

Abstract
The pharmacokinetics of sublingual artemether (ArTiMist) was investigated in 91 young African children with severe malaria or who could not tolerate oral antimalarial therapy. Each received 3.0 mg/kg of body weight of artemether at 0, 8, 24, 36, 48, and 60 h or until the initiation of oral treatment. Few blood samples were drawn postdose. Plasma artemether and dihydroartemisinin (DHA) levels were measured using liquid chromatography-mass spectrometry, and the data were analyzed using established population compartmental pharmacokinetic models. Parasite clearance was prompt (median parasite clearance time, 24 h), and there were no serious adverse events. Consistent with studies in healthy adults (S. Salman, D. Bendel, T. C. Lee, D. Templeton, and T. M. E. Davis, Antimicrob Agents Chemother 59:3197–3207, 2015, http://dx.doi.org/10.1128/AAC.05013-14), the absorption of sublingual artemether was biphasic, and multiple dosing was associated with the autoinduction of the metabolism of artemether to DHA (which itself has potent antimalarial activity). In contrast to studies using healthy volunteers, pharmacokinetic modeling indicated that the first absorption phase did not avoid first-pass metabolism, suggesting that the drug is transferred to the upper intestine through postdose fluid/food intake. Simulations using the present data and those from an earlier study in older Melanesian children with uncomplicated malaria treated with artemether-lumefantrine tablets suggested that the bioavailability of sublingual artemether was at least equivalent to that after conventional oral artemether-lumefantrine (median [interquartile range] areas under the concentration-time curve for artemether, 3,403 [2,471 to 4,771] versus 3,063 [2,358 to 4,514] μg · h/liter, respectively; and for DHA, 2,958 [2,146 to 4,278] versus 2,839 [1,812 to 3,488] μg · h/liter, respectively; P ≥ 0.42). These findings suggest that sublingual artemether could be used as prereferral treatment for sick children before transfer for definitive management of severe or moderately severe malaria.
INTRODUCTION
Although parenteral artesunate is the recommended initial treatment for severe malaria (1), intramuscular (i.m.) artemether is an acceptable and practical alternative (1, 2). Artemether is also a recommended first-line oral therapy in combination with the longer half-life partner drug lumefantrine for uncomplicated Plasmodium falciparum (3) and Plasmodium vivax (4) infections. There are, however, few detailed studies assessing the pharmacokinetics of artemether and its active metabolite dihydroartemisinin (DHA) in either of these settings in children.
Artemether-lumefantrine is a safe and effective treatment for uncomplicated pediatric malaria (3, 4), but there is evidence of significant between-dose variability in absorption even when coadministered with a small amount of fat to improve bioavailability (5). In addition, the nausea and vomiting that are frequently associated with malaria, together with an unwell child's refusal to feed or take medications by mouth (6), can reduce the effectiveness of oral treatment through reduced adherence to the World Health Organization (WHO) recommended 3-day regimen (7). In cases of more severe malaria, there is evidence of substantial between-patient variability in the absorption of i.m. artemether (8, 9), with some acidotic children likely exposed to subtherapeutic concentrations when the recommended doses are given (9). Rectal artesunate administration is associated with more rapid absorption and initial parasite clearance than is i.m. artemether administration in severely ill children (8). However, there is also marked between-patient variability in the dispositions of artesunate and DHA, and there is evidence that some children are able to expel artesunate suppositories even in the context of close monitoring as part of a formal pharmacokinetic evaluation (10). This might help explain why artesunate suppositories do not improve mortality compared to that with a placebo in older relative to younger pediatric age groups (11).
There is a clear need for a prereferral formulation of an artemisinin derivative that can be easily administered and adequately absorbed in a child who may be unconscious or uncooperative or in whom nausea and vomiting preclude oral dosing. ArTiMist (Essential Nutrition Ltd., Brough, England) is an artemether formulation in neutral oil that can be administered as a metered sublingual spray and which is more rapidly and completely absorbed than is artemether given in tablet form in healthy adult volunteers (see accompanying paper [12]). In the present study, we assessed the pharmacokinetics of ArTiMist in African children with severe malaria or in whom gastrointestinal symptoms prevented the administration of artemether-lumefantrine tablets.
MATERIALS AND METHODS
Study site, approvals, and patients.
The present study was conducted in two parts. Study 1 was an open-label randomized comparative trial of an artemether sublingual spray (ArTiMist; Essential Nutrition Ltd., Brough, England) and intravenous (i.v.) quinine, which was conducted in Rwanda (Rwinkwavu District Hospital). Study 2 was a phase III, randomized, open-label, and multicenter superiority trial of ArTiMist versus i.v. quinine, which was carried out at three different sites in Rwanda (Ruhuha Health Center, Eastern Province), Ghana (Navrongo Health Research Centre, Upper East Region), and Burkina Faso (Centre National de Recherche et de Formation sur le Paludisme, Ouagadougou). Full details of trial procedures and the clinical and parasitologic outcomes will be published separately. The present data relate only to the pharmacokinetics of ArTiMist in those children allocated to this form of treatment.
Children weighing between 5 and 15 kg were eligible for either study 1 or 2 if (i) they had falciparum malaria confirmed by blood film microscopy showing a P. falciparum density of >500 parasites/μl of whole blood (including those positive for other plasmodial species), (ii) they had severe or complicated malaria based on the WHO criteria (13) or uncomplicated malaria but were unable to tolerate oral medication as a result of nausea, vomiting, or diarrhea, (iii) they had received any antimalarial therapy within the 7 days prior to the first study drug administration, (iv) they had evidence of significant comorbidity, including other infections, (v) they had a contraindication or allergy or had a history of intolerance to either artemether or quinine, and (vi) their parents or attendant relatives/guardians gave witnessed informed consent and, where possible, the child assented to participate.
