

Abstract
BK polyomavirus (BKPyV)-associated hemorrhagic cystitis (PyVHC) complicates 5 to 15% of allogeneic hematopoietic stem cell transplantations. Targeted antivirals are still unavailable. Brincidofovir (BCV; previously CMX001) has shown inhibitory activity against diverse viruses, including BKPyV in a primary human renal tubule cell culture model of polyomavirus-associated nephropathy. We investigated the effects of BCV in BKPyV-infected and uninfected primary human urothelial cells (HUCs), the target cells of BKPyV in PyVHC. The BCV concentrations causing 50 and 90% reductions (EC50 and EC90) in the number of intracellular BKPyV genome equivalents per cell (icBKPyV) were 0.27 μM and 0.59 μM, respectively. At 0.63 μM, BCV reduced viral late gene expression by 90% and halted progeny release. Preinfection treatment for only 24 h reduced icBKPyV similarly to treatment from 2 to 72 h postinfection, while combined pre- and postinfection treatment suppressed icBKPyV completely. After investigating BCV's effects on HUC viability, mean selectivity indices at 50 and 90% inhibition (SI50 and SI90) calculated for cellular DNA replication were 2.7 and 2.9, respectively, those for mitochondrial activity were 8.9 and 10.4, those for total ATP were 8.6 and 8.2, and those for membrane integrity were 25.9 and 16.7. The antiviral and cytostatic effects, but less so the cytotoxic effects, were inversely related to cell density. The cytotoxic effects at concentrations of ≥10 μM were rapid and likely related to BCV's lipid moiety. After carefully defining the antiviral, cytostatic, and cytotoxic properties of BCV in HUCs, we conclude that a preemptive or prophylactic approach in PyVHC is likely to give the best results.
INTRODUCTION
The ubiquitous and usually quiescent BK polyomavirus (BKPyV) causes polyomavirus-associated nephropathy (PyVAN) and hemorrhagic cystitis (PyVHC) following kidney and hematopoietic stem cell transplantation (HSCT), respectively (1).
PyVHC afflicts 5 to 15% of HSCT patients after engraftment (2–5). The pathogenesis of PyVHC is incompletely characterized but is believed to involve initial iatrogenic damage to the bladder mucosa during pretransplantation conditioning, which is subsequently aggravated by lytic BKPyV replication in urothelial cells in the absence of immune surveillance. This leads to exposure of both viral and host epitopes to the foreign, engrafting lymphoid cells and may contribute to graft-versus-host disease (GVHD) (6–10). Inflammation and the resultant denudation of the urothelium cause hemorrhagic cystitis ranging from microhematuria to macrohematuria, clot-related urinary retention, and postrenal failure (reviewed in reference 5).
PyVAN occurs in transplanted kidneys in 1 to 10% of recipients and often results in graft loss via a combination of lytic and inflammatory destruction of the renal parenchyma (2, 5). The initial reactivation signal is unknown, but mathematical modeling supports initial reactivation in the tubular epithelial compartment, followed by viral efflux with amplification in the urothelial compartment and subsequent recruitment of new tubular foci via a viral efflux-reflux cycle (11; reviewed in reference 5). Controlling viral replication in the urothelial cellular compartment is therefore pivotal for recovery in both PyVHC and PyVAN.
Due to the lack of specific antiviral treatment, management of PyVAN centers on reduction of the immunosuppressive regimen to facilitate reestablishment of virus-specific T-cell control. Reducing immunosuppression after allogeneic HSCT is a difficult procedure due to the threat of exacerbating GVHD. Thus, management of PyVHC is mostly supportive, and the disease wanes as immunological control of viral replication is regained and the urothelium regenerates.
Brincidofovir (BCV) was developed under a mandate from the U.S. government in response to the perceived threat of variola virus release (12). It is an ether-lipid ester conjugated prodrug of cidofovir (CDV). The lipid moiety of BCV is important for its pharmacokinetic properties, facilitating rapid uptake by cells and allowing oral administration. The antiviral properties of BCV are, however, attributable to CDV, which is an acyclic nucleoside phosphonate analogue of dCMP and acts as an inhibitor of DNA synthesis. Cidofovir is licensed by the U.S. FDA for the treatment of cytomegalovirus (CMV) retinitis in patients with AIDS (Drug Information Database [http://www.drugs.com/pro/vistide.html]). Cidofovir's characterized mechanistic interface in CMV infection is the viral DNA polymerase, which incorporates CDV into the nascent DNA strand, slowing or halting DNA synthesis in subsequent steps (13). Compared to the few human DNA polymerases tested, the herpesvirus polymerases have shown greater affinity for CDV, providing a molecular basis for selectivity (14–16). Since polyomaviruses utilize host cell DNA polymerases for their genome replication, there is no known selectivity for CDV-mediated inhibition. However, reduced viral replication has been seen with BCV and CDV in in vitro systems modeling polyomavirus diseases (17–19), and symptomatic improvement has been documented in a number of case studies involving topical CDV treatment of trichodysplasia spinulosa (20–22) and intravesicular CDV treatment of PyVHC (reviewed in reference 23). For the latter indication, CDV is currently entering phase I clinical trials (ClinicalTrials.gov [https://clinicaltrials.gov/ct2/show/NCT01816646]). Brincidofovir has also recently been used for oral prophylaxis and treatment of BKPyV disease (24–26).
When administered intravenously, CDV is nephrotoxic, necessitating simultaneous probenecid administration. This side effect is apparently abrogated by lipid conjugation in BCV (12). In addition, comparative in vitro studies show a 24- to 400-fold reduction in the concentrations required to give 50 and 90% inhibition of viral replication (EC50 and EC90) (19, 27). This has been attributed to the rapid cellular uptake conferred by the lipid moiety (27).
Given the important role of urothelial cells in the pathogenesis and course of both PyVAN and PyVHC, we investigated the effects of BCV in BKPyV-infected and uninfected primary human urothelial cells (HUCs). The results may provide a rationale for planning clinical trials for BCV treatment of PyVHC and PyVAN.
MATERIALS AND METHODS
Cell culture, BKPyV infection, and BCV treatment.
Primary human urothelial cells (HUCs) were acquired from ScienCell Research Laboratories and maintained in serum-free urothelial cell medium (UCM; ScienCell) as described by the manufacturer. All experiments were performed with HUCs at passage 3. Gradient-purified BKPyV Dunlop from Vero cells was used for all infections. The number of fluorescence-forming units (FFU) per milliliter was calculated as previously described (28) before HUCs were infected at a multiplicity of infection (MOI) of 2 to 5 FFU/cell. After 2 h, cells were washed twice with UCM and replenished with fresh UCM containing 0 to 31.6 μM BCV. Brincidofovir (Chimerix, Inc.) was prepared as a 1.78 to 3.56 mM stock solution in buffer (methanol-water-ammonium hydroxide with a volume-to-volume ratio of 50:50:2) and stored at 4°C (29). Prior to each experiment, dilution series were carried out in buffer before introduction into UCM, so that all wells received the same amount of buffer.
Immunofluorescence staining, microscopy, and image processing.
Infected-cell monolayers were fixed at 72 hpi with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min and permeabilized with 100% methanol for 10 min. The cells were then blocked with 3% goat serum in PBS for 30 min, followed by incubation with primary (37°C) and secondary antibodies (room temperature) for 30 min each. The primary antibodies used were mouse monoclonal anti-simian virus 40 (SV40) large T antigen (LTag) (Pab416; 1:100; Abcam) and rabbit polyclonal antiserum directed against agnoprotein (1:1,000) (30, 31). The secondary antibodies were anti-mouse IgG conjugated with Alexa Fluor 568 and anti-rabbit IgG conjugated with Alexa Fluor 488 (1:500; Molecular Probes). Nuclear DNA was labeled with DRAQ5 (Biostatus). A Nikon TE2000 microscope and NIS Elements basic research software, version 4 (Nikon Corporation), were used for image acquisition and processing.
DNA extraction.
Cell culture supernatants were harvested at 72 hpi (24 hpi for measurement of input BKPyV DNA load) and frozen at −70°C. Shortly before qPCR, supernatants were thawed, diluted 1:100 in Accugene molecular-grade water (Lonza), and boiled for 5 min. For the determination of intracellular BKPyV loads, cells were washed and lysed in G2 buffer from the MagAttract DNA M48 minikit (Qiagen) and frozen at −70°C until robotic extraction with either the GenoM-48 (Qiagen) or the NucliSENS easyMAG (bioMérieux) system.
Quantification of extracellular and intracellular BKPyV.
The BKPyV DNA load was determined in both supernatants and cell extracts by quantitative PCR (qPCR) using primers and a probe targeting the BKPyV LTag gene (32). To express intracellular BKPyV DNA as genome equivalents (Geq)/cell, each sample was analyzed simultaneously by qPCR (multiplexed) for the aspartoacylase (ACY) gene (33). For a description of this multiplex PCR, see reference (17). Each experiment encompassed either two or three parallels, and each parallel was measured by qPCR in triplicate.
