin | >32 | >32 | 32 to >32 |
| Levofloxacin | 0.015 | 0.03 | 0.015–0.03 |
| A. baumannii (19) | |||
| Kibdelomycin | ≤0.015 | 0.125 | ≤0.015 to >32 |
| Linezolid | >16 | >16 | >16 |
| Vancomycin | >32 | >32 | >32 |
| Levofloxacin | 8 | >16 | 0.03 to >16 |
MICs were determined by the CLSI broth microdilution method.
TABLE 4.
MICs for key Gram-negative bacteria with systematic changes in permeability and efflux
| Species | MIC (μg/ml)a |
Source or reference | |||||||
|---|---|---|---|---|---|---|---|---|---|
| KBD | RIF | NOVO | CAM | ERY | IMI | CIPRO | TET | ||
| E. coli BW25113 wild type | >128 | 16 | >128 | 32 | 128 | 0.25 | 0.016 | 4 | E. coli Genetic Stock Center |
| E. coli BW25113 ΔtolC | 32 | 16 | 4 | 8 | 4 | 0.5 | 0.004 | 1 | This study |
| E. coli BW25113 ΔtolC DmukB::kan | 4 | 4 | 0.5 | 1 | 2 | 0.125 | 0.004 | 0.5 | This study |
| E. coli BW25113 ΔtolC DmukF::kan | 16 | 8 | 1 | 2 | 4 | 0.5 | 0.008 | 1 | This study |
| E. coli MB4903 wild type | >128 | 16 | >128 | 16 | 128 | 0.25 | 0.031 | >128 | 29 |
| E. coli MB4902 lpxC101 | >128 | 0.0625 | 64 | 4 | 2 | 0.063 | 0.008 | 64 | 29 |
| E. coli MB5747 tolC::Tn10 | 128 | 8 | 2 | 2 | 4 | 0.5 | 0.004 | 64 | 30 |
| E. coli MB5746 tolC::Tn10 lpxC101 | 32 | 0.5 | 2 | 2 | 0.25 | 0.5 | 0.156 | 128 | 30 |
| P. aeruginosa MB5919 (CB0046) wild type | 32 | 32 | >128 | >128 | 128 | 8 | 1 | 32 | 31 |
| P. aeruginosa MB5890 (CB1101) Efflux mutantb | 64 | 32 | 128 | 2 | 16 | 1 | 0.004 | 0.5 | This study |
| P. aeruginosa ATCC 12055 wild type | 16 | 8 | >128 | 128 | 128 | 1 | 0.125 | 16 | ATCC |
| P. aeruginosa ATCC 35151 hypersusceptible | 1 | 0.5 | 2 | 4 | 2 | 0.25 | 0.063 | 0.5 | ATCC |
| A. baumannii ATCC 19606 wild type | 16 | 2 | 8 | 128 | 64 | 0.25 | 1 | 4 | ATCC |
| A. baumannii ATCC 19606 ΔlpxC | 0.031 | 0.0005 | 0.625 | 16 | 0.25 | 0.063 | 0.25 | 0.5 | This study |
| A. baumannii NCIMB12457 wild type | 32 | 4 | 16 | >128 | 32 | 0.5 | 0.5 | 8 | Microbiologics Inc. |
| A. baumannii NCIMB12457 ΔlpxA | 0.125 | 0.0005 | 0.063 | 64 | 0.25 | 0.063 | 0.25 | 0.5 | This study |
MICs were determined by the CLSI broth microdilution method. Abbreviations: KBD, kibdelomycin; RIF, rifampin; NOVO, novobiocin; CAM, chloramphenicol; ERY, erythromycin; IMI, imipenem; CIPRO, ciprofloxacin; TET, tetracycline.
mexAB-oprM mexCD-oprJ mexXY mexJKL mexHI-opmD opmH.
Determination of MICs.
MICs were determined by either the broth microdilution or the agar dilution method recommended by the Clinical and Laboratory Standards Institute in Mueller-Hinton II broth (Becton Dickinson, NJ) (S. B. Singh, J. D. Polishook, D. L. Zink, O. Genilloud, M. A. Goetz, and F. Vicente, U.S. patent application WO2011/079034A1). The MIC was defined as the lowest concentration of an antibacterial agent that inhibited visible growth after incubation.
Determination of time-kill kinetics.