Studies 1 and 2 were approved by the University Teaching Hospital Kigali Research Ethics Committee (EC/CHUK/002/09 and EC/CHUK/015/10, respectively, and study 2 was also approved by the Navrongo Health Research Centre institutional review board (NHRCIRB107) and Centre National de Recherche et de Formation sur le Paludisme Comité Institutionnel de Bioéthique (AEP-002/02/2011/CIB-CNRFP). During the course of study 2, the treatment recommendations for severe childhood malaria were updated (1) following the publication of a large randomized controlled trial showing a significant mortality benefit with i.v. artesunate compared to that with i.v. quinine (14). With the approval of each ethics committee, local regulatory authorities, and investigators, i.v. quinine was retained as the comparator regimen since (i) i.v. quinine remains an acceptable alternative treatment in the updated WHO treatment guidelines (1), and (ii) i.v. artesunate was neither on national treatment guidelines nor available in a good manufacturing practice formulation in any country involved in the present studies at the time.
Study procedures.
In study 1, children were randomized by a computer-generated schedule to receive either ArTiMist or i.v. quinine. ArTiMist was administered at a dose of 3.0 mg/kg of body weight to the nearest 3.0 mg using the most appropriate combination of 3.0-mg/actuation and 6-mg/actuation delivery devices (which have equivalent bioavailabilities in adult volunteer studies [11]) at 0, 8, 24, 36, 48, and 60 h or until the initiation of oral antimalarial therapy. The exact timing of each dose was recorded. Venous blood samples (5 ml) were collected via direct venipuncture under a sparse sampling protocol. The first sample was collected immediately prior to the first dose, with further samples collected at five out of 12 possible times (0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, and 7.8 h) using a balanced randomization schedule. A similar randomization schedule was used to select the final 5 collection times from 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, 8.0, 9.0, 10.0, and 11.8 h after the third dose.
In study 2, children were also randomized using a computer-generated schedule to receive ArTiMist or i.v. quinine. ArTiMist was administered at a dose of 3.0 mg/kg (to the nearest 6.0 mg) using 6.0-mg/actuation delivery devices at 0, 8, 24, 36, 48, and 60 h. Following the initial six doses, patients could, at the discretion of the local investigator, receive a further four daily doses of 3 mg/kg of ArTiMist to complete a 7-day treatment course or be changed to another suitable treatment, typically artemisinin combination therapy (ACT), in accordance with the respective national treatment guidelines. A sparse sampling protocol was used. All patients had a baseline blood sample followed by (i) three samples selected at random from 0.25, 0.5, 0.75, 1.0, 1.5, or 2.0 h after the first dose, (ii) five samples selected at random from 0.5, 0.75, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 7.0, 7.5, 8.0, or 11.5 h after the third dose, and (iii) immediately before and 1.5 h after the sixth dose in all patients. A maximum of 10 blood samples were drawn from any child (total volume, <20 ml). In both studies 1 and 2, the weight of the delivery device was measured before and after each dose so that the actual dose could be calculated.
Drug assays.
Plasma artemether and DHA concentrations were measured using a high-performance liquid chromatography-tandem mass spectrometric method based on that of Shi et al. (15), described in the accompanying paper (12). The calibration curves were linear for both artemether and DHA from 2 to 200 ng/ml (r > 0.995, P < 0.0001), with between-day and within-day precision of <11% for both analytes at the limit of quantification (LOQ), as well as at low, medium, and high plasma concentrations. The LOQ was 2 ng/ml (signal-to-noise ratio, >5), and the limit of detection was 1 ng/ml (signal-to-noise ratio, >3) for both artemether and DHA.
Pharmacokinetic modeling.
Loge plasma concentration-time data sets for artemether and DHA were analyzed by nonlinear mixed-effects modeling using NONMEM (version 7.2.0; Icon Development Solutions, Ellicott City, MD, USA) with an Intel Visual Fortran 10.0 compiler, as described previously (12). The first-order conditional estimation with interaction (FOCE-I) estimation method was used. The minimum objective function value (OFV) and visual predictive checks was used to choose suitable models during the model-building process. A P value of <0.05 was set as the significance level for a comparison of the nested models. Allometric scaling was employed a priori, with the volume terms multiplied by (body weight [BW]/70)1.0 and clearance terms by (BW/70)0.75 (16). Residual variability (RV) was estimated as the additive error for the log-transformed data. Secondary pharmacokinetic parameters, including area under the concentration-time curve from time zero to infinity (AUC0–∞) and the elimination half-life (t1/2), for the participants were obtained from post hoc Bayesian predictions in NONMEM using the final model parameters. The base models were parameterized using ka (absorption rate constant), Vc (central volume of distribution), CL (clearance), and Vp and Q (peripheral volumes of distribution[s] and their respective intercompartmental clearance[s]). The data from the two studies were pooled for pharmacokinetic modeling.
Initially, only the artemether data set was used, with one-, two- and three-compartment models (ADVAN2, -4, and -12, respectively) being tested. As a double peak was noted in many of the individual time-concentration curves for ArTiMist, as was seen in a study conducted in healthy volunteers (12), a number of absorption models were tested, and once a suitable structural model for artemether had been established, the plasma DHA concentration data were included. Custom general linear disposition models were constructed using ADVAN5. The modeling of artemether and DHA was performed simultaneously. To allow identifiability in the parent-metabolite model, the complete conversion of artemether to DHA was assumed. Thus, all artemether parameters were relative to bioavailability (F), while all DHA parameters were relative to F × metabolic conversion (F*). One and two additional compartments were tested for DHA, as well as models estimating the degree of first-pass (FP) metabolism of artemether to DHA. Once the structure of the models was established, interindividual variability (IIV) and interoccasion variability (IOV), as well as correlations between the IIV terms, were evaluated for each suitable parameter and included where supported by the data. IIV was modeled exponentially for all parameters, with the exception of the degree of FP metabolism, for which a probit distribution (with variability fixed to 1) was utilized to ensure estimates between 0 and 1.