Western blotting.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitor (Complete Mini EDTA-free; Roche) at 24, 48, and 72 hpi and stored at −70°C until measurement of total protein using the EZQ protein quantitation kit (Invitrogen) and Tecan Infinite F200PRO reader (TECAN) before separation of 5.2 μg total protein per well with a NuPAGE 4 to 12% bis-Tris gel (Invitrogen) and blotting onto a polyvinyl difluoride membrane (LI-COR Biosciences). The membrane was blocked with Odyssey blocking buffer (LI-COR Biosciences) and incubated with the following primary antibodies: rabbit polyclonal antiserum directed against the N-terminal region common to LTag and small T antigen (sTag) (1:2,000) (30), VP1 (1:10,000) (34), or agnoprotein (1:10,000) (30, 31) and mouse monoclonal antibody directed against LTag (PAb416; 1:500 Abcam) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Ab8245; 1:5,000; Abcam). The secondary antibodies were anti-rabbit and anti-mouse IgG infrared dye-labeled antibodies (IRDye 680LT goat anti-mouse [1:10,000] and IRDye 800CW goat anti-rabbit [1:7,500]; LI-COR). The LI-COR Odyssey system and the corresponding software program were used for detection and quantification.
Viability assays.
The electrical impedance of the cell monolayer was measured using E plates and the xCELLigence system (ACEA Biosciences). The background impedance of the wells was measured after addition of 200 μl UCM. Subsequently, 100 μl UCM was removed, and 100 μl cell suspension was seeded. For BKPyV infection and BCV treatment, 24 h after seeding, 100 μl of the medium was removed and replenished with fresh UCM containing 0 to 63.2 μM BCV (final concentration, 0 to 31.6 μM), with or without purified BKPyV Dunlop. The cells were grown for 96 to 120 h, and impedance was measured every 15 min for the first hour and then every 30 min. Impedance was expressed as an arbitrary number called the cell index. As an alternative method for continuously monitoring cell viability, the Nikon Biostation IM was used as previously described (28), with the exception that a cyanine dye from the CellTox Green cytotoxicity assay (Promega) was added at a final concentration of 1:1,000 to allow visualization of loss of membrane integrity.
For endpoint and semikinetic assays, 10,000 to 40,000 cells/cm2 were seeded and, 24 h later, treated with BCV at 0 to 15 μM. A 100% lysis control was included using the lysis buffer from the CellTox Green cytotoxicity assay. Cellular DNA synthesis between 24 and 72 h posttreatment (hpt) was quantified by colorimetric measurement of bromodeoxyuridine (BrdU) incorporation into DNA using a cell proliferation BrdU enzyme-linked immunosorbent assay (ELISA) (Roche) according to the manufacturer's instructions. Mitochondrial activity was measured at 72 hpt in the same wells, after incubation for 3 h with resazurin, by the colorimetric measurement of resazurin reduction using the TOX8 resazurin-based in vitro toxicology assay (Sigma) according to the manufacturer's instructions. As an alternative measure of metabolic activity, the total ATP content of the cells was measured at 72 hpt using the CellTiter-Glo luminescent cell viability assay (Promega) as described earlier (35). To assess the cytotoxic properties of BCV, the semikinetic CellTox Green cytotoxicity assay was used, and fluorescence was measured at 0.5, 24, 48, and 72 hpt using the Tecan Infinite F200PRO reader (Tecan) as previously described (35).
Time-lapse microscopy.
For continuous microscopic monitoring of BCV-treated and untreated HUCs, a Nikon Biostation was used. Cells were seeded at 15,000 cells/cm2 in Hi-Q4 culture dishes (Ibidi) in 500 μl UCM including a 1:500 dilution of the green cyanine dye from the CellTox Green cytotoxicity assay. Approximately 24 h after seeding, 300 μl of fresh UCM with BCV was added, giving a final BCV concentration of 0 to 10 μM. Pictures were taken at 9 points in each of 4 wells every 10 min from 4.5 h before treatment until 72 hpt. The ImageJ software with the stitcher plugin was used to make the movies (36).
Calculation of the effective concentrations and the selective indices.
The Microsoft Excel-based plug-in XLfit was used for curve fitting to determine the 50% and 90% effective concentrations (EC50 and EC90) and the 50% and 90% cytostatic or cytotoxic concentration (CC50 and CC90) and their respective 95% confidence intervals. These results were used to calculate the selective indices (SI50 and SI90).
RESULTS
Effect of BCV treatment on BKPyV replication in HUCs.
We previously investigated the effect of BCV on BKPyV replication in renal proximal tubule epithelial cells (RPTECs) by measuring the extracellular BKPyV DNA load (19). However, considering that BKPyV is less efficiently released from HUCs than from RPTECs (28), we decided to investigate the effect of BCV on both intracellular and extracellular BKPyV DNA at 72 hpi. Cells were seeded (20,000 cells/cm2) and infected 24 h later at semiconfluence, and growth medium containing BCV at concentrations from 0.156 to 5 μM was added at 2 hpi. Since quantification of extracellular BKPyV DNA revealed a contribution of less than 1% of the total BKPyV DNA (data not shown), confirming our earlier published results (28), we decided to focus solely on the intracellular DNA. A dose-dependent response with a steep decline in the number of intracellular BKPyV genome equivalents (Geq) per cell (icBKPyV) was found when BCV concentrations between 0.31 μM and 1.25 μM were used (Fig. 1A). Approximately 90% inhibition was achieved at a mean concentration of 0.63 μM, while concentrations ≥1.25 μM reduced the icBKPyV to the input level.
FIG 1.
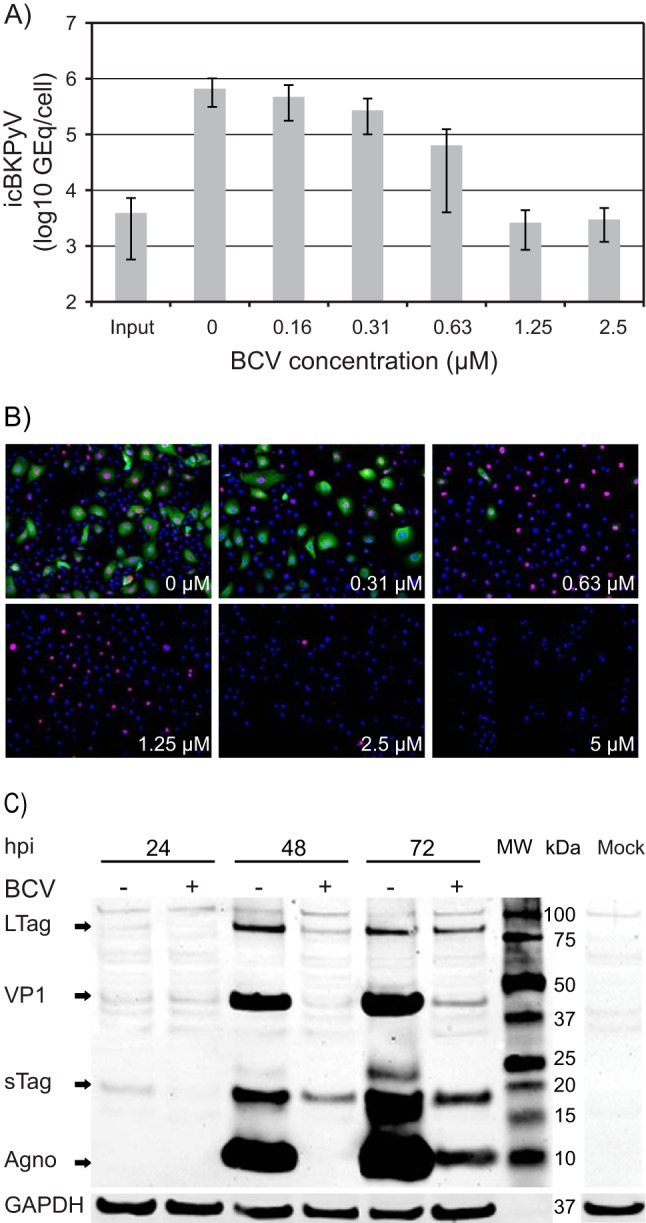
Effect of brincidofovir (BCV) on BKPyV replication in HUCs. (A) Effect of BCV on BKPyV DNA replication. Cells were seeded at 20,000 cells/cm2 and infected 24 h later with purified BKPyV at 2 FFU/cell. Infected cells, treated with the indicated concentrations of BCV, were harvested 72 hpi (70 hpt), and icBKPyV (number of genome equivalents [Geq]/cell) was measured by qPCR. The mean icBKPyV values from three independent experiments conducted and analyzed in triplicate (27 quantification reactions per mean) are presented. Error bars indicate the standard deviations of means between experiments. (B) Effect of BCV on BKPyV expression of early and late proteins at 72 hpi. Immunofluorescence micrographs of BKPyV-infected HUCs treated with the indicated BCV concentrations are shown. The cells were fixed and subsequently stained using polyclonal rabbit antiagnoprotein serum (green) for visualization of agnoprotein and the SV40 LTag monoclonal Pab416 for the visualization of early LTag (red). Cell nuclei (blue) were stained with DRAQ5. Pictures were taken with a fluorescence microscope (10× objective). (C) Effect of BCV on temporal expression of BKPyV early and late proteins. Western blot of lysates of BKPyV infected HUCs treated with 0.63 μM BCV (+) and buffer only (−). Treatment was initiated 2 h after infection, and cells were lysed at the indicated times postinfection. Electrophoresis was performed with 5.2 μg total protein per well. The membrane was stained with polyclonal rabbit anti-LTag serum, anti-agnoprotein, and anti-VP1 serum and with a mouse monoclonal antibody directed against the housekeeping protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Lane MW, molecular weight markers.