The time-kill kinetics of kibdelomycin were determined by the broth microdilution method in accordance with CLSI guidelines (18) with S. aureus ATCC 29213 at the MIC and 2, 4, and 8 times the MIC; the corresponding concentrations are 1, 2, 4, and 8 μg/ml, respectively. The MIC was determined with an inoculum of 5 × 105 CFU/ml prior to commencing the time-kill experiment. Briefly, a stock kibdelomycin solution of 5.12 mg/ml of dimethyl sulfoxide was prepared. The inoculum suspension was prepared from growth on a tryptic soy agar (TSA)–5% sheep blood plate to equal the turbidity of a 0.5 McFarland standard in Mueller-Hinton II broth (MHBII; Becton Dickinson, Sparks, MD), diluted 1:3, grown in fresh MHBII at 35°C, and incubated while shaking at 150 rpm. After approximately 2 h, the suspension was again adjusted to equal a 0.5 McFarland standard and diluted 1:2 in fresh MHBII, and 1 ml was used to inoculate each 125-ml Erlenmeyer flask containing 8.75 ml of sterile MHBII. The final target cell density was approximately 106 to 107 CFU/ml. Just prior to T0, 0.25 ml of the appropriate concentration of kibdelomycin solution was added to each flask and gently mixed, and 1.0 ml of diluted inoculum was added. Thus, test drug vessels contained 8.75 ml of MHBII, 1.0 ml of inoculum, and 0.25 ml of drug solution (40×). A total of 22 vessels were prepared in this fashion and immediately mixed by swirling, and 0.03 ml was removed from the initial 0.5 McFarland standard tube for determination of the viable count at the baseline (T0) by serial 10-fold dilution in MHBII. All vessels were incubated at 35°C in a New Brunswick Scientific Series 25 Incubated Shaker rotating at 150 rpm to provide gentle mixing. The vessels were sampled at 2 h (T2), 4 h (T4), 6 h (T6), 8 h (T8), and 24 h (T24) for determination of viable counts. Viable counts were determined by removing 0.3 ml from each vessel, serially diluting it 10-fold in 0.27 ml of MHBII with the Biomek 2000, and then plating duplicate 10-μl samples onto TSA–5% sheep blood plates by the track dilution method (19). In brief, 10-μl aliquots were spotted in duplicate across the top of a 100-mm-square TSA–5% sheep blood plate. The plate was then tilted at a 45 to 90° angle to allow the 10-μl aliquot to track across the agar surface to the opposite side of the plate. All plates were incubated for 20 to 24 h at 35°C. Colonies were counted manually, and the number of CFU/ml was determined as the average count of duplicate plates, followed by calculation of the log10 number of CFU/ml. A bactericidal effect was defined as a 3-log10 CFU/ml decrease in the viable count at 24 h relative to that of the starting inoculum.
Deletion of EctolC, EcmukB, and EcmukF.
E. coli tolC (EctolC) was knocked out of strain BW25113 by using the linear PCR product generated by amplification from pKD4 (20) with primers P1 and P2 (Table 5). Transformation and selection on kanamycin were performed as described previously (21), yielding strain BW25113ΔtolC::kan. Transformation with pFlp2 (22) allowed the subsequent excision of the kanamycin resistance cassette through Flp-mediation recombination, leaving an 84-nucleotide scar between the first and last six codons of EctolC, thus generating strain BW25113ΔtolC.
TABLE 5.
Primers used in this study
| Primer | Sequence |
|---|---|
| P1 | TTTACAGTTTGATCGCGCTAAATACTGCTTCACCACAAGGAATGCAAATGGTGCAGGCTGGAGCTGCTTC |
| P2 | CAGACGGGGCCGAAGCCCCGTCGTCGTCATCAGTTACGGAAAGGGTTATGCATATGAATATCCTCCTTAG |
| P3 | ACGCTGATTAACTGGAACGGCTTTTTTGCCCGAACTTTTGACCTTGACGAGTGCAGGCTGGAGCTGCTTC |
| P4 | TGGAAGCGTTTCAGGGAGTTGCGGCGCAAATCCTCGCAGGCCGACGACATCATATGAATATCCTCCTTAG |
| P5 | CTGGTTGCCTGGGCCAGAAAAAATGACTTCTCCATCTCGCTGCCGGTAGAGTGCAGGCTGGAGCTGCTTC |
| P6 | GGCTCCGTAATCATTAATCGGCTGCCATTTCGCTGGCAGTCCGGTGAAATCATATGAATATCCTCCTTAG |
| P7 | ACTCTTCCTTTTTCAATATTATTGAAGC |
| P8 | TAACTGTCAGACCAAGTTTACTCATATATAC |