The time-dependent kinetics of artemether were assessed using two different approaches. The first assumed that the CL of artemether increased with subsequent doses, an approach used previously for children with malaria treated with artemether-lumefantrine (5, 17). The second approach considered the time-dependent kinetics to be attributable to an increase in the FP metabolism of artemether. As previously noted (18), this may be attributable to the autoinduction of enzymatic activity. For both approaches, a different value was estimated for each day of dosing. For the FP approach, models incorporating FP metabolism for both absorption phases and for the second phase only were tested.
Potential covariate relationships were investigated first by an inspection of individual parameter versus covariate plots and using the generalized additive model within Xpose (19). The identified relationships were then tested within NONMEM using a stepwise forward and backwards approach (P < 0.05 for forward steps and P < 0.01 for backwards steps). The tested covariates were sex, age, fever on admission, log10 (baseline parasitemia), severe versus uncomplicated malaria, vomiting documented on admission, and Blantyre coma score. The effect size (%) of the categorical data was assessed, while both linear and power relationships were evaluated for continuous covariates. The following equations were used: for effect size:
| (1) |
for linear relationships:
| (2) |
and for power relationships:
| (3) |
For the model evaluation, plots of observed versus individual- and population-predicted values, as well as time versus weighted residuals (WRES), were assessed. A bootstrap using Perl speaks NONMEM (PSN) with 200 samples was performed, and the parameters derived from this analysis were summarized as the median and 2.5th and 97.5th percentiles (95% empirical confidence interval [CI]) to facilitate an evaluation of the final model parameter estimates. In addition, prediction-corrected visual predictive checks (pcVPCs) were performed with 1,000 data sets simulated from the final models. The observed 10th, 50th, and 90th percentiles were plotted with their respective simulated 95% CIs to assess the predictive performance of the model and to evaluate any major bias. The VPCs were plotted against the time from the last dose and against the time from the first dose. Shrinkage of the population variability parameters and residual variability were incorporated as measures to help determine whether the models were overparameterized and to determine the reliability of the diagnostic plots (20).
Simulation study.
Using the final model, the concentration-time curves for artemether and DHA after ArTiMist dosing were simulated for 1,000 children based on the sample studied. The same simulated population was also used to obtain concentration-time curves for artemether and DHA after the recommended artemether-lumefantrine dosing using a population pharmacokinetic model previously reported for children (5). The AUC contribution of the first dose was calculated using standard pharmacokinetic formulae.
Statistical analysis.
Statistical analysis was performed using R version 2.14.2 (R Foundation for Statistical Computing, Vienna, Austria). Two-sample comparisons for nonnormally distributed variables were assessed by the Mann-Whitney U test. Unless otherwise stated, all P values are two-tailed and unadjusted for multiple comparisons.
RESULTS
Patient characteristics.
The baseline characteristics of the participants in each study are summarized in Table 1. There were 16 patients recruited to study 1 who received ArTiMist, but one was withdrawn after the second dose due to a protocol violation (incorrect drug storage). In study 2, a total of 76 patients were allocated to ArTiMist from the three sites (27, 25, and 24 children from Rwanda, Ghana, and Burkina Faso, respectively). All patients responded to ArTiMist with prompt parasite clearance (median parasite clearance time in both studies, 24 h), and there were no treatment-related serious adverse events (data not shown).
TABLE 1.
Baseline characteristics of participants in studies 1 and 2
| Characteristic | Study 1 | Study 2 | Combined |
|---|---|---|---|
| n | 15 | 76 | 91 |
| Wt (median [IQR]) (kg)a | 10.3 (9.8–14.0) | 12.0 (10.0–13.7) | 12.0 (10.0–13.8) |
| Age (median [IQR]) (yr) | 3.4 (1.6–4.0) | 2.7 (1.8–3.5) | 2.8 (1.8–3.5) |
| Male (no. [%]) | 7 (44) | 36 (47) | 43 (47) |
| Ht (median [IQR]) (cm) | 82 (75–92) | 87 (81–93) | 87 (79–93) |
| Baseline parasitemia (median [IQR]) (no. of parasites/μl) | 19,660 (13,710–26,310) | 52,775 (20,047–193,669) | 41,383 (16,805–141,834) |
| Admission temp (median [IQR]) (°C) | 38.1 (37.6–38.9) | 38.7 (37.8–39.5) | 38.7 (37.8–39.4) |
| Severe malaria (no. [%]) | 10 (63) | 48 (63) | 58 (63) |
| Vomiting at admission (no. [%]) | 2 (13) | 46 (61) | 48 (52) |
| Blantyre coma score (no. [%]) of: | |||
| 1 | 0 (0) | 2 (3) | 2 (2) |
| 2 | 0 (0) | 0 (0) | 0 (0) |
| 3 | 1 (6) | 7 (9) | 8 (9) |
| 4 | 2 (13) | 7 (9) | 9 (10) |
| 5 | 13 (81) | 60 (79) | 73 (79) |
IQR, interquartile range.
Pharmacokinetic modeling.
There were 824 and 788 individual plasma concentrations for artemether and DHA, respectively, and 4 (0.5%) and 72 (9.1%) of the artemether and DHA concentrations, respectively, were below the limit of quantification (BLQ). Given that BLQ data comprised <10% of the total data, these were excluded from the analysis using method M1 for dealing with BLQ data (21).