Next, we investigated the effect of the different BCV concentrations on BKPyV gene expression in HUC monolayers at 72 hpi. In agreement with the observed effect on icBKPyV, immunofluorescence staining of BCV treated BKPyV-infected cells demonstrated a clear and substantial decline in the number of cells expressing the late agnoprotein when BCV concentrations of ≥0.63 μM were used. Of note, expression of LTag was less affected at this concentration but also declined steeply with concentrations above 1.25 μM (Fig. 1B). From 2.5 μM, the total cell number was also clearly reduced. For subsequent fixed-concentration investigations, we decided to use BCV at a concentration of 0.63 μM.
To further evaluate the temporal effect of BCV on viral gene expression, we performed Western blotting with lysates harvested at 24, 48, and 72 hpi from untreated cells and cells treated with 0.63 μM BCV. In untreated cells, expression of sTag and LTag was weakly detectable from 24 hpi but increased from 48 hpi. In BCV-treated cells, early expression was delayed and reduced, first becoming evident at 48 hpi and at no point reaching the levels seen in untreated cells. This delayed early expression suggests that BCV may also inhibit some step in the BKPyV replication cycle prior to DNA replication. In agreement with our immunofluorescence microscopy findings, synthesis of the late proteins agnoprotein and VP1 was inhibited to a greater degree than that of the early proteins. More than 99% reduction in VP1 expression was found at 48 hpi, and approximately 90% reduction was seen at 72 hpi.
We concluded that BCV reduced BKPyV DNA replication in a concentration-dependent manner and that the reduction of late gene expression was much greater than that of early gene expression, similar to results in RPTECs (19). Differing slightly from these earlier findings, however, inhibition of viral early gene expression was evident from as early as 24 hpi.
The BCV EC50 and EC90 and the influence of cell density.
In order to determine the EC50 and EC90, curve fitting based on the data in Fig. 1A and some additional experiments were performed. An EC50 of 0.28 μM (95% CI, ±0.02) and an EC90 of 0.59 μM (95% CI, ±0.03) were calculated (Fig. 2A [20,000 cells/cm2] and Table 1). Compared to previous titration experiments in RPTECs, there appeared to be more interexperiment variation between independent repeats in HUCs, while intraexperiment variation between parallels of each experiment was low (see Fig. S1 in the supplemental material). Since routine microscopic observation had revealed differences in seeding efficiency, the effects of varying the cell density on the EC50 and EC90 were investigated. The BCV titrations were repeated at half and double the original cell density. While 10,000 cells/cm2 proliferated throughout the experiment, 40,000 cells/cm2 were confluent from 1 day postseeding (data not shown). The results encompassed the spectrum of values obtained earlier, giving an EC90 range of 0.46 to 0.80 μM (95% CI, 0.33 to 0.86 μM) (Fig. 2A and Table 1), confirming the suspicion that variable seeding efficiency led to variation between titration experiments. All quantification results were then pooled for recalculation of the EC50 and EC90, which were essentially unchanged at 0.27 μM (95% CI, ±0.03) and 0.59 μM (95% CI, ±0.06), respectively (Fig. 2B and Table 1). We concluded that the antiviral effect of BCV is dependent upon the number and/or proliferative state of cells.
FIG 2.
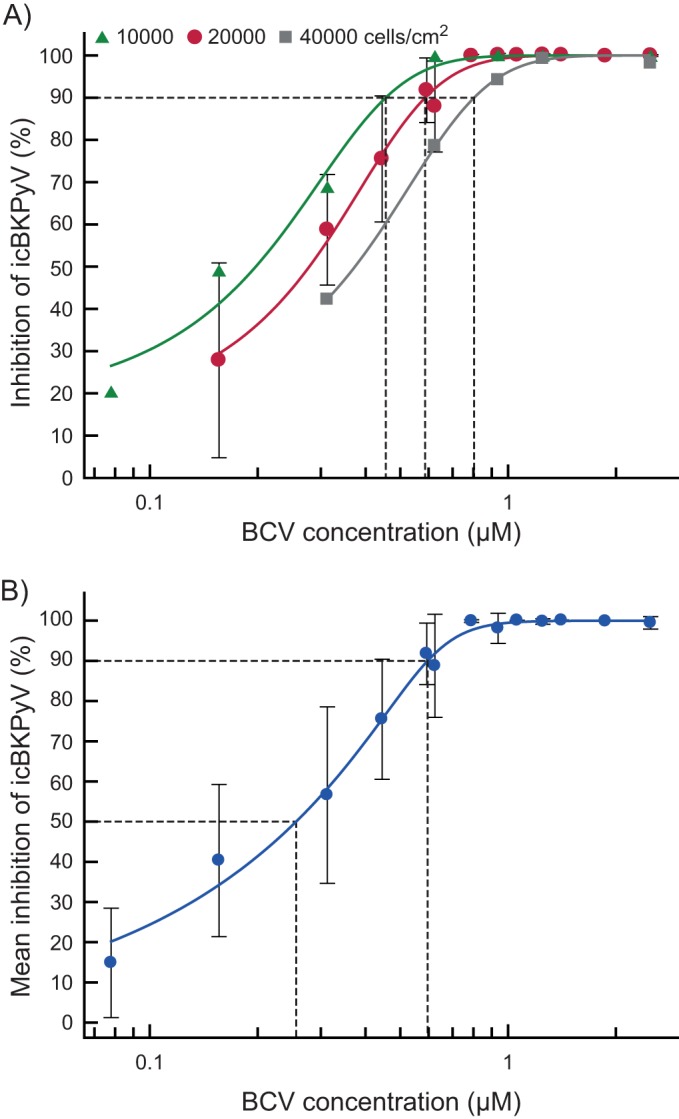
Calculation of the 50% and 90% effective concentrations (EC50 and EC90) of BCV. (A) Effect of cell density on the EC90. Cells were seeded at 10,000, 20,000, and 40,000 cells/cm2 and infected 24 h later with purified BKPyV at 2 FFU/cell. Infected cells, treated with various concentrations of BCV 2 hpi, were harvested 72 hpi (70 hpt), and the icBKPyV was measured by qPCR. Each dilution was tested between 2 and 12 times in independent experiments with 2 or 3 replicates, each of which was amplified in triplicate, giving between 12 and 63 individual quantification results per point in the plot. The mean percent inhibition of the icBKPyV is plotted against the BCV concentration. The value for medium containing only buffer was set as 0% inhibition, and that for the input viral load was set as 100% inhibition. Dotted vertical lines indicate the EC90 values resulting from the different cell densities. The error bars shown represent standard deviations of the mean percent inhibition between experiments carried out with 20,000 cell/cm2 are shown. Error bars for other cell densities were excluded for clarity. (B) Combined titration results at all three cell densities presented in panel A. As described above, the mean percent inhibition of the icBKPyV is plotted against the BCV concentration. The value for medium containing only buffer was set as 0% inhibition, and that for the input viral load was set as 100% inhibition. Error bars indicate the standard deviations of means between experiments. The EC50 and EC90 are indicated by dotted lines.
TABLE 1.
