

Abstract
β-Lactamase production is one of the most important strategies for Gram-negative bacteria to combat β-lactam antibiotics. Studies of the regulation of β-lactamase expression have largely been focused on the class C β-lactamase AmpC, whose induction by β-lactams requires LysR-type regulator AmpR and permease AmpG-dependent peptidoglycan recycling intermediates. In Shewanella, which is ubiquitous in aquatic environments and is a reservoir for antibiotic resistance, production of the class D β-lactamase BlaA confers bacteria with natural resistance to many β-lactams. Expression of the blaA gene in the genus representative Shewanella oneidensis is distinct from the AmpC paradigm because of the lack of an AmpR homologue and the presence of an additional AmpG-independent regulatory pathway. In this study, using transposon mutagenesis, we identify proteins that are involved in blaA regulation. Inactivation of mrcA and lpoA, which encode penicillin binding protein 1a (PBP1a) and its lipoprotein cofactor, LpoA, respectively, drastically enhances blaA expression in the absence of β-lactams. Although PBP1b and its cognate, LpoB, also exist in S. oneidensis, their roles in blaA induction are dispensable. We further show that the mrcA-mediated blaA expression is independent of AmpG.
INTRODUCTION
Beta-lactams are the most widely used antibiotics for treatment of bacterial infections. They mimic the dipeptide d-Ala-d-Ala and bind covalently to the serine active center of penicillin binding proteins (PBPs), resulting in inactivation of the latter, which are responsible for peptidoglycan synthesis and hydrolysis (1, 2). As one of the major approaches that evolved in bacteria to cope with the threat of β-lactams, β-lactamases are produced to directly degrade this group of antibiotics (3, 4). A variety of β-lactamases have been isolated, and their biochemical characteristics are intensively studied. Among them, some are found to be induced by β-lactams, but studies of the underlying regulatory mechanisms are largely focused on the class C β-lactamase AmpC in several Gram-negative bacteria, including Enterobacter cloacae, Citrobacter freundii, and Pseudomonas aeruginosa (5–13). Because of the discrepancy of the locations between β-lactam action and β-lactamase production, intermediate molecular signals must be produced to guide AmpC induction. Many studies have demonstrated that these molecular cues are derived from peptidoglycan recycling and that multiple proteins are involved in their production. AmpG is an inner membrane-bound permease that is responsible for the transportation of signal precursors, and its loss results in enhanced sensitivity to β-lactams because of the interruption of signal transduction (12–16). AmpR, a LysR family transcriptional regulator, alternates its conformation by binding to two different ligands to activate or repress the transcription of ampC (7, 17–21).
As the primary targets for β-lactams, PBPs are no doubt potential candidates to sense the stress from antibiotics in the periplasm. PBPs are classified into two main categories: high-molecular-weight PBPs (HMW PBPs) and low-molecular-weight PBPs (LMW PBPs) (22, 23). In total, five HMW PBPs have been identified in Escherichia coli, and they belong to either class A or class B. Class A PBPs (PBP1a, PBP1b, and PBP1c) are bifunctional enzymes containing both glycosyltransferase (GTase) and transpeptidase (TPase) domains, whereas class B PBPs (PBP2 and PBP3) consist of a TPase domain only and thus are monofunctional. In E. coli, PBP1s are organized into multienzyme peptidoglycan-synthesizing complexes with other components. PBP1a interacts with PBP2 for cell elongation, while PBP1b interacts with PBP3 and FtsZ for cell division (24–26). Loss of PBP1a reduces both the growth rate in minimal medium and the cell diameter (24), but depletion of PBP1b results in a hypersensitivity to some β-lactams (27). Despite these phenotypic differences, removal of both PBP1s together causes synthetic lethality, implying that there is a functional complementation between them (27, 28). Importantly, the functionality of PBP1a and PBP1b requires the outer membrane lipoproteins LpoA and LpoB, respectively, as essential cofactors. Each lipoprotein forms a complex with its cognate PBP and stimulates both the GTase and TPase activities (27–29).
Shewanella species are gammaproteobacteria that are widely distributed in various habitats and renowned for the ability to use a diverse range of electron acceptors for respiration (30, 31). The members of the genus are regarded as emerging human pathogens and as a reservoir for antibiotic resistance, given that a number of β-lactamases and Qnr-type determinants have been isolated from them (32–37). The representative species Shewanella oneidensis possesses seven genes that are predicted to encode β-lactamases (38, 39). The Amber class D β-lactamase BlaA (SO0837), also named OXA-54, confers on the bacterium a natural resistance to many β-lactams, while others were found to be dispensable by a mutational analysis (39). BlaA, along with its homologues from other Shewanella members, is proposed to be the progenitor of the carbapenem-hydrolyzing enzyme oxacillinase, which has been acquired by the clinically relevant Gram-negative species Acinetobacter baumannii and Klebsiella pneumoniae (34, 37, 40). Our previous studies showed that the blaA gene can be induced by ampicillin at high (>12.5 μg/ml) but not low (<5 μg/ml) levels, and its induction is also intimately linked to peptidoglycan recycling (39, 41). However, a homologue of AmpR is absent in S. oneidensis, and the impacts of major peptidoglycan recycling enzymes on blaA expression are distinct from the ampR-ampC paradigm. In particular, the loss of AmpG increases blaA expression, in contrast to the effect observed in previously studied cases (41).