The structural model derived for the artemether and DHA dispositions was the same as that in used in the study with healthy volunteers (12). A two-compartment model for artemether was most appropriate, with no benefit obtained from additional compartments (P > 0.05). A two-phase absorption was superior to single-phase absorption (P < 0.001). This was represented by two separate lag times (LAG1 and LAG2), followed by first-order absorption with two separate absorption rates (ka1 and ka2). The ratio of the fraction of the dose absorbed with the first phase versus the second phase (RATIO) was estimated within NONMEM. The second phase of ArTiMist absorption occurred slightly >1 h after drug administration. A model estimating a bolus input was also tested; however, this was estimated at <1% of the total dose and did not improve the overall fit. A single additional compartment was suitable for characterizing the disposition of DHA. LAG1 was fixed to a value obtained in the final artemether-only model for the simultaneous modeling of both the parent drug and the metabolite to enable successful minimization.
Models with increasing FP metabolism for subsequent doses were better than those with increasing clearance for subsequent doses in describing the time-dependent kinetics of artemether. A model with FP metabolism influencing the second, but not the first, phase of absorption, as pertains in healthy adult volunteers (12), resulted in a worse fit of the data.
The population maximum of FP metabolism for the first day was estimated at 18%, increasing to 84.0% on the second day and to 96% on the last day of dosing. The ratio of the population maximum to the population minimum of FP metabolism was 1.5. IIV was estimable for artemether (ARM) clearance (CLARM), DHA clearance (CLDHA), and ka1, with values of 33%, 29%, and 121%, respectively. As in the studies using healthy adults (12), IIV and IOV could not be estimated simultaneously for RATIO, with the estimate for IOV being much higher (197%), indicating that most of the variability was between occasions in the same individual rather than between different individuals. The IOV of relative bioavailability was high, at 110%. No significant covariate relationships were identified.
The final model parameter estimates and the bootstrap results are summarized in Table 2. The bias was <10% for all fixed- and random-model parameters. Figures 1, 2, and 3 show goodness-of-fit plots and pcVPCs, which demonstrate the reasonable predictive performance of the model. The half-life and AUC0–∞ values derived from post hoc individual parameters are shown in Table 3. These are presented along with equivalent post hoc individual parameters from a previous study in Melanesian children 5 to 10 years old with uncomplicated malaria who were treated with conventional artemether-lumefantrine tablets (5). The study with Melanesian children was selected for comparison with the present study because (i) individual patient data were available for that study, (ii) as in the present study, blood sampling in that study included time points beyond those associated with trough concentrations, the lack of which has been a limitation of previous studies (17), (iii) a comparison with pediatric studies of artesunate, whether given parenterally or rectally, would be problematic given that it is a different artemisinin derivative with distinct pharmacologic properties, (iv) i.m. artemether has quite different pharmacological properties from those of orally administered artemether, and (v) a proportion of the children in the present study did not fulfill the WHO criteria for severe malaria and would have been suitable for oral artemether-lumefantrine were it not for gastrointestinal symptoms and/or a refusal to take the tablets.
TABLE 2.
Final population pharmacokinetic variable estimates and bootstrap results of artemether and DHA for study 1a
| Parameterb | Mean | RSE%c | Bootstrap (median [95% CI]) |
|---|---|---|---|
| Objective function value | 1,458.776 | 1,409.466 (1,249.640–1,596.160) | |
| Structural model parameters | |||
| ka1 (/h) | 4.15 | 30 | 4.01 (1.25–12.5) |
| ka2 (/h) | 0.81 | 15 | 0.80 (0.52–1.19) |
| LAG1 (h) | 0.20 | 6 | 0.20 (0.14–0.23) |
| LAG2 (h) | 1.24 | 9 | 1.24 (0.98–1.40) |
| RATIO | 0.82 | 29 | 0.80 (0.44–2.28) |
| CL/FARM (liters/h/70 kg) | 112 | 9 | 111 (93.4–136) |
| Vc/FARM (liters/70 kg) | 318 | 13 | 319 (216–425) |
| Q/FARM (liters/h/70 kg) | 45.0 | 16 | 46.2 (34.1–66.8) |
| Vp/FARM (liters/70 kg) | 1,347 | 30 | 1,345 (828–3,404) |
| CL/F*DHA (liters/h/70 kg) | 336 | 10 | 342 (270–424) |
| V/F*DHA (liters/70 kg) | 779 | 15 | 801 (502–1,017) |
| FPmax, day1 (%) | 0.178 | 17 | 0.169 (0.112–0.227) |
| FPmax, day2 (%) | 0.840 | 8 | 0.799 (0.681–0.901) |
| FPmax, day3 (%) | 0.959 | 5 | 0.935 (0.803–0.990) |
| FPmax-to-FPmin ratio | 1.50 | 17 | 1.37 (1.01–1.98) |
| Variability model parameters (shrinkage %) | |||
| IIV in CL/FARM | 33 (27) | 22 | 33 (17–53) |
| IIV in CL/F*DHA | 29 (36) | 25 | 29 (12–43) |
| IIV in ka1 | 121 (14) | 14 | 121 (79–152) |
| r(CL/FARM, CL/F*DHA) | 0.620 | 58 | 0.656 (−0.395–0.981) |
| IOV in F | 109 (16, 22, 36, 22) | 10 | 108 (90–132) |
| IOV in RATIO | 197 (33, 21) | 13 | 195 (154–235) |
| RV for artemether | 56 (21) | 6 | 55 (51–62) |
| RV for DH | 83 (11) | 4 | 83 (76–89) |
Fixed in final model; RSE% and 95% CI are from earlier modeling.
ka1, absorption rate constant for first absorption phase of ArTiMist; ka2, absorption rate constant for second absorption phase of ArTiMist; LAG1, lag time for first absorption phase of ArTiMist; LAG2, lag time for second absorption phase of ArTiMist; RATIO, ratio between the first and second absorption phases of ArTiMist; CL/FARM, artemether clearance; Vc/FARM, artemether central volume of distribution; Q/FARM, artemether intercompartmental clearance; Vp/FARM, artemether first peripheral volume of distribution; CL/F*DHA, DHA clearance; Vc/F*DHA, DHA central volume of distribution; FPmax, day1, FPmax, day2, and FPmax, day3, population maximum first-pass metabolism on days 1, 2, and 3, respectively; FPmax-to-FPmin ratio, the ratio of the population maximum to the population minimum of FP; r, correlation coefficient; FARM, relative bioavailability of artemether; F*DHA, FARM × metabolic conversion of artemether to DHA; IOV, interoccasion variability; IIV, interindividual variability; RV, residual variability. IOV and IIV are presented as 100% × the square root of the variability estimate.