Calculated EC50, EC90, CC50, CC90, SI50, and SI90 values
| Assay | Cell density (cells/cm2) | EC50 (μM) (95% CI)b | EC90 (μM) (95% CI)b | ||
|---|---|---|---|---|---|
| icBKPyVa | 10,000 | 0.20 (0.16–0.24) | 0.46 (0.33–0.58) | ||
| 20,000 | 0.28 (0.26–0.30) | 0.59 (0.55–0.62) | |||
| 40,000 | 0.37 (0.33–0.41) | 0.80 (0.75–0.86) | |||
| Pooled | 0.27 (0.24–0.29) | 0.59 (0.53–0.65) | |||
| CC50 (μM) (95% CI)b | CC90 (μM) (95% CI)b | SI50 (CI)c | SI90 (CI)c | ||
| BrdU | 20,000 | 0.2 (0.1–0.3) | 1.3 (1.1–1.5) | 0.8 (0.5–1.2) | 2.2 (1.8–2.7) |
| 40,000 | 1.7 (1.3–2.0) | 2.8 (2.1–3.5) | 4.5 (3.2–6.1) | 3.5 (2.4–4.7) | |
| Resazurin | 20,000 | 1.1 (0.6–1.6) | 5.4 (3.5–7.4) | 4.0 (1.9–6.2) | 9.3 (5.7–13.3) |
| 40,000 | 5.1 (4.6–5.6) | 9.3 (8.2–10.5) | 13.8 (11.2–17.1) | 11.6 (9.5–14.0) | |
| Total ATP | 10,000 | 1.3 (1.1–1.6) | 3.4 (2.6–4.1) | 6.7 (4.5–10.1) | 7.4 (4.5–12.4) |
| 20,000 | 2.4 (1.8–3.1) | 5.2 (3.5–6.9) | 8.8 (6.1–11.8) | 8.8 (5.6–12.4) | |
| 40,000 | 3.9 (3.4–4.3) | 6.8 (5.8–7.7) | 10.4 (8.3–13.0) | 8.4 (6.8–10.3) | |
| Membrane integrity | 10,000 | 5.6 (5.4–5.8) | 8.1 (7.4–8.9) | 28.4 (22.5–37.2) | 17.9 (12.9–26.5) |
| 20,000 | 7.9 (7.6–8.3) | 11.1 (10.6–11.6) | 28.5 (25.7–31.6) | 18.9 (17.0–20.9) | |
| 40,000 | 7.8 (7.3–8.2) | 10.9 (10.2–11.5) | 21.0 (17.7–25.0) | 13.5 (11.8–15.4) |
icBKPyV refers to the number of intracellular BKPyV genome equivalents per cell.
95% confidence intervals were calculated using the curve-fitting software XLfit with a 4-parameter logistic model (model 208).
Confidence intervals for SI90 and SI50 values were calculated as follows: lower limit = (CC90 − 95% CI)/(EC90 + 95% CI); upper limit = (CC90 + 95% CI)/(EC90 − 95%CI).
Infectious-progeny release from BCV-treated HUCs.
To determine the effect of BCV treatment on infectious-progeny release, fresh cultures of HUCs (20,000 cells/cm2) were inoculated with supernatants from titration experiments carried out at two different seeding densities (10,000 and 40,000 cells/cm2), and immunofluorescence staining for BKPyV LTag and agnoprotein was performed at 72 hpi. While a high percentage of cells inoculated with supernatants from untreated cells were infected, only a few cells in wells inoculated with supernatants from BCV-treated cells were infected (Fig. 3). Moreover, the reduction of infectious progeny by BCV was apparently greater in semiconfluent than confluent cells. These results indicate that the effect of BCV on viral replication and gene expression is also reflected in progeny release. In fact, the reduction of infectious progeny release appeared to occur at lower BCV concentrations than seen for both icBKPyV and in situ BKPyV gene expression during the first replication cycle (Fig. 1A and B). The results also support the observation that increasing cell density reduces the antiviral effect of BCV. We concluded that both the observed BCV mediated inhibition of viral DNA replication and its dependency on cell density are reflected in progeny release.
FIG 3.
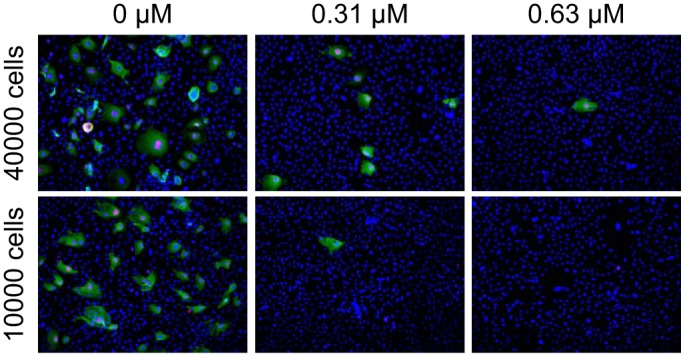
Effect of BCV on release of infectious progeny. Supernatants harvested at 72 hpi from HUCs seeded at 40,000 and 10,000 cells/cm2 and infected with 2 FFU/cell were diluted 1:10 and inoculated into fresh cultures of HUCs, seeded at 20,000 cells/cm2. The cells were fixed at 72 hpi and stained using polyclonal rabbit anti-agnoprotein serum (green) for visualization of agnoprotein and the SV40 LTag monoclonal Pab416 for LTag (red). Cell nuclei (blue) were stained with DRAQ5. Pictures were taken with a fluorescence microscope (10× objective).
BCV preinfection treatment of HUCs.
In order to examine whether prophylactic treatment with BCV could prevent or reduce BKPyV replication in HUCs, icBKPyV and viral protein production in 3 treatment groups were compared to that in untreated cells. Twenty thousand cells per well were seeded and treated with 0.63 μM BCV for 24 h prior to infection, from 2 to 72 h postinfection, or both. The greatest reduction in icBKPyV was observed with combined pre- and postinfection treatment, which was much more effective than pre- or postinfection treatment alone and gave an almost complete reduction of 99% (Fig. 4A). Twenty-four hours of preinfection treatment resulted in an 82% reduction, while 70 h of postinfection treatment yielded only a 74% reduction. The surprisingly weak effect of 70 h of postinfection treatment with 0.63 μM, considering our earlier determined EC90 of 0.59 μM, could be ascribed to an increase in cell density during the extra 24 h of incubation prior to infection and treatment. Immunofluorescence staining for viral antigens supported the qPCR results, showing a clear reduction of cells producing LTag and a greater reduction of cells producing agnoprotein in wells treated both pre- and postinfection (Fig. 4B). The reduction in the number of cells producing BKPyV proteins was also slightly greater in wells that had been pretreated than in those that had been treated postinfection. Of note, DRAQ5 staining revealed a progressive decrease in total cell numbers, from untreated to postinfection treated to preinfection treated and finally to cells treated both pre- and postinfection. We concluded that preinfection treatment was slightly more inhibitory for BKPyV replication than postinfection treatment but that combined pre- and postinfection treatment had the strongest effect on both BKPyV protein expression and viral replication. The latter regimen also had the strongest negative effect on cell proliferation and/or viability.
FIG 4.
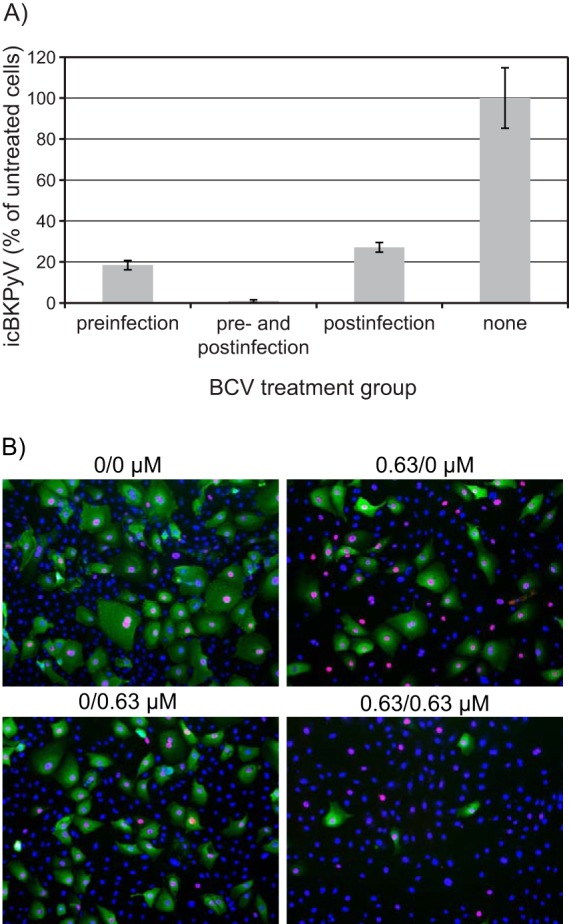
Effect of pre- and postinfection treatment with BCV. HUCs were seeded at 20,000 cells/cm2, incubated for 24 h, and then treated with 0.63 μM BCV or buffer only for 24 h prior to infection with purified BKPyV at 2 FFU/cell for 2 h. Following infection, the cells were washed with medium and then replenished with medium containing 0.63 μM BCV or buffer only. Cells were harvested for extraction of DNA (A) or fixed at 72 hpi (B). (A) The icBKPyV is plotted as a percentage of that in wells treated with buffer only (100%). Bars represent means from 3 individual experiments conducted and quantified in triplicate, giving 27 quantification reactions per mean. Error bars represent standard deviations. (B) Immunofluorescence micrographs of HUCs fixed at 72 hpi and stained using polyclonal rabbit anti-agnoprotein serum (green) for visualization of agnoprotein and the SV40 LTag monoclonal Pab416 for the visualization of early LTag (red). Cell nuclei (blue) were stained with DRAQ5. Pictures were taken with a fluorescence microscope (10× objective).