The goal of the work reported here was to identify proteins involved in regulation of the blaA gene in S. oneidensis. We found that the loss of PBP1a/LpoA results in enhanced expression by designing and performing a colony color-based transposon mutagenesis assay. We then showed that PBP1b and LpoB had no effect on the expression of blaA. In addition, we showed that elevated blaA expression due to PBP1a depletion was independent of AmpG.
MATERIALS AND METHODS
Bacterial stains, plasmids, and culture conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. Information for all of the primers used in this study is available upon request. For genetic manipulation purposes, S. oneidensis and E. coli were cultivated aerobically in Luria-Bertani (LB) medium (Difco, Detroit, MI) at 30°C and 37°C, respectively. Where needed, the growth medium was supplemented with chemicals at the following concentrations: 2,6-diaminopimelic acid, 0.3 mM; ampicillin (AMP), 100 μg/ml; kanamycin (Km), 50 μg/ml; and gentamicin (Gm), 15 μg/ml. All chemicals were acquired from Sigma-Aldrich (Shanghai, China) unless otherwise noted.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli strains | ||
| DH5α | Host strain for plasmids | Lab stock |
| WM3064 | Donor strain for conjugation; ΔdapA | W. Metcalf, UIUC |
| S. oneidensis strains | ||
| MR-1 | Wild type | Lab stock |
| HG0280 | ΔmrcA strain derived from MR-1 | This study |
| HG0300 | ΔlpoA strain derived from MR-1 | This study |
| HG0633 | ΔmrcB strain derived from MR-1 | This study |
| HG0837 | ΔblaA strain derived from MR-1 | 39 |
| HG1060 | ΔlpoB strain derived from MR-1 | This study |
| HG3814 | ΔampG strain derived from MR-1 | 41 |
| HG0280-0837 | ΔmrcA ΔblaA strain derived from MR-1 | This study |
| HG0300-0837 | ΔlpoA ΔblaA strain derived from MR-1 | This study |
| HG0280-3814 | ΔmrcA ΔampG strain derived from MR-1 | This study |
| WT/PblaA-lacZ | Wild type harboring the PblaA-lacZ fusion | This study |
| Plasmids | ||
| pHGM01 | Apr Gmr Cmr; suicide vector | 42 |
| pHG101 | Promoterless vector for complementation | 43 |
| pHGC02 | pHG101 harboring the PBAD promoter | This study |
| pFAC | Mariner-based transposon vector | 44 |
| pHGT02 | pFAC removing the Apr gene | This study |
| pHGEI01-PblaA | pHGEI01 containing the blaA promoter | 41 |
In-frame mutagenesis and complementation.
In-frame deletion strains of S. oneidensis were constructed by the att-based fusion PCR method (42). In brief, two fragments flanking the targeted gene were amplified by PCR with primers containing attB and gene-specific sequences and then were joined by a second round of PCR (fusion PCR). The fusion fragments were introduced into plasmid pHGM01 by site-specific recombination, using BP Clonase (Invitrogen), and then transformed into E. coli WM3064. The resulting mutagenesis vectors were transferred from E. coli into the correct S. oneidensis strains via conjugation. Integration of the mutagenesis constructs into the chromosome was selected by resistance to gentamicin and verified by PCR. The correct transconjugants were grown in LB broth in the absence of NaCl and plated onto LB medium supplemented with 10% sucrose. Gentamicin-sensitive and sucrose-resistant colonies were screened by PCR for deletion of the targeted gene. The deletion mutations were then verified by sequencing.
Plasmid pHG101 was used for genetic complementation of mutants (43). A fragment containing the gene of interest and its native promoter was amplified by PCR and cloned into pHG101. To control the levels of gene expression, the araBAD promoter (PBAD) along with araC was ligated into pHG101 to construct the arabinose-inducible plasmid pHGC02. The gene of interest, generated by PCR, was introduced into pHGC02 and verified by sequencing, and the resulting vectors were transferred into the relevant strains via conjugation.
Transposon mutagenesis.