RSE, relative standard error.
FIG 1.

Goodness-of-fit plots for artemether. The observed plasma concentration has been plotted against population-predicted (upper left) and individual-predicted (upper right) plasma concentrations, and conditional weighted residuals against time (lower left) and population-predicted plasma concentrations (lower right). The solid lines in the upper two graphs represent the lines of identity and the dashed lines are the least-squares regression lines.
FIG 2.

Goodness-of-fit plots for dihydroartemisinin. The observed plasma concentration has been plotted against population-predicted (upper left) and individual-predicted (upper right) plasma concentrations, and conditional weighted residuals against time (lower left) and population-predicted plasma concentrations (lower right). The solid lines in the upper two graphs represent the lines of identity and the dashed lines are the least-squares regression lines.
FIG 3.
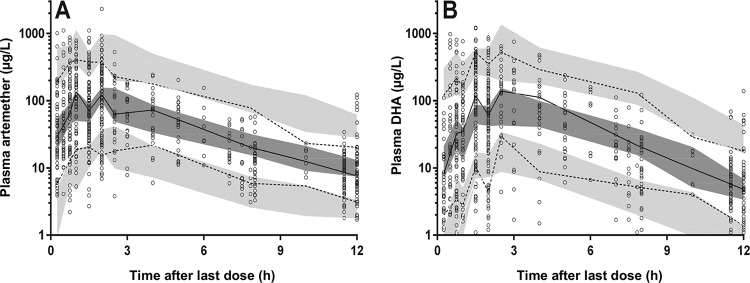
Prediction-corrected visual predictive check for plasma artemether (A) and dihydroartemisinin (DHA) (B) (in micrograms per liter on a log10 scale) plotted against time after the last dose, with observed 50th (solid line) and 10th and 90th (dotted lines) percentiles within their simulated 95% CI (gray shaded areas) overlying the data points (○).
TABLE 3.
Derived secondary pharmacokinetic parameters from post hoc individual parameters for the present studies compared with those from a previous study (5) of artemether-lumefantrine in Melanesian children 5 to 10 years old with uncomplicated malaria
| Parametera | Present studies | Prior oral artemether study | P value |
|---|---|---|---|
| Artemether | |||
| t1/2α (h) | 0.841 (0.747–0.926) | 0.62 (0.60–0.64)b | <0.001 |
| t1/2β (h) | 18.3 (17.7–19.9) | 16.4 (15.7–16.8)b | <0.001 |
| AUC0–∞ (μg · h/liter) | |||
| Dose 1 | 894 (473–1,420) | 983 (371–1,770) | 0.88 |
| Dose 6 | 232 (147–380) | 164 (145–254) | 0.15 |
| Total | 3,403 (2,471–4,771) | 3,063 (2,358–4,514) | 0.63 |
| Total normalized to dose | 1.18 (0.85–1.64) | 1.63 (1.26–2.44) | <0.001 |
| Dihydroartemisinin | |||
| t1/2 (h) | 0.987 (0.859–1.11) | 0.80 (0.76–0.82) | <0.001 |
| AUC0–∞ (μg · h/liter) | |||
| Dose 1 | 355 (206–626) | 362 (136–652) | 0.77 |
| Dose 6 | 448 (277–690) | 311 (214–440) | 0.09 |
| Total | 2,958 (2,146–4,278) | 2,839 (1,812–3,488) | 0.42 |
| Total normalized to dose | 1.04 (0.71–1.42) | 1.52 (0.98–1.89) | 0.052 |
| Dihydroartemisinin-to-artemether ratio | |||
| Dose 1 | 0.42 (0.38–0.46) | 0.37 (0.37–0.37) | <0.001 |
| Dose 6 | 1.74 (1.57–2.01) | 1.86 (0.92–2.68) | 0.97 |
| Total | 0.85 (0.74–1.04) | 0.93 (0.59–0.94) | 0.46 |
Data are presented as the median (interquartile range). t1/2α, first half-life; t1/2β, second half-life; AUC0–∞, area under the concentration-time curve from time 0 to infinity.
For the first dose.
Between the two studies, there were significant differences in t1/2 values, corresponding to the difference in weights and ages between the subjects of the present and Melanesian studies (12.0 versus 19.0 kg and 2.8 versus 7.7 years, respectively; P < 0.001 in each case). There was no significant difference in the dose 1, dose 6, and total AUC0–∞ of artemether or DHA or in the total DHA-to-artemether ratios. However, the children in the present study received a higher dose of 2.9 mg/kg versus 1.9 mg/kg in the previous study (P < 0.001). The distribution and terminal t1/2 for artemether were 0.84 and 18 h, respectively, and the elimination t1/2 for DHA was 0.99 h.