Cytostatic and cytotoxic effects of BCV.
Given the lack of a known virus-selective target for CDV in the polyomavirus proteome, the inherent genotoxic properties of CDV (37, 38), and our observations of decreased cell numbers, it was thought pertinent to characterize the cytostatic and cytotoxic effects in some detail.
Temporal effects.
In order to first gain insight into the temporal effects of BCV on cellular metabolism in HUCs, we used the xCELLigence system for continuous monitoring of electrical impedance in cell culture monolayers. Shortly after addition of 0.63 μM BCV to BKPyV-infected and mock-infected cells, a reduction in the slope of the cell index curve in treated wells relative to untreated wells was observed (Fig. 5A). This suggests that BCV causes reductions in cellular proliferation, cell size, and/or cell attachment of both BKPyV-infected and uninfected cells. In BKPyV-infected untreated cells, the cell index peaked at 48 hpi, thereafter declining rapidly to baseline, suggesting a sudden onset of cell detachment or cell death. Brincidofovir seemed to impede this process, radically reducing the maximum cell index and apparently postponing or preventing the death of infected cells. Increasing concentrations of BCV caused successive, lasting decreases in the cell index slope in mock-infected cells (Fig. 5B), indicating dose-dependent cytostatic effects. At the highest concentrations, 10 and 32 μM, the cell index fell to subseeding levels within 20 hpt. We concluded that BCV reduces cellular proliferation, size, and/or attachment in a rapid and dose-dependent manner in both infected and uninfected cells and that the effect lasted for at least 120 h. Based on these results, endpoint viability analyses were set to 72 hpi.
FIG 5.

Cytostatic and cytotoxic effects of BCV. (A and B) Concentration-dependent temporal effects of BCV on the cell index (a measure of electrical impedance) in infected and mock-infected HUCs. HUCs were seeded and incubated for 24 h prior to either infection with purified BKPyV at 5 FFU/cell or mock infection and simultaneous treatment with BCV or buffer only. Electrical impedance in cell culture monolayers was measured using the xCELLigence system from seeding, 24 h before infection, until 96 to 120 hpi/hpt. Cell index was normalized at the point of infection/treatment. Experiments were repeated 7 times in triplicate, and results of a representative experiment are presented. (A) Comparison of cell index for infected and mock-infected, BCV- and buffer-only-treated HUCs seeded at 60,000 cells/cm2. (B) Temporal effects of increasing concentrations of BCV on the cell index of mock-infected HUCs seeded at 30,000 cells/cm2. (C, D, E, and F) Concentration-dependent effects of BCV on viability of HUCs in endpoint assays. Inhibition of cellular DNA replication (BrdU incorporation assay) (C) and inhibition of mitochondrial activity (resazurin reduction assay) (D) both at 20,000 (red) and 40,000 (gray) cells/cm2, as well as inhibition of total ATP levels (CellTiter-Glo luminescent cell viability assay) (E) and loss of membrane integrity (CellTox Green cytotoxicity assay) (F) both at 10,000 (green), 20,000 (red), and 40,000 (gray) cells/cm2, are shown. The curve for inhibition of icBKPyV (blue) is included for reference. Dotted lines indicate EC90 and CC90. All viability measurements were performed at 72 hpt in uninfected HUCs. Mean values from 3 experiments carried out with 6 parallels are presented. Error bars represent standard deviations between experiments. (G) Daily measurement of the loss of membrane integrity in uninfected HUCs with increasing concentrations of BCV. Cells were seeded at 20,000 cells/cm2 and incubated for 24 h before addition of CellTox Green cyanine dye and treatment with the indicated concentrations of BCV, buffer only, or lysis buffer. Fluorescence, indicating nuclear penetration and binding to the DNA by the cyanine dye, was recorded at 0.5, 24, 48, and 72 hpt. Mean values from 3 experiments carried out with 6 parallels are presented. Error bars represent standard deviations between experiments.
Cellular DNA replication.
The active metabolite of BCV and CDV, cidofovir diphosphate (CDVpp), competes with dCTP for DNA polymerase-mediated incorporation into DNA (13, 15, 16). BKPyV DNA replication is dependent on cellular DNA polymerases, giving no clear mechanism for selective interaction with CDVpp. Therefore, the effect of BCV on cellular DNA replication in HUCs was investigated by measurements of BrdU incorporation occurring between 24 and 72 hpt. Cellular DNA replication was affected in a concentration-dependent manner with a 90% inhibitive concentration (CC90) of 1.3 μM (95% CI, ±0.2) and 2.8 μM (95% CI, ±0.7) for 20,000 and 40,000 cells/cm2, respectively, and a 50% inhibitive concentration (CC50) of 0.2 (95% CI, ±0.1) and 1.7 (95% CI, ±0.3) for 20,000 and 40,000 cells/cm2, respectively (Fig. 5C and 6A and Table 1). Comparing the cellular DNA replication with the viral DNA replication, the fraction CC90/EC90 gave selectivity indices at 90% inhibition (SI90) of 2.2 (CI, 1.8 to 2.7) and 3.5 (CI, 2.4 to 4.7) for 20,000 and 40,000 cells/cm2, respectively (Fig. 6B and Table 1). The fraction CC50/EC50 gave selectivity indices at 50% inhibition (SI50) of 0.8 (CI, 0.5 to 1.2) and 4.5 (CI, 3.2 to 6.1) for 20,000 and 40,000 cells/cm2, respectively (Fig. 6B and Table 1). This trend of increasing selective index with increasing cell density was significant for the SI50, indicating that increased cell density protects cells and, to a lesser degree, virus from inhibition by BCV. We concluded that BCV provides marginal selectivity for inhibition of viral DNA replication and that selectivity tends to increase with increasing cell density.
FIG 6.

Graphic representation of the selectivity profile of BCV for inhibition of BKPyV in HUCs. (A) EC90, EC50, CC90, and CC50 of BCV. (B) SI50 and SI90. The icBKPyV and endpoint viability were measured at 72 hpi/hpt. EC90 and CC90 values and their 95% confidence intervals were calculated using the curve-fitting software XLfit with a 4-parameter logistic model (model 208). Selectivity indices were calculated by dividing CC by EC. Confidence intervals for selectivity indices were calculated as follows: lower limit = (CCx – 95% CI)/(ECx + 95% CI); upper limit = (CCx + 95% CI)/(ECx – 95% CI).
Overall cell metabolism and viability.
Next we investigated the effect of BCV on metabolism in HUCs by measurement of resazurin hydrolysis and total ATP at 72 hpt. The resazurin reduction assay demonstrated a dose-dependent reduction of the metabolic activity with a CC90 of 5.4 μM (95% CI, ±1.9) and 9.3 μM (95% CI, ±1.1) for 20,000 and 40,000 cells/cm2, respectively, and a CC50 of 1.1 (95% CI, ±0.5) and 5.1 (95% CI, ±0.5) for 20,000 and 40,000 cells/cm2, respectively (Fig. 5D and 6A and Table 1). The corresponding SI90 values were 9.3 (CI, 5.7 to 13.3) and 11.6 (CI, 9.5 to 14.0) and the corresponding SI50 values were 4.0 (CI, 1.9 to 6.2) and 13.8 (CI, 11.2 to 17.1) (Fig. 6B and Table 1). Also the total ATP was reduced in a concentration-dependent manner with a CC90 of 3.4 μM (95% CI, ±0.8), 5.2 μM (95% CI, ±1.7) and 6.8 μM (95% CI, ±0.9) for 10,000, 20,000, and 40,000 cells/cm2, respectively, and a CC50 of 1.3 (95% CI, ±0.3), 2.4 (95% CI, ±0.6), and 3.9 (95% CI, ±0.4) for 10,000, 20,000, and 40,000 cells/cm2, respectively (Fig. 5E and 6A and Table 1). The corresponding SI90 values were 7.4 (range, 4.5 to 12.4), 8.8 (range, 5.6 to 12.5) and 8.4 (range, 6.8 to 10.3) and the corresponding SI50 values were 6.7 (range, 4.5 to 10.1), 8.8 (range, 6.1 to 11.8), and 10.4 (range, 8.3 to 13.0) (Fig. 6B and Table 1). We concluded that the BCV-induced reduction in metabolic activity occurs at significantly higher concentrations than reduction of DNA replication.