| P9 | TGAAAAAGGAAGAGTATGATTGAACAAGATGGATTG |
| P10 | TTGGTCTGACAGTTAGAAGAACTCGTCAAGAAGGCG |
| P11 | TGCCCTGAATGAACTGCAAGACGAGGCAGCGC |
| P12 | TTGGTCTGACAGTTAGAAGAACTCGTCAAGAAGGCG |
| P13 | CCAAGCTTGCATGCCTGCAGTTTGCGTAAATAGAATATTATGACCG |
| P14 | CCACGCTCTGATTGTTCAAGAGGTAGAATGGATTAAATCGTG |
| P15 | CACGATTTAATCCATTCTACCTCTTGAACAATCAGAGCGTGG |
| P16 | CGGTACCCGGGGATCCTCTGAGCAGCAGCCTAGCGCACTAACAACC |
| P17 | CCAAGCTTGCATGCCTGCAGGCGAAAGGCGTATTAATTAACATTAC |
| P18 | CACGTATGGAATTGGACAGTCCACACGATTGAGAGTACGCTG |
| P19 | CAGCGTACTCTCAATCGTGTGGACTGTCCAATTCCATACGTG |
| P20 | CGGTACCCGGGGATCCCATAAAAACAGGAAACCTTACGTTTCTAAC |
EcmukB and EcmukF were deleted from strain BW25113ΔtolC by the aforementioned one-step λ Red method in conjunction with the linear PCR products generated by amplification from pKD4 (20) with primers P3 and P4 for EcmukB and P5 and P6 for EcmukF. Transformation and selection on kanamycin were performed as described previously (21), yielding strains BW25113ΔtolCΔmukB::kan and BW25113ΔtolCΔmukF::kan, respectively. As previously described, the E. coli mukB and mukF knockouts were temperature sensitive (23), so in order to facilitate downstream MIC testing, single-step temperature-insensitive mutants were selected by plating on cation-adjusted Mueller-Hinton agar (CAMHA) containing 30 μg/ml kanamycin and isolating colonies that grew overnight at 37°C.
Deletion of A. baumannii lpxA and lpxC.
Unmarked lpxA and lpxC deletion mutants of NCIMB 12457 and ATCC 19606, respectively, were constructed with a version of the pEX18Ap suicide vector (22) in which the ampicillin resistance cassette was replaced with the kanamycin resistance cassette from pKD4 (20). To create this pEX18Km vector, the pEX18Ap vector backbone was amplified with primers P7 and P8 and the kanamycin resistance cassette was amplified in two sections to remove an internal PstI site with primers P9 and P10 for the 5′ portion and P11 and P12 for the 3′ portion. In-Fusion Clonase (Clontech) was then used to fuse these three fragments into the final pEX18Km vector.
Flanking regions of the A. baumannii pxA (AblpxA) and AblpxC genes were fused together by splicing by overlap extension. Primer pairs P13/P14 and P17/18 were used to amplify 500-bp regions upstream of lpxA and lpxC, respectively, and primer pairs P15/P16 and P19/P20 were used to amplify 500-bp regions downstream of lpxA and lpxC, respectively. In a second-round PCR, up- and downstream products were combined and amplified with primers P13/P16 for lpxA and P17/P20 for lpxC. These PCR products were subsequently digested with restriction enzymes PstI and BamHI and ligated into a similarly digested pEX18Km vector. The resulting pEX18KmΔlpxA and pEX18KmΔlpxC vectors were mobilized into A. baumannii from E. coli RHO3 (24) as described previously (21). To resolve cointegrants to double-crossover knockouts, single colonies were restreaked onto CAMHA with 10% (wt/vol) sucrose and 10 μg/ml colistin to select for loss of the sacB-containing vector backbone. This generated the final lpxA and lpxC knockout strains NCIMB12457ΔlpxA and ATCC 19606ΔlpxC.
RESULTS
Kibdelomycin is a broad-spectrum aerobic antibiotic.
Kibdelomycin was shown to inhibit the growth of Gram-positive bacteria S. aureus (wild-type and MRSA), Streptococcus pneumoniae, and Enterococcus faecalis and the Gram-negative bacterium Haemophilus influenzae with MICs of <2 μg/ml as tested by the CLSI broth microdilution method (9). MIC measurements by the agar diffusion method confirm these antibacterial activities and help extend the prior observed antibacterial spectrum (Table 1). The agar diffusion kibdelomycin MIC of 0.25 μg/ml for S. aureus and S. pneumoniae strains was 4- to 8-fold better than that measured by the broth microdilution method. Kibdelomycin inhibited the growth of E. faecium on agar with an MIC of 1 μg/ml.