The results of the simulation study are presented in Fig. 4 as box-and-whisker plots demonstrating the 90% prediction intervals (PI) of the artemether, DHA, and combined AUCs after the first dose of ArTiMist and artemether-lumefantrine. Both demonstrate wide prediction intervals due to the large variability in the bioavailability of artemether in the population pharmacokinetic models. The median (90% PI) combined AUCs for ArTiMist and artemether-lumefantrine were 1,380 (201 to 8,689) μg · h/liter and 1,039 (219 to 5,390) μg · h/liter, respectively.
FIG 4.
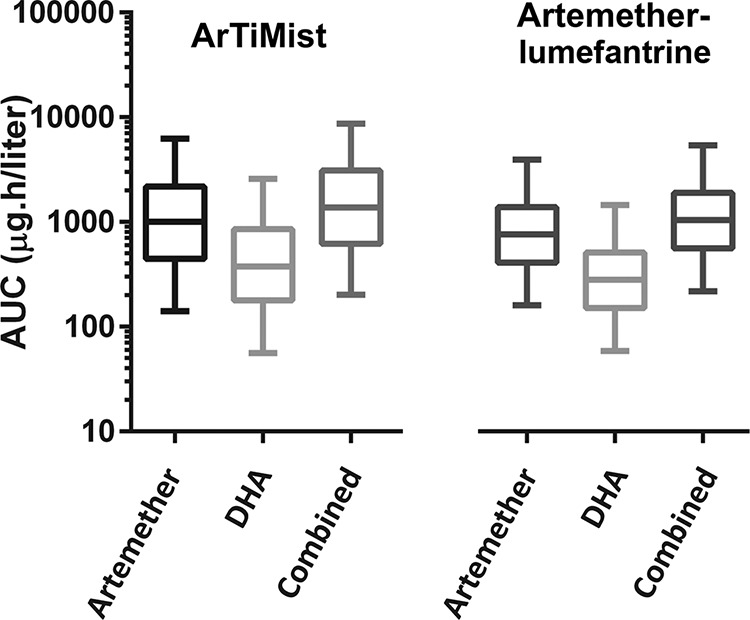
Results of the simulation study presented as box-and-whisker plots demonstrating the 90% prediction intervals for artemether AUCs, dihydroartemisinin (DHA) AUCs, and combined AUCs after the first doses of ArTiMist and artemether-lumefantrine tablets. The horizontal line in each box represents the median and the box indicates the interquartile range within each 95% prediction interval (vertical lines).
DISCUSSION
The present data show that ArTiMist given at a dose of 3.0 mg/kg twice daily for 3 days to young African children who have severe malaria or who are unable to tolerate conventional oral therapy is promptly and adequately absorbed, regardless of the indices of severity, including consciousness level and even in patients with a history of vomiting at presentation. As in healthy adult volunteers (12), there is a biphasic absorption of the drug. However, in contrast to our adult data suggesting the avoidance of FP metabolism during the first phase, there was model-derived evidence of FP metabolism in both absorption phases in children with malaria. These data suggest that the amount of artemether absorbed directly through the oral mucosa in this clinical setting is less than that in healthy volunteers. Previous models of the time-dependent pharmacokinetics of artemether have assumed that declining plasma concentrations reflect increasing drug CL (5, 17). However, the present study, utilizing a greater number of plasma concentration-time coordinates across doses and absorption phases offers support for a reduction in F through increased FP metabolism via the induction of enzymatic activity in the liver and intestines (18, 22).
Pharmacokinetic studies with intensive sampling schedules are challenging in small children because of the relatively large volume of blood that might need to be taken and the difficulty in obtaining, and the discomfort associated with, venous access and/or repeated venesections (23). The use of sparse sampling and a population modeling approach is a better alternative. We employed this approach in the present study and found that the final structural models characterizing sublingual artemether and DHA dispositions in our young African children were similar to those in healthy adult volunteers who underwent intensive postdose blood sampling (12). This provides evidence supporting the validity of the pharmacokinetic parameters derived in the present study.
The only difference in the ArTiMist dispositions between the present studies and our investigations in healthy adult volunteers (12) related to the first phase of absorption. There was evidence of FP metabolism during the first phase of absorption in the children with malaria, while the ka1 in the pediatric study was faster than that in the adults (4.15/h versus 1.46/h, respectively) (12). This suggests that the retention time in the sublingual space is longer in healthy adults than in sick young children; also, in the young children, some artemether was swallowed soon after administration and absorbed through the upper gastrointestinal tract into the portal venous system rather than exhibiting the more substantial sublingual absorption as seen in the adult volunteers. There was no requirement to restrict fluids or food after ArTiMist dosing in the present study. Since rehydration is an important part of the management of acute malaria in children, it is possible that a significant proportion of the children were given water after dosing, and this promptly transferred the drug into the stomach. The slower second absorption phase paralleled that in adults and may represent drug in the buccal cavity that enters the stomach more slowly in swallowed saliva.
Although there are age-related differences in the factors affecting sublingual drug absorption, this route of delivery is used for the administration of analgesics, immunotherapy, and other treatments in children (24). However, of potential relevance to the present data is the observation that there is abnormal sublingual microcirculatory density and function in pediatric sepsis (25, 26), which may be exacerbated by the microvascular sequestration of P. falciparum (27) in malaria. These factors might have attenuated the buccal absorption of ArTiMist in our children but may not have been influential in the upper small intestine.
Young children between 1 and 6 years of age, such as those who formed the majority of patients in the present studies, may have increased body weight-normalized plasma drug clearance and a need for higher milligram-per-kilogram doses than those in older children and adults (28). Thus, although ArTiMist has increased bioavailability relative to that of tablets containing the same dose in healthy adults (12), repeated 3.0-mg/kg ArTiMist doses in the African children in the present study achieved a similar drug exposure (AUC0–∞) to only 1.7 mg/kg/dose by artemether-lumefantrine tablets in older children from Papua New Guinea (PNG) (5) (see Table 3). This finding emphasizes the value of age-specific pharmacokinetic studies as part of drug development.