Cytotoxicity.
In order to investigate potential cytotoxic effects of BCV, the semikinetic CellTox Green cytotoxicity assay for measuring loss of membrane integrity was used. The CC90 values for membrane integrity were 8.1 (95% CI, ±0.7), 11.1 (95% CI, ±0.5), and 10.9 μM (95% CI, ±0.7) for 10,000, 20,000 and 40,000 cells/cm2, respectively (Fig. 5F and 6A and Table 1). The CC50 values were 5.6 (95% CI, ±0.2), 7.9 (95% CI, ±0.3), and 7.8 (95% CI, ±0.5) (Fig. 5F and 6A and Table 1). There was no difference in cytotoxicity between the two highest cell densities. The corresponding SI90 values were 17.9 (CI, 12.9 to 26.5), 18.9 (CI, 17.0 to 20.9), and 13.5 (CI, 11.8 to 15.4), and the corresponding SI50 values were 28.4 (CI, 22.5 to 37.2), 28.5 (CI, 25.7 to 31.6), and 21.0 (CI, 17.7 to 25.0) (Fig. 6B and Table 1). To explore the basis for the rapid changes in electrical impedance observed, membrane integrity of HUCs treated with BCV at concentrations from 0.16 to 15 μM was measured at 0.5, 24, 48, and 72 hpt. At 0.5 hpt, no cytotoxic effect was seen, but by 24 hpt, cytotoxicity was observed with BCV concentrations of ≥5.0 μM. With 15 μM BCV, cytotoxicity essentially equivalent to complete lysis was seen from 24 hpt (Fig. 5G), explaining the fall in cell index to subseeding levels during the first 24 hpt. The rapid cytotoxic effect at high BCV concentrations was further confirmed using time-lapse photography of cells after addition of the green cyanine dye. At 10 μM BCV, cell mobility and morphological changes occurred within 1 h, and increased cell death was apparent within 5 h (see Movie S1 in the supplemental material). In contrast, cells treated with 0.63 μM BCV demonstrated viability (see Movie S2) similar to untreated cells (see Movie S3). We concluded that BCV has a rapid cytotoxic effect in HUCs at high concentrations, which is relatively independent of cell density.
In summary, the cytostatic effects of BCV were, like the inhibition of viral replication, inversely proportional to cell density. This was most pronounced for DNA replication and mitochondrial activity. In contrast, cytotoxicity, expressed as loss of membrane integrity, was comparatively stable at the different cell densities.
DISCUSSION
Specific antiviral treatment for PyVHC and PyVAN is still an unmet clinical need in transplantation medicine (1). Here we report the effects of BCV on BKPyV-infected and uninfected HUCs, the cells which are targeted by BKPyV in PyVHC and which facilitate the spread of BKPyV between nephrons in PyVAN (11).
We found that BCV addition 2 hpi inhibited BKPyV DNA replication by 90% at 0.46 to 0.80 μM, depending on the cell density. At 0.63 μM, a 90 to 99% reduction in viral late gene expression and an almost complete arrest of progeny release was observed. Treatment with BCV for 24 h prior to infection inhibited BKPyV DNA replication slightly more than postinfection treatment for 70 h, but a combination of pre- and postinfection treatment was the most effective, though also relatively cytostatic. With 20,000 to 40,000 cells/cm2, BCV gave 90% inhibition of cellular DNA replication at 1.3 to 2.8 μM (95% CI, 1.1 to 3.5), mitochondrial activity and total ATP at 5.2 to 9.3 μM (95% CI, 3.5 to 10.5), and membrane integrity at 10.9 to 11.1 μM (95% CI, 10.2 to 11.6). Since both antiviral and cytostatic effects were dependent on cell density, a spectrum of selectivity indices ranging from 0.8 to 28.5 was generated by investigating three different cell densities with four different viability assays. Selectivity was modest when viral DNA replication was compared with cellular DNA replication and greater when viral DNA replication was compared with mitochondrial activity, total ATP content, and membrane integrity.
Considering that BCV is an analog of dCMP, inhibition of viral DNA replication was expected. Since newly synthesized BKPyV genomes serve as templates for late transcription and thereby amplify the production of late proteins during lytic infection (39), inhibition of late expression and progeny production was also expected. Of note, infectious-progeny production was inhibited at a lower BCV concentration than BKPyV DNA replication. A possible potentiating factor could be major structural derangements of the viral genome inhibiting its packaging, as shown for poxvirus in CDV-treated cells (40). Furthermore, progeny genomes produced in the presence of CDVpp may be poor templates for further replication, as template-incorporated CDV has been shown to inhibit the viral polymerase in both CMV and vaccinia virus (13, 41). Apparently, this has not been investigated for the human DNA polymerases utilized by polyomavirus. Interestingly, mutations in the funnel and docking domains of the viral RNA polymerase of CDV-resistant monkeypox virus have been observed (42), indicating that viral RNA polymerases but potentially also cellular RNA polymerases may be directly targeted by CDV. Such an additional mechanism of action could possibly cause the observed inhibition of early gene expression before 24 hpi and thereby amplify the effects on viral DNA replication, which inhibit late gene expression.
In agreement with previous observations of CDV in animal studies (43) and BCV in in vitro studies (19), we found that preinfection treatment for 24 h gave slightly greater inhibition of icBKPyV and viral gene expression than 70 h of postinfection treatment despite the 3-fold-shorter treatment time. This may translate to a considerable postantimicrobial effect in vivo. Additionally we found that combined pre- and postinfection treatment had the strongest effect. These results can be explained by the kinetics of cellular uptake and metabolism of BCV and the lag before BKPyV DNA replication starts (28, 44). First, since BCV is rapidly absorbed and stored in membranes (45), postinfection replenishment with BCV at the initial concentration is effectively a second dose, and therefore combined treatment has the strongest effect. Second, it takes time to build up intracellular concentrations of the active metabolite CDVpp (45). In preinfection treatment, the intracellular CDVpp concentration is probably high but diminishing during BKPyV DNA replication, which starts at around 24 to 36 hpi (28, 44), i.e., 48 to 60 hpt, as opposed to being low but increasing in postinfection treatment. Therefore, preinfection treatment has a slightly greater effect than postinfection treatment.
We observed low selectivity of inhibition of viral versus cellular DNA replication. This was not surprising, since BKPyV utilizes cellular DNA polymerases in genome replication. In fact, with a cell density of 20,000 cells/cm2 at 50% inhibition, no selective inhibition could be shown. Selectivity improved, however, with increasing cell density. One explanation for this is that uninfected cells exit the cell cycle when confluent, providing protection from BCV. Conversely, BKPyV LTag interacts with retinoblastoma (Rb) proteins to free the transcription factor E2F, thereby driving infected cells into the replicative S phase (reviewed in reference 46). Similar to our results, the cytostatic effect of BCV in RPTECs was found to be inversely proportional to cell density (19). Another possible explanation for a stronger cytostatic effect in infected than uninfected cells could be that the DNA damage response is dysregulated in polyomavirus-infected cells (reviewed in reference 47). Analogous to this, primary keratinocytes with an intact DNA damage response were protected from CDV incorporation, while cancer cells failed to respond to DNA damage and incorporated more CDV (37). The viability of HUCs was much less affected than DNA replication by BCV, indicating that quiescent cells, such as the terminally differentiated umbrella cells of the urothelium, may tolerate BCV well. However, high BCV concentrations induced cell death within 5 to 24 h (Fig. 5B; also, see Movie S1 in the supplemental material) independent of the cell density of ≥20,000 cells/cm2 (Fig. 5F and 6A). Before incorporation of CDV can occur, de-esterification, two phosphorylation steps, and translocation to the nucleus must take place. In human lung fibroblasts (MRC-5), intracellular BCV has been found to increase linearly during the first 6 h and progressively until 24 hpt (45). Together, these results imply that the rapid cell death observed at high BCV concentrations was caused by mechanisms other than inhibition of DNA replication and is possibly a detergent-like effect. Amphipathic small molecules like BCV are known to disrupt membranes (48). This mechanism may underlie the dose-limiting adverse effect of diarrhea observed in a recent clinical trial of oral BCV (49).
Compared to previous BKPyV studies using BCV, our ECs were between 1.5- and 3-fold higher than those reported for lung fibroblasts (50) and RPTECs (19). Interestingly a, 30-fold-lower EC90 (0.0197 μM) was found for the related polyomavirus JC (JCPyV) in human progenitor-derived astrocytes. However, in the same study, JCPyV in the monkey kidney cell line COS-7 exhibited an EC90 of 0.74 μM (18), which is within the range of our EC90 spectrum. Our SIs were at the lower end of the spectrum, 1.1-fold greater than the lowest values and 150-fold lower than the highest values found in the above-mentioned studies. Alternative methods and time points investigated may partly account for the observed differences.