Kibdelomycin showed potent growth inhibition of the Gram-negative bacterium A. baumannii (agar MIC, 2 μg/ml), similar to that observed with H. influenzae, but unlike the case of these two Gram-negative strains, kibdelomycin did not inhibit the growth of E. coli or that of most P. aeruginosa PAO1 strains (MICs, >16 μg/ml). A similar trend (up to16× lower activity against E. coli and 64× lower activity against P. aeruginosa than against A. baumannii) has been reported for a few new bacterial topoisomerase inhibitors binding to gyrase A/ParC, a binding site entirely different from that of kibdelomycin.(25, 26) The desmethyl congener kibdelomycin A and acetate analogs have been shown to be significantly less active in antibacterial assays (10).
In order to understand better whether the lack of Gram-negative activity of kibdelomycin was due to weak target engagement or a lack of cellular accumulation caused by either efflux or poor membrane-permeating ability, we tested the activity of kibdelomycin against a series of hypersusceptible Gram-negative mutants (Table 4). In E. coli, kibdelomycin activity was measured after knocking out tolC, which encodes the outer membrane protein component of the major efflux pump. Additionally, because it has been reported that E. coli mutants deficient in the chromosome-partitioning complex MukBEF are hypersusceptible to novobiocin (23), an inhibitor of the GyrB ATPase domain like kibdelomycin, it was of interest to test kibdelomycin against mukB and mukF knockout strains. Kibdelomycin was tested against P. aeruginosa strain ATCC 35151, which has been described as being hypersusceptible to most classes of antibiotics (27), presumably because of multiple defects in membrane permeability. Finally, we tested kibdelomycin against two lipopolysaccharide (LPS)-deficient mutants of A. baumannii. Such mutants completely devoid of LPS biosynthesis have been described to be exquisitely more permeable and deficient in efflux (28), as the outer membrane protein components of the efflux pumps no longer have the correct membrane composition in which to fold and function.
In all cases, the mutants were more susceptible to kibdelomycin than were the wild-type parental strains, indicating that the spectrum of activity is influenced by the ability to enter and accumulate in the cell. Depending on the organism, kibdelomycin activity appears to be limited by different factors. In E. coli, efflux appears to be a contributor to the lack of activity, as the MB4902 lpxC mutant with membrane permeability defects alone is not more susceptible to kibdelomycin but tolC knockout strains (BW25113ΔtolC, MB5747, MB5746) are slightly more susceptible. The activity of kibdelomycin can be improved further through mechanism-based synergy, as both mukB and mukF mutants are more susceptible, as is the case with novobiocin. Conversely, in P. aeruginosa, efflux does not appear to be the main contributor to the lack of activity as the MB5890 strain, in which six major efflux systems have been knocked out, is not more susceptible to kibdelomycin; instead, hyperpermeable strain ATCC 35151 is more susceptible to inhibition. Finally, in A. baumannii, kibdelomycin activity is greatly enhanced in both LPS-deficient mutants (NCIMB12457ΔlpxA and ATCC 19606ΔlpxC) by >2 orders of magnitude.
Absence of cross-resistance to kibdelomycin and other gyrase inhibitors.
We have shown earlier that kibdelomycin is still potently active (no shift in MIC) against novobiocin-resistant S. aureus harboring a single mutation in GyrB (D89G) and coumermycin-resistant S. aureus with three mutations in GyrB (Q136E, I175T, L455I) shows only moderate cross-resistance (4-fold MIC shift) (9). While the lack of cross-resistance to these antibiotics is important for mechanistic analysis, analysis of quinolone-resistant strains for cross-resistance is more critical from a drug development perspective. Kibdelomycin was evaluated against a variety of well-characterized quinolone-resistant S. aureus strains selected through multiple rounds of mutations against quinolones. Mutant strains were sequenced to determine nucleotide changes, and mutations were mapped to gyrA, parC, and grlA (Table 2). The MICs (0.25 to 0.5 μg/ml) of kibdelomycin were not affected by these mutations, whereas the MICs of ciprofloxacin were shifted 4- to >512-fold, indicating a lack of cross-resistance to kibdelomycin. A quinolone-resistant S. pneumoniae strain with mutations in parC and gyrA also retained a wild-type level susceptibility to kibdelomycin, similar to S. aureus (Table 2).
Bacterial profiling.