The median and range of artemether and DHA AUC0–∞ values across the six doses (as evidenced by the interquartile ranges in Table 3) were generally similar in African and PNG children, albeit after different doses. The simulation study produced a similar result. Importantly, the lower bounds of the 90% PI are almost identical, indicating that there is no increased risk of underdosing artemether with the current ArTiMist regimen compared to the recommended doses of artemether-lumefantrine tablets. The high degree of between-subject variability parallels that with intramuscular artemether (8, 9). However, sublingual/oral dosing has the added benefit that the swallowed fraction may undergo FP metabolism of artemether to DHA, with higher relative amounts of the more potent metabolite being present (29).
Given the WHO recommendations regarding the need for artemisinin drugs to be administered in combination with a longer half-life partner (such as lumefantrine), except in situations in which initial oral therapy cannot be given safely and reliably (30), ArTiMist is likely to have application as one- or two-dose prereferral treatment, as sick children are transferred for parenteral artemisinin therapy or oral ACT. In view of potential pharmacokinetic (8), cultural (31), and efficacy (11) concerns regarding artesunate suppositories in this situation, ArTiMist appears to be a valuable alternative. The present study is the first clinical evaluation of this novel antimalarial treatment strategy, and the population models developed from the data have provided initial pharmacokinetic insights. Further studies should examine in greater detail the effects of factors, such as the ingestion of oral fluids and food, on ArTiMist disposition in sick children with malaria, as well as compare the pharmacokinetics and efficacy of sublingual artemether administration to those after parenteral administration of artemisinin derivatives in similar groups of patients.
ACKNOWLEDGMENTS
We thank the children in this study and their parents/guardians for their participation. We also thank the medical, nursing, and laboratory staff at the Rwinkwavu District Hospital in Rwanda, the Ruhuha Health Centre in Rwanda, the Navrongo Health Research Centre in Ghana, and the Centre National de Recherche et de Formation sur le Paludisme in Burkina Faso for their invaluable assistance in conducting the studies. We also thank Proto Pharma Ltd., Nowrich, United Kingdom, for study management and Suda Ltd., Osborne Park, Western Australia, for funding.
T.M.E.D. is supported by a National Health and Medical Research Council of Australia Practitioner Fellowship.
D.B., T.C.L., and D.T. received funding from Suda Ltd. via Proto Pharma Ltd. for performing the clinical studies. T.M.E.D. has received funding from Suda Ltd. for consultations and advisory board participation.
REFERENCES
- 1.World Health Organization. 2010. Guidelines for the treatment of malaria, 2nd ed World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Kyu HH, Fernandez E. 2009. Artemisinin derivatives versus quinine for cerebral malaria in African children: a systematic review. Bull World Health Organ 87:896–904. doi: 10.2471/BLT.08.060327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adjei GO, Goka BQ, Binka F, Kurtzhals JA. 2009. Artemether-lumefantrine: an oral antimalarial for uncomplicated malaria in children. Expert Rev Anti Infect Ther 7:669–681. doi: 10.1586/eri.09.53. [DOI] [PubMed] [Google Scholar]
- 4.Bassat Q. 2011. The use of artemether-lumefantrine for the treatment of uncomplicated Plasmodium vivax malaria. PLoS Negl Trop Dis 5:e1325. doi: 10.1371/journal.pntd.0001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salman S, Page-Sharp M, Griffin S, Kose K, Siba PM, Ilett KF, Mueller I, Davis TM. 2011. Population pharmacokinetics of artemether, lumefantrine, and their respective metabolites in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother 55:5306–5313. doi: 10.1128/AAC.05136-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tumwesigire S, Watson S. 2002. Health seeking behavior by families of children suspected to have malaria in Kabale: Uganda. Afr Health Sci 2:94–98. [PMC free article] [PubMed] [Google Scholar]
- 7.Banek K, Lalani M, Staedke SG, Chandramohan D. 2014. Adherence to artemisinin-based combination therapy for the treatment of malaria: a systematic review of the evidence. Malar J 13:7. doi: 10.1186/1475-2875-13-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karunajeewa HA, Reeder J, Lorry K, Dabod E, Hamzah J, Page-Sharp M, Chiswell GM, Ilett KF, Davis TM. 2006. Artesunate suppositories versus intramuscular artemether for treatment of severe malaria in children in Papua New Guinea. Antimicrob Agents Chemother 50:968–974. doi: 10.1128/AAC.50.3.968-974.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mithwani S, Aarons L, Kokwaro GO, Majid O, Muchohi S, Edwards G, Mohamed S, Marsh K, Watkins W. 2004. Population pharmacokinetics of artemether and dihydroartemisinin following single intramuscular dosing of artemether in African children with severe falciparum malaria. Br J Clin Pharmacol 57:146–152. doi: 10.1046/j.1365-2125.2003.01986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karunajeewa HA, Ilett KF, Dufall K, Kemiki A, Bockarie M, Alpers MP, Barrett PH, Vicini P, Davis TM. 2004. Disposition of artesunate and dihydroartemisinin after administration of artesunate suppositories in children from Papua New Guinea with uncomplicated malaria. Antimicrob Agents Chemother 48:2966–2972. doi: 10.1128/AAC.48.8.2966-2972.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okebe J, Eisenhut M. 2014. Pre-referral rectal artesunate for severe malaria. Cochrane Database Syst Rev 5:CD009964. doi: 10.1002/14651858.CD009964.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salman S, Bendel D, Lee TC, Templeton D, Davis TME. 2015. Pharmacokinetics of a novel sublingual spray formulation of the antimalarial drug artemether in healthy adults. Antimicrob Agents Chemother 59:3197–3207. doi: 10.1128/AAC.05013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.World Health Organization (WHO). 2000. Severe falciparum malaria. World Health Organization, Communicable Diseases Cluster. Trans R Soc Trop Med Hyg 94(Suppl 1):S1–S90. doi: 10.1016/S0035-9203(00)90300-6. [DOI] [PubMed] [Google Scholar]