Our results indicate that BCV can efficiently inhibit replication of BKPyV in HUCs without cytotoxic effects when used at low concentration but the question remains as to whether BCV can be useful in clinical treatment of BKPyV diseases. Primary HUCs divide about once every 2 to 3 days, while the urothelium in vivo has a very slow basal turnover rate but regenerates rapidly following injury (51). Considering the cell density-dependent effects, BCV is likely to have an optimal antiviral effect when administered early in the course of BKPyV infection, while virus-induced urothelial damage is still limited and BCV-mediated inhibition of cellular DNA replication is less likely to hinder mucosal regeneration. As such, either a prophylactic or a preemptive approach for patients with increasing urine BKPyV loads may be better than waiting for symptoms to arise. Since orally administered BCV is preferentially distributed to the gastrointestinal tract, lung, liver, and spleen but not to the urinary tract in quantitative drug distribution studies in mice (52), for treatment of diagnosed PyVHC, intravesicular administration of BCV is likely to give better results than oral administration. Intravesicular treatment of PyVHC with CDV has been successfully used in some patients (reviewed in reference 23), but this has been questioned recently (53). Due to BCV's rapid transporter-independent absorption into all exposed cells, including urothelial cells as shown here, we speculate that intravesicular BCV is likely to be more efficient than intravesicular CDV. In addition, the reported gastrointestinal side effects will be avoided with this administration form. We also envision that a combination of intravenous CDV and intravesicular BCV might be useful in the treatment of PyVAN, as BCV has the potential to curtail the urothelial amplification of BKPyV, thereby inhibiting the feedback infection of the allograft nephrons (11).
In conclusion, our results demonstrate that BCV efficiently inhibits BKPyV DNA replication, late protein expression, and progeny production in HUCs. The antiviral mechanism seems to be closely linked to the cytostatic effects of BCV, which appear to affect infected cells more than confluent uninfected cells. Our results suggest that BCV could be useful in prophylactic or preemptive treatment of PyVHC. However, considering BCV's rapid absorption kinetics, intravesicular treatment of early diagnosed PyVHC is likely to be a more effective approach, but this needs to be carefully studied in well-designed clinical trials.
Supplementary Material
ACKNOWLEDGMENTS
We thank Biswa Nath Sharma and Stian Henriksen at the Department of Microbiology and Infection Control at the University Hospital of North Norway for helpful comments, instruction, and invaluable technical assistance.
This work was supported by the Northern Norway Regional Health Authority Medical Research Program.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00238-15.
REFERENCES
- 1.Rinaldo CH, Tylden GD, Sharma BN. 2013. The human polyomavirus BK (BKPyV): virological background and clinical implications. APMIS 121:728–745. doi: 10.1111/apm.12134. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch HH, Randhawa P. 2013. BK polyomavirus in solid organ transplantation. Am J Transplant 13(Suppl 4):179–188. doi: 10.1111/ajt.12110. [DOI] [PubMed] [Google Scholar]
- 3.Rorije NM, Shea MM, Satyanarayana G, Hammond SP, Ho VT, Baden LR, Antin JH, Soiffer RJ, Marty FM. 2014. BK virus disease after allogeneic stem cell transplantation: a cohort analysis. Biol Blood Marrow Transplant 20:564–570. doi: 10.1016/j.bbmt.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Dropulic LK, Jones RJ. 2008. Polyomavirus BK infection in blood and marrow transplant recipients. Bone Marrow Transplant 41:11–18. doi: 10.1038/sj.bmt.1705886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirsch H. 2010. Polyoma and papilloma virus infections after hematopoietic stem cell or solid organ transplantation, p 465–482. In Bowden R, Ljungman P, Snydman DR (ed), Transplant infections, 3rd ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Leung AY, Mak R, Lie AK, Yuen KY, Cheng VC, Liang R, Kwong YL. 2002. Clinicopathological features and risk factors of clinically overt haemorrhagic cystitis complicating bone marrow transplantation. Bone Marrow Transplant 29:509–513. doi: 10.1038/sj.bmt.1703415. [DOI] [PubMed] [Google Scholar]
- 7.Azzi A, Cesaro S, Laszlo D, Zakrzewska K, Ciappi S, De Santis R, Fanci R, Pesavento G, Calore E, Bosi A. 1999. Human polyomavirus BK (BKV) load and haemorrhagic cystitis in bone marrow transplantation patients. J Clin Virol 14:79–86. doi: 10.1016/S1386-6532(99)00055-4. [DOI] [PubMed] [Google Scholar]
- 8.Arthur RR, Shah KV, Baust SJ, Santos GW, Saral R. 1986. Association of BK viruria with hemorrhagic cystitis in recipients of bone marrow transplants. N Engl J Med 315:230–234. doi: 10.1056/NEJM198607243150405. [DOI] [PubMed] [Google Scholar]
- 9.Binet I, Nickeleit V, Hirsch HH. 2000. Polyomavirus infections in transplant recipients. Curr Opin Organ Transplant 5:210–216. doi: 10.1097/00075200-200009000-00007. [DOI] [Google Scholar]
- 10.Leung AY, Yuen KY, Kwong YL. 2005. Polyoma BK virus and haemorrhagic cystitis in haematopoietic stem cell transplantation: a changing paradigm. Bone Marrow Transplant 36:929–937. doi: 10.1038/sj.bmt.1705139. [DOI] [PubMed] [Google Scholar]
- 11.Funk GA, Gosert R, Comoli P, Ginevri F, Hirsch HH. 2008. Polyomavirus BK replication dynamics in vivo and in silico to predict cytopathology and viral clearance in kidney transplants. Am J Transplant 8:2368–2377. doi: 10.1111/j.1600-6143.2008.02402.x. [DOI] [PubMed] [Google Scholar]
- 12.Lanier R, Trost L, Tippin T, Lampert B, Robertson A, Foster S, Rose M, Painter W, O'Mahony R, Almond M, Painter G. 2010. Development of CMX001 for the treatment of poxvirus infections. Viruses 2:2740–2762. doi: 10.3390/v2122740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong X, Smith JL, Chen MS. 1997. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation. Antimicrob Agents Chemother 41:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho HT, Woods KL, Bronson JJ, De Boeck H, Martin JC, Hitchcock MJ. 1992. Intracellular metabolism of the antiherpes agent (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine. Mol Pharmacol 41:197–202. [PubMed] [Google Scholar]
- 15.Xiong X, Smith JL, Kim C, Huang ES, Chen MS. 1996. Kinetic analysis of the interaction of cidofovir diphosphate with human cytomegalovirus DNA polymerase. Biochem Pharmacol 51:1563–1567. doi: 10.1016/0006-2952(96)00100-1. [DOI] [PubMed] [Google Scholar]
- 16.Cherrington JM, Allen SJ, McKee BH, Chen MS. 1994. Kinetic analysis of the interaction between the diphosphate of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, ddCTP, AZTTP, and FIAUTP with human DNA polymerases beta and gamma. Biochem Pharmacol 48:1986–1988. doi: 10.1016/0006-2952(94)90600-9. [DOI] [PubMed] [Google Scholar]
- 17.Bernhoff E, Gutteberg TJ, Sandvik K, Hirsch HH, Rinaldo CH. 2008. Cidofovir inhibits polyomavirus BK replication in human renal tubular cells downstream of viral early gene expression. Am J Transplant 8:1413–1422. doi: 10.1111/j.1600-6143.2008.02269.x. [DOI] [PubMed] [Google Scholar]
- 18.Gosert R, Rinaldo CH, Wernli M, Major EO, Hirsch HH. 2011. CMX001 (1-O-hexadecyloxypropyl-cidofovir) inhibits polyomavirus JC replication in human brain progenitor-derived astrocytes. Antimicrob Agents Chemother 55:2129–2136. doi: 10.1128/AAC.00046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rinaldo CH, Gosert R, Bernhoff E, Finstad S, Hirsch HH. 2010. 1-O-hexadecyloxypropyl cidofovir (CMX001) effectively inhibits polyomavirus BK replication in primary human renal tubular epithelial cells. Antimicrob Agents Chemother 54:4714–4722. doi: 10.1128/AAC.00974-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Meijden E, Janssens RW, Lauber C, Bouwes Bavinck JN, Gorbalenya AE, Feltkamp MC. 2010. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog 6:e1001024. doi: 10.1371/journal.ppat.1001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sperling LC, Tomaszewski MM, Thomas DA. 2004. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol 50:318–322. doi: 10.1016/S0190-9622(03)01490-7. [DOI] [PubMed] [Google Scholar]