Kibdelomycin was profiled against 196 bacterial strains collected from various infection sites and tissue samples collected from patients throughout the world. These strains were selected randomly and included antibiotic-susceptible and -resistant isolates. Only key Gram-positive and selected Gram-negative species that were susceptible to kibdelomycin were selected for this larger study, including A. baumannii. Linezolid, vancomycin, and levofloxacin were used as comparators. The MIC50, MIC90, and MIC range of all four antibiotics for 196 diverse strains are presented in Table 3. Kibdelomycin showed strong activity against S. aureus, with an MIC50 and MIC90 of 1 and 2 μg/ml, respectively. Many strains showed susceptibility at 0.25 μg/ml, as was observed in the agar dilution assay. The kibdelomycin MICs for linezolid-, vancomycin-, and levofloxacin-resistant strains were unaffected. The activity of kibdelomycin against a small panel of S. epidermis and coagulase-negative Staphylococcus isolates was somewhat weaker. Kibdelomycin showed good activity against S. pneumoniae, with a narrow MIC range and the same MIC50 and MIC90 (2 μg/ml). Its activity against S. pyogenes was 2-fold lower than that against S. pneumoniae, with the same MIC50 and MIC90 of 4 μg/ml. It showed good activity against E. faecalis, with an MIC50 of 0.5 μg/ml and an MIC90 of 2 μg/ml, and reasonable activity against E. faecium, with an MIC50 and MIC90 of 4 μg/ml. Most importantly, the activity of kibdelomycin was indifferent to the vancomycin resistance status of E. faecalis and E. faecium strains. Kibdelomycin showed potent activity against the Gram-negative bacteria M. catarrhalis and A. baumannii, with MIC90s of 0.5 and 0.125 μg/ml, respectively. Of the 19 strains of the nonfermenter A. baumannii tested, only 1, collected in the United States, demonstrated an MIC of >32 μg/ml and that strain also showed resistance to levofloxacin. A second strain with an MIC of 8 μg/ml was susceptible to levofloxacin (MIC, 0.25 μg/ml). The kibdelomycin MICs for the remaining 17 strains were <0.12 μg/ml. The strains resistant to levofloxacin in this group showed susceptibility to kibdelomycin. The differences between the MICs of kibdelomycin for clinical strains and laboratory strains of A. baumannii (Tables 1, 3, and 4) are perplexing. They may be due to differences between laboratory strains IID876 (Table 1), ATCC 19606, and NCIMB12457 (Table 4) and clinical strains (Table 3), compounded by the chelating tetramic acid structural moiety of kibdelomycin affected by differences in the contents of agar (Table 1) and broth media (Tables 3 and 4) and need further exploration. Kibdelomycin showed good activity against H. influenzae, with an MIC50 of 2 μg/ml and an MIC90 of 4 μg/ml. As expected, linezolid and vancomycin did not show much activity against Gram-positive bacteria other than the moderate activity shown by linezolid against M. catarrhalis.
Time-kill kinetics of kibdelomycin.
The time-kill kinetics of kibdelomycin were determined with S. aureus strain ATCC 29213 at a final cell density of 106 to 107 CFU/ml at the MIC and 2, 4, and 8 times the MIC and incubation for up to 24 h. Kibdelomycin showed slow bactericidal activity at 4 and 8 times the MIC, with a 3-log drop in the viable bacterial count at 24 h (Fig. 2). Regrowth was observed in cultures treated with kibdelomycin at 2 times the MIC (no regrowth with ciprofloxacin at 2 times the MIC). Although the culture at 24 h was not tested for increased MICs, resistance would probably not have been selected for under the kill curve conditions used since the frequency of resistance was previously determined to be <10−10 (9). More likely, regrowth was due to a difference in the conditions under which the MIC was measured (broth microdilution) and the kill curve was determined, resulting in a possible inoculum effect. Alternative explanations could be a lack of compound stability in the medium and a decreased concentration because of precipitation, chelation, or some other mechanism. Ciprofloxacin was used as a positive control that showed rapid bactericidal activity at 2, 4, and 8 times the MIC with a 3-log drop in viable bacterial counts at 2 to 24 h (Fig. 2).
FIG 2.