- 14.Dondorp AM, Fanello CI, Hendriksen IC, Gomes E, Seni A, Chhaganlal KD, Bojang K, Olaosebikan R, Anunobi N, Maitland K, Kivaya E, Agbenyega T, Nguah SB, Evans J, Gesase S, Kahabuka C, Mtove G, Nadjm B, Deen J, Mwanga-Amumpaire J, Nansumba M, Karema C, Umulisa N, Uwimana A, Mokuolu OA, Adedoyin OT, Johnson WBR, Tshefu AK, Onyamboko MA, Sakulthaew T, Ngum WP, Silamut K, Stepniewska K, Woodrow CJ, Bethell D, Wills B, Oneko M, Peto TE, von Seidlein L, Day NP, White NJ, AQUAMAT Group. 2010. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet 376:1647–1657. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi B, Yu Y, Li Z, Zhang L, Zhong Y, Su S, Liang S. 2006. Quantitative analysis of artemether and its metabolite dihydroartemisinin in human plasma by LC with tandem mass spectrometry. Chromatographia 64:523–530. doi: 10.1365/s10337-006-0064-y. [DOI] [Google Scholar]
- 16.Anderson BJ, Holford NH. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24:25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- 17.Hietala SF, Martensson A, Ngasala B, Dahlstrom S, Lindegardh N, Annerberg A, Premji Z, Farnert A, Gil P, Bjorkman A, Ashton M. 2010. Population pharmacokinetics and pharmacodynamics of artemether and lumefantrine during combination treatment in children with uncomplicated falciparum malaria in Tanzania. Antimicrob Agents Chemother 54:4780–4788. doi: 10.1128/AAC.00252-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Agtmael MA, Cheng-Qi S, Qing JX, Mull R, van Boxtel CJ. 1999. Multiple dose pharmacokinetics of artemether in Chinese patients with uncomplicated falciparum malaria. Int J Antimicrob Agents 12:151–158. doi: 10.1016/S0924-8579(99)00063-1. [DOI] [PubMed] [Google Scholar]
- 19.Jonsson EN, Karlsson MO. 1999. Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58:51–64. [DOI] [PubMed] [Google Scholar]
- 20.Savic RM, Karlsson MO. 2009. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J 11:558–569. doi: 10.1208/s12248-009-9133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beal SL. 2001. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 28:481–504. doi: 10.1023/A:1012299115260. [DOI] [PubMed] [Google Scholar]
- 22.van Agtmael MA, Gupta V, van der Graaf CA, van Boxtel CJ. 1999. The effect of grapefruit juice on the time-dependent decline of artemether plasma levels in healthy subjects. Clin Pharmacol Ther 66:408–414. doi: 10.1053/cp.1999.v66.a101946. [DOI] [PubMed] [Google Scholar]
- 23.Kern SE. 2009. Challenges in conducting clinical trials in children: approaches for improving performance. Expert Rev Clin Pharmacol 2:609–617. doi: 10.1586/ecp.09.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam JK, Xu Y, Worsley A, Wong IC. 2014. Oral transmucosal drug delivery for pediatric use. Adv Drug Deliv Rev 73:50–62. doi: 10.1016/j.addr.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Paize F, Sarginson R, Makwana N, Baines PB, Thomson AP, Sinha I, Hart CA, Riordan A, Hawkins KC, Carrol ED, Parry CM. 2012. Changes in the sublingual microcirculation and endothelial adhesion molecules during the course of severe meningococcal disease treated in the paediatric intensive care unit. Intensive Care Med 38:863–871. doi: 10.1007/s00134-012-2476-5. [DOI] [PubMed] [Google Scholar]
- 26.Top AP, Ince C, de Meij N, van Dijk M, Tibboel D. 2011. Persistent low microcirculatory vessel density in nonsurvivors of sepsis in pediatric intensive care. Crit Care Med 39:8–13. doi: 10.1097/CCM.0b013e3181fb7994. [DOI] [PubMed] [Google Scholar]
- 27.Moxon CA, Grau GE, Craig AG. 2011. Malaria: modification of the red blood cell and consequences in the human host. Br J Haematol 154:670–679. doi: 10.1111/j.1365-2141.2011.08755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van den Anker JN. 2010. Developmental pharmacology. Dev Disabil Res Rev 16:233–238. doi: 10.1002/ddrr.122. [DOI] [PubMed] [Google Scholar]
- 29.Teja-Isavadharm P, Nosten F, Kyle DE, Luxemburger C, Ter Kuile F, Peggins JO, Brewer TG, White NJ. 1996. Comparative bioavailability of oral, rectal, and intramuscular artemether in healthy subjects: use of simultaneous measurement by high performance liquid chromatography and bioassay. Br J Clin Pharmacol 42:599–604. doi: 10.1111/j.1365-2125.1996.tb00115.x. [DOI] [PubMed] [Google Scholar]
- 30.World Health Organization. 2011. World malaria report 2011. World Health Organization, Geneva, Switzerland: http://www.who.int/malaria/world_malaria_report_2011/9789241564403_eng.pdf. [Google Scholar]
- 31.Hinton RL, Auwun A, Pongua G, Oa O, Davis TM, Karunajeewa HA, Reeder JC. 2007. Caregivers' acceptance of using artesunate suppositories for treating childhood malaria in Papua New Guinea. Am J Trop Med Hyg 76:634–640. [PubMed] [Google Scholar]