- 22.Osswald SS, Kulick KB, Tomaszewski MM, Sperling LC. 2007. Viral-associated trichodysplasia in a patient with lymphoma: a case report and review. J Cutan Pathol 34:721–725. doi: 10.1111/j.1600-0560.2006.00693.x. [DOI] [PubMed] [Google Scholar]
- 23.Mackey MC. 2012. Intravesicular cidofovir for the treatment of polyomavirus-associated hemorrhagic cystitis. Ann Pharmacother 46:442–446. doi: 10.1345/aph.1Q430. [DOI] [PubMed] [Google Scholar]
- 24.Reisman L, Habib S, McClure GB, Latiolais LS, Vanchiere JA. 2014. Treatment of BK virus-associated nephropathy with CMX001 after kidney transplantation in a young child. Pediatr Transplant 18:E227-231. doi: 10.1111/petr.12340. [DOI] [PubMed] [Google Scholar]
- 25.Papanicolaou G, Kolitsopoulos Y, Young J, Boruchov A, Sankaranarayanan N, Hull D, Glezerman I, Mommeja-Marin H. 2013. BK virus-associated nephropathy (BKVN), an under-recognized cause of renal dysfunction in severely immunosuppressed hematopoietic stem cell transplant (HSCT) patients: report of 5 cases of Bkvn and the potential role of CMX001 for treatment. Biol Blood Marrow Transplant 19:S302–S303. doi: 10.1016/j.bbmt.2012.11.442. [DOI] [Google Scholar]
- 26.Mommeja-Marin HMF, Boeckh M, Winston D, Rowley SD, Godkin S, Margolskee D. 2012. CMX001, a novel broad spectrum antiviral, may mitigate signs of BK virus (BKV) associated bladder and kidney end-organ damage, abstr P010.034. Abstr 24th International Congress of The Transplantation Society, Berlin, Germany. [Google Scholar]
- 27.Painter GR, Hostetler KY. 2004. Design and development of oral drugs for the prophylaxis and treatment of smallpox infection. Trends Biotechnol 22:423–427. doi: 10.1016/j.tibtech.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Li R, Sharma BN, Linder S, Gutteberg TJ, Hirsch HH, Rinaldo CH. 2013. Characteristics of polyomavirus BK (BKPyV) infection in primary human urothelial cells. Virology 440:41–50. doi: 10.1016/j.virol.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 29.Jiang ZG, Cohen J, Marshall LJ, Major EO. 2010. Hexadecyloxypropyl-cidofovir (CMX001) suppresses JC virus replication in human fetal brain SVG cell cultures. Antimicrob Agents Chemother 54:4723–4732. doi: 10.1128/AAC.00837-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hey AW, Johnsen JI, Johansen B, Traavik T. 1994. A two fusion partner system for raising antibodies against small immunogens expressed in bacteria. J Immunol Methods 173:149–156. doi: 10.1016/0022-1759(94)90294-1. [DOI] [PubMed] [Google Scholar]
- 31.Rinaldo CH, Traavik T, Hey A. 1998. The agnogene of the human polyomavirus BK is expressed. J Virol 72:6233–6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirsch HH, Mohaupt M, Klimkait T. 2001. Prospective monitoring of BK virus load after discontinuing sirolimus treatment in a renal transplant patient with BK virus nephropathy. J Infect Dis 184:1494–1495, 1495–1496. doi: 10.1086/324425. [DOI] [PubMed] [Google Scholar]
- 33.Randhawa PS, Vats A, Zygmunt D, Swalsky P, Scantlebury V, Shapiro R, Finkelstein S. 2002. Quantitation of viral DNA in renal allograft tissue from patients with BK virus nephropathy. Transplantation 74:485–488. doi: 10.1097/00007890-200208270-00009. [DOI] [PubMed] [Google Scholar]
- 34.Rinaldo CH, Myhre MR, Alstad H, Nilssen O, Traavik T. 2003. Human polyomavirus BK (BKV) transiently transforms and persistently infects cultured osteosarcoma cells. Virus Res 93:181–187. doi: 10.1016/S0168-1702(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 35.Sharma BN, Marschall M, Rinaldo CH. 2014. Antiviral effects of artesunate on JC polyomavirus replication in COS-7 cells. Antimicrob Agents Chemother 58:6724–6734. doi: 10.1128/AAC.03714-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Schutter T, Andrei G, Topalis D, Naesens L, Snoeck R. 2013. Cidofovir selectivity is based on the different response of normal and cancer cells to DNA damage. BMC Med Genomics 6:18. doi: 10.1186/1755-8794-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wutzler P, Thust R. 2001. Genetic risks of antiviral nucleoside analogues—a survey. Antiviral Res 49:55–74. doi: 10.1016/S0166-3542(00)00139-X. [DOI] [PubMed] [Google Scholar]
- 39.Cole CN. 1996. Polyomavirinae: the viruses and their replication, p 1997–2025. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, 3rd ed, vol 2 Lippincott-Raven, Philadelphia, PA. [Google Scholar]
- 40.Jesus DM, Costa LT, Goncalves DL, Achete CA, Attias M, Moussatche N, Damaso CR. 2009. Cidofovir inhibits genome encapsidation and affects morphogenesis during the replication of vaccinia virus. J Virol 83:11477–11490. doi: 10.1128/JVI.01061-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magee WC, Aldern KA, Hostetler KY, Evans DH. 2008. Cidofovir and (S)-9-[3-hydroxy-(2-phosphonomethoxy)propyl]adenine are highly effective inhibitors of vaccinia virus DNA polymerase when incorporated into the template strand. Antimicrob Agents Chemother 52:586–597. doi: 10.1128/AAC.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farlow J, Ichou MA, Huggins J, Ibrahim S. 2010. Comparative whole genome sequence analysis of wild-type and cidofovir-resistant monkeypoxvirus. Virol J 7:110. doi: 10.1186/1743-422X-7-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H, Datema R. 1991. Prolonged and potent therapeutic and prophylactic effects of (S)-1-[(3-hydroxy-2-phosphonylmethoxy)propyl]cytosine against herpes simplex virus type 2 infections in mice. Antimicrob Agents Chemother 35:1596–1600. doi: 10.1128/AAC.35.8.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howley PM. 1980. Molecular biology of SV40 and the human polyomaviruses BK and JC, p 489–537. In Klein G. (ed), Viral oncology. Raven Press, New York, NY. [Google Scholar]
- 45.Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. 2003. Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-(14)C]cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol Pharmacol 63:678–681. doi: 10.1124/mol.63.3.678. [DOI] [PubMed] [Google Scholar]
- 46.White MK, Khalili K. 2006. Interaction of retinoblastoma protein family members with large T-antigen of primate polyomaviruses. Oncogene 25:5286–5293. doi: 10.1038/sj.onc.1209618. [DOI] [PubMed] [Google Scholar]
- 47.White MK, Gordon J, Reiss K, Del Valle L, Croul S, Giordano A, Darbinyan A, Khalili K. 2005. Human polyomaviruses and brain tumors. Brain Res Brain Res Rev 50:69–85. doi: 10.1016/j.brainresrev.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 48.Maher P, Singer SJ. 1984. Structural changes in membranes produced by the binding of small amphipathic molecules. Biochemistry 23:232–240. doi: 10.1021/bi00297a010. [DOI] [PubMed] [Google Scholar]
- 49.Marty FM, Winston DJ, Rowley SD, Vance E, Papanicolaou GA, Mullane KM, Brundage TM, Robertson AT, Godkin S, Mommeja-Marin H, Boeckh M. 2013. CMX001 to prevent cytomegalovirus disease in hematopoietic-cell transplantation. N Engl J Med 369:1227–1236. doi: 10.1056/NEJMoa1303688. [DOI] [PubMed] [Google Scholar]
- 50.Randhawa P, Farasati NA, Shapiro R, Hostetler KY. 2006. Ether lipid ester derivatives of cidofovir inhibit polyomavirus BK replication in vitro. Antimicrob Agents Chemother 50:1564–1566. doi: 10.1128/AAC.50.4.1564-1566.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khandelwal P, Abraham SN, Apodaca G. 2009. Cell biology and physiology of the uroepithelium. Am J Physiol Renal Physiol 297:F1477-1501. doi: 10.1152/ajprenal.00327.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quenelle DC, Lampert B, Collins DJ, Rice TL, Painter GR, Kern ER. 2010. Efficacy of CMX001 against herpes simplex virus infections in mice and correlations with drug distribution studies. J Infect Dis 202:1492–1499. doi: 10.1086/656717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koskenvuo M, Dumoulin A, Lautenschlager I, Auvinen E, Mannonen L, Anttila VJ, Jahnukainen K, Saarinen-Pihkala UM, Hirsch HH. 2013. BK polyomavirus-associated hemorrhagic cystitis among pediatric allogeneic bone marrow transplant recipients: treatment response and evidence for nosocomial transmission. J Clin Virol 56:77–81. doi: 10.1016/j.jcv.2012.09.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.