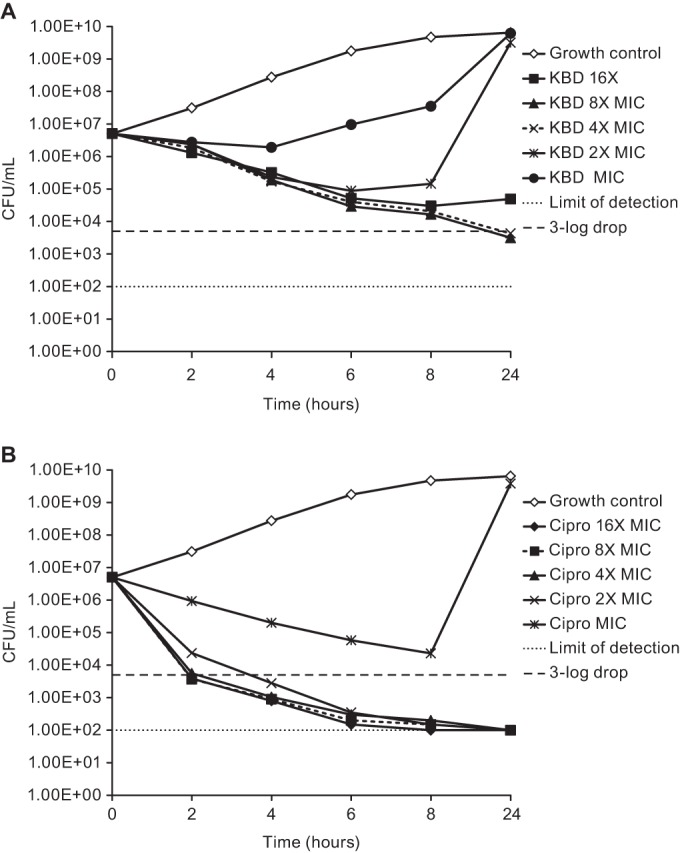
Time-kill kinetics of kibdelomycin (KBD, A) and ciprofloxacin (Cipro, B) at multiples of the MIC evaluated against S. aureus MMX 0100 (ATCC 29213).
DISCUSSION
Kibdelomycin is a novel natural product from Kibdelosporangium sp. recently discovered through an innovative S. aureus fitness test-based screening method (9). Kibdelomycin selectively inhibits bacterial DNA synthesis by specifically inhibiting the ATPase activity of bacterial gyrase B (GyrB) and topoisomerase IV (ParE). It binds the GyrB and ParE enzymes in a unique U-shaped binding mode with multipoint contacts (12). Kibdelomycin selectively inhibited C. difficile without affecting many other anaerobic gut bacteria, including Bacteroides species (11). It shows potent bactericidal activity against S. aureus, although it is slower than fluoroquinolones (Fig. 2). Most significantly, kibdelomycin did not show cross-resistance to other known gyrase inhibitors, regardless of their mode of binding, including ATPase inhibitors such as novobiocin and coumermycin, and DNA cleavage/resealing gyrase A/ParC inhibitors such as levofloxacin and other quinolones. We have already shown that kibdelomycin showed no MIC change between wild-type and novobiocin-resistant (harboring single D89G mutation in GyrB) S. aureus strains. There was a moderate (4-fold) shift in its MIC for coumermycin-resistant S. aureus harboring three mutations in GyrB (Q136E, I175T, L455I) (9). These findings were nicely explained by several kibdelomycin-bound crystal structures of GyrB and ParE (12). Most importantly, this study, kibdelomycin did not show a shift in its MIC for many well-characterized highly quinolone-resistant S. aureus strains harboring multiple mutations in GrlA and GyrA and S. pneumoniae strains harboring mutations in ParC and GyrA (Table 2). As expected, there was no cross-resistance to it and antibiotics to which bacteria are resistant by other mechanisms, such as linezolid and vancomycin (Table 3).
Kibdelomycin shows a broad spectrum of activity against clinically relevant Gram-positive bacteria. It also shows a broad spectrum of activity against select Gram-negative bacteria, including potent activity against clinical strains of H. influenzae, M. catarrhalis, and A. baumannii (Table 3); moderate activity against some strains of P. aeruginosa; and poor activity against E. coli. The differences in the potency of kibdelomycin against various strains of bacteria did not appear to be target dependent. Significant homology between gyrase enzymes from bacterial strains exists, and binding interactions appear to be preserved in all of the enzymes. The difference in the activity of kibdelomycin against Gram-negative is dependent on various factors (Table 4). In E. coli, both efflux and membrane permeability appear to play roles in the lack of activity (and the two may be synergistic), whereas in A. baumannii and P. aeruginosa, the permeability barrier is more critical, which may be related to physical properties of the compound and potentially addressable by structure-based chemical modification. Indeed, it is somewhat surprising that the effects of efflux deletion were so different in E. coli and P. aeruginosa. Generally, compounds that are subject to the resistance-nodulation-division efflux pumps show similar tendencies for AcrAB/TolC (E. coli) and MexAB/OprM (P. aeruginosa). Kibdelomycin may be used as a tool compound to explore further the properties that allow compounds to become substrates for this clinically important family of efflux pumps.
In summary, our previous studies have shown that kibdelomycin is a candidate for potential development for the treatment of C. difficile-associated diarrhea and current studies show that the potential exists for X-ray structure-guided chemical modification of certain parts of the molecule to produce compounds with potentially broad-spectrum activities addressing many of the ESKAPE pathogens.
ACKNOWLEDGMENTS
We acknowledge Micromyx for providing time-kill kinetic data and Eurofins for MIC50 and MIC90 data and thank Lynn L. Silver for careful reading of the manuscript.
REFERENCES
- 1.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Singh SB, Barrett JF. 2006. Empirical antibacterial drug discovery—foundation in natural products. Biochem Pharmacol 71:1006–1015. doi: 10.1016/j.bcp.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 4.Walsh CT. 2003. Antibiotics: actions, origin, resistance. ASM Press, Washington, DC. [Google Scholar]
- 5.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang Y, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Alloco J, Basilio Á, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes J, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. 2006. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 6.Singh SB, Jayasuriya H, Ondeyka JG, Herath KB, Zhang C, Zink DL, Tsou NN, Ball RG, Basilio A, Genilloud O, Diez MT, Vicente F, Pelaez F, Young K, Wang J. 2006. Isolation, structure, and absolute stereochemistry of platensimycin, a broad spectrum antibiotic discovered using an antisense differential sensitivity strategy. J Am Chem Soc 128:11916–11920. doi: 10.1021/ja062232p. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Kodali S, Lee SH, Galgoci A, Painter R, Dorso K, Racine F, Motyl M, Hernandez L, Tinney E, Colletti S, Herath K, Cummings R, Salazar O, Gonzalez I, Basilio A, Vicente F, Genilloud O, Pelaez F, Jayasuriya H, Young K, Cully D, Singh SB. 2007. Platencin is a dual FabF and FabH inhibitor with potent in vivo antibiotic properties. Proc Natl Acad Sci U S A 104:7612–7616. doi: 10.1073/pnas.0700746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayasuriya H, Herath KB, Zhang C, Zink DL, Basilio A, Genilloud O, Diez MT, Vicente F, Gonzalez I, Salazar O, Pelaez F, Cummings R, Ha S, Wang J, Singh SB. 2007. Isolation and structure of platencin: a novel FabH and FabF dual inhibitor with potent broad spectrum antibiotic activity produced by Streptomyces platensis MA7339. Angew Chem Int Ed Engl 46:4684–4688. doi: 10.1002/anie.200701058. [DOI] [PubMed] [Google Scholar]
- 9.Phillips JW, Goetz MA, Smith SK, Zink DL, Polishook J, Onishi R, Salowe S, Wiltsie J, Allocco J, Sigmund J, Dorso K, Lee S, Skwish S, de la Cruz M, Martin J, Vicente F, Genilloud O, Lu J, Painter RE, Young K, Overbye K, Donald RG, Singh SB. 2011. Discovery of kibdelomycin, a potent new class of bacterial type II topoisomerase inhibitor by chemical-genetic profiling in Staphylococcus aureus. Chem Biol 18:955–965. doi: 10.1016/j.chembiol.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 10.Singh SB, Goetz MA, Smith SK, Zink DL, Polishook J, Onishi R, Salowe S, Wiltsie J, Allocco J, Sigmund J, Dorso K, de la Cruz M, Martin J, Vicente F, Genilloud O, Donald RG, Phillips JW. 2012. Kibdelomycin A, a congener of kibdelomycin, derivatives and their antibacterial activities. Bioorg Med Chem Lett 22:7127–7130. doi: 10.1016/j.bmcl.2012.09.071. [DOI] [PubMed] [Google Scholar]
- 11.Miesel L, Hecht DW, Osmolski JR, Gerding D, Flattery A, Li F, Lan J, Lipari P, Polishook JD, Liang L, Liu J, Olsen DB, Singh SB. 2014. Kibdelomycin is a potent and selective agent against toxigenic Clostridium difficile. Antimicrob Agents Chemother 58:2387–2392. doi: 10.1128/AAC.00021-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, Patel S, Sharma N, Soisson SM, Kishii R, Takei M, Fukuda Y, Lumb KJ, Singh SB. 2014. Structures of kibdelomycin bound to Staphylococcus aureus GyrB and ParE showed a novel U-shaped binding mode. ACS Chem Biol 9:2023–2031. doi: 10.1021/cb5001197. [DOI] [PubMed] [Google Scholar]
- 13.Morita Y, Kodama K, Shiota S, Mine T, Kataoka A, Mizushima T, Tsuchiya T. 1998. NorM, a putative multidrug efflux protein, of Vibrio parahaemolyticus and its homolog in Escherichia coli. Antimicrob Agents Chemother 42:1778–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukuda H, Hori S, Hiramatsu K. 1998. Antibacterial activity of gatifloxacin (AM-1155, CG5501, BMS-206584), a newly developed fluoroquinolone, against sequentially acquired quinolone-resistant mutants and the norA transformant of Staphylococcus aureus. Antimicrob Agents Chemother 42:1917–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukuda H. 2000. Genetic study of the mechanisms of action of fluoroquinolones in Streptococcus pneumoniae. Jpn J Chemother 48:243–250. [Google Scholar]
- 16.Fukuda H, Hosaka M, Hirai K, Iyobe S. 1990. New norfloxacin resistance gene in Pseudomonas aeruginosa PAO. Antimicrob Agents Chemother 34:1757–1761. doi: 10.1128/AAC.34.9.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chow JW, Satishchandran V, Snyder TA, Harvey CM, Friedland IR, Dinubile MJ. 2005. In vitro susceptibilities of aerobic and facultative gram-negative bacilli isolated from patients with intra-abdominal infections worldwide: the 2002 Study for Monitoring Antimicrobial Resistance Trends (SMART). Surg Infect (Larchmt) 6:439–448. doi: 10.1089/sur.2005.6.439. [DOI] [PubMed] [Google Scholar]
- 18.National Committee for Clinical Laboratory Standards. 1999. Methods for determining bactericidal activity of antimicrobial agents; approved guideline. NCCLS document M26-A National Committee for Clinical Laboratory Standards, Wayne, PA. [Google Scholar]
- 19.Jett BD, Hatter KL, Huycke MM, Gilmore MS. 1997. Simplified agar plate method for quantifying viable bacteria. Biotechniques 23:648–650. [DOI] [PubMed] [Google Scholar]
- 20.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balibar CJ, Hollis-Symynkywicz MF, Tao J. 2011. Pantethine rescues phosphopantothenoylcysteine synthetase and phosphopantothenoylcysteine decarboxylase deficiency in Escherichia coli but not in Pseudomonas aeruginosa. J Bacteriol 193:3304–3312. doi: 10.1128/JB.00334-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 23.Sawitzke JA, Austin S. 2000. Suppression of chromosome segregation defects of Escherichia coli muk mutants by mutations in topoisomerase I. Proc Natl Acad Sci U S A 97:1671–1676. doi: 10.1073/pnas.030528397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.López CM, Rholl DA, Trunck LA, Schweizer HP. 2009. Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl Environ Microbiol 75:6496–6503. doi: 10.1128/AEM.01669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh SB, Kaelin DE, Wu J, Miesel L, Tan CM, Meinke PT, Olsen D, Lagrutta A, Bradley P, Lu J, Patel S, Rickert KW, Smith RF, Soisson S, Wei C, Fukuda H, Kishii R, Takei M, Fukuda Y. 2014. Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med Chem Lett 5:609–614. doi: 10.1021/ml500069w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh SB, Kaelin DE, Wu J, Miesel L, Tan CM, Black T, Nargund R, Meinke PT, Olsen DB, Lagrutta A, Lu J, Patel S, Rickert KW, Smith RF, Soisson S, Sherer E, Joyce LO, Wei C, Peng X, Wang X, Fukuda H, Kishii R, Takei M, Takano H, Shibasaki M, Yajima M, Nishimura A, Shibata T, Fukuda Y. 24 March 2015. Tricyclic 1,5-naphthyridinone oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents-SAR of left-hand-side moiety (part-2). Bioorg Med Chem Lett doi: 10.1016/j.bmcl.2015.03.044. [DOI] [PubMed] [Google Scholar]
- 27.Zimmermann W. 1979. Penetration through the Gram-negative cell wall: a co-determinant of the efficacy of beta-lactam antibiotics. Int J Clin Pharmacol Biopharm 17:131–134. [PubMed] [Google Scholar]
- 28.Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, Henry R, Crane B, St Michael F, Cox AD, Adler B, Nation RL, Li J, Boyce JD. 2010. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother 54:4971–4977. doi: 10.1128/AAC.00834-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young K, Silver LL. 1991. Leakage of periplasmic enzymes from envA1 strains of Escherichia coli. J Bacteriol 173:3609–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kodali S, Galgoci A, Young K, Painter R, Silver LL, Herath KB, Singh SB, Cully D, Barrett JF, Schmatz D, Wang J. 2005. Determination of selectivity and efficacy of fatty acid synthesis inhibitors. J Biol Chem 280:1669–1677. doi: 10.1074/jbc.M406848200. [DOI] [PubMed] [Google Scholar]
- 31.Robertson GT, Doyle TB, Du Q, Duncan L, Mdluli KE, Lynch AS. 2007. A novel indole compound that inhibits Pseudomonas aeruginosa growth by targeting MreB is a substrate for MexAB-OprM. J Bacteriol 189:6870–6881. doi: 10.1128/JB.00805-07. [DOI] [PMC free article] [PubMed] [Google Scholar]