Plasmid pHGT02, derived from the widely used mariner transposon-containing plasmid pFAC (44), was used to perform transposon mutagenesis. As pFAC contains an AMP resistance (Apr) gene that interferes with screening, it was digested by ApaLI and then self-ligated to remove the Apr gene, giving pHGT02. The resulting plasmid was transferred into S. oneidensis via conjugation, and transconjugants were plated onto medium containing Gm and 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-Gal). Mutants with a blue colony phenotype were selected for further studies. The locations of the transposon insertion in the selected mutants were determined by arbitrarily primed PCR (AP-PCR) as described previously (45).
Synthetic lethality test.
To identify synthetically lethal interactions between PBP1 and Lpo proteins, a conditional ΔmrcA ΔlpoA double-deletion mutant was constructed. The mutagenesis vector for the lpoA gene, obtained by constructing an lpoA in-frame deletion strain, was introduced into the ΔmrcA strain by conjugation. pHGC02-mrcA was introduced after chromosomal integration of the mutagenesis construct. The remaining steps of the in-frame deletion procedure were carried out in the presence of arabinose. For the synthetic lethality test, late-logarithmic-phase cultures (optical density at 600 nm [OD600] ≈ 0.6) were used to prepare a decimal dilution series. Three microliters of each dilution was placed on an LB medium plate supplemented with or without 0.002% (vol/vol) arabinose. The plates were incubated for 18 h at 30°C and then photographed.
Antibiotic susceptibility assays.
The MICs of the antibiotics were determined by the broth microdilution method as recommended by the Clinical and Laboratory Standards Institute (CLSI) (46). Antibiotics used in the susceptibility assay were AMP, cefotaxime (CTX), and imipenem (IPM). All MICs were determined at least in triplicate.
Promoter activity assay.
The activity of the PblaA promoter was assayed by use of a markerless integrative lacZ reporter system as described previously (41). The PblaA-lacZ fusion was integrated into the degenerated nrfCD locus as described before (47). Cells grown with or without AMP for 3 h were harvested, and their β-galactosidase activity was determined by monitoring color development at 420 nm as described previously (43).
BlaA β-lactamase activity assay.
The specific BlaA β-lactamase activity was measured directly by nitrocefin hydrolysis as described previously (41). BlaA β-lactamase activity was expressed in nanomoles of nitrocefin hydrolyzed per minute per milligram of protein. All experiments were performed in triplicate, and the results presented are averages for the three experiments.
Statistical analyses.
Experimental values were subjected to statistical analyses and are presented as means ± standard deviations. Student's t test was performed for pairwise comparisons of groups.
RESULTS
Screening for mutants with enhanced blaA expression.
We have previously shown that expression of the blaA gene is substantially induced by AMP at high (>12.5 μg/ml) but not low (<5 μg/ml) levels in S. oneidensis (39, 41). We speculated that an S. oneidensis strain harboring the E. coli lacZ gene under the control of the blaA promoter would form white and blue colonies on X-Gal plates in the presence of AMP at different levels. To this end, a PblaA-lacZ fusion was integrated into the chromosome of the wild type at the degenerated nrfCD locus (WT/PblaA-lacZ) by homologous recombination (39, 47). As expected, blue colonies appeared on X-Gal plates supplemented with AMP at 50 but not 2.5 μg/ml (Fig. 1A; see Fig. S1 in the supplemental material). Therefore, the expression of blaA in the fusion strain was mirrored by the colony color.
FIG 1.
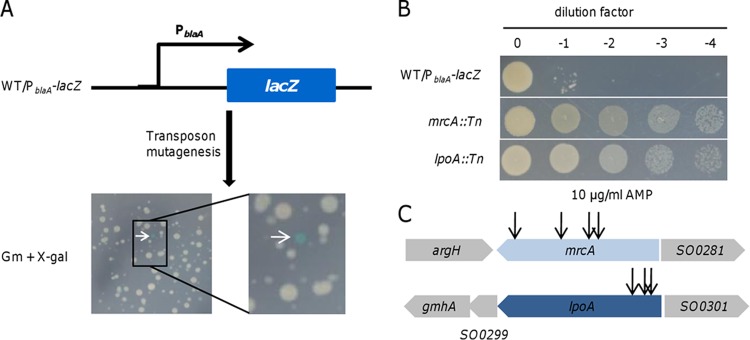
Screening for mutants with enhanced blaA expression by transposon mutagenesis. (A) Colonies of transposon mutants grown on a Gm plate harboring X-Gal. The arrow indicates a blue colony with enhanced blaA expression. (B) AMP susceptibility assay of transposon mutants. (C) Relative insertional sites of transposon mutants with hyperresistance to AMP.
To identify genes involved in regulating blaA expression, pHGT02, a mariner-based transposon vector, was used to generate a random mutant library in the background of the WT/PblaA-lacZ strain. pHGT02 was derived from pFAC by removing the AMP resistance gene, which interferes with screening (44). Apart from their application for construction of transposon insertion libraries, these vectors can also be used to unveil cryptic operons because of an embedded promoter in the transposable sequence (47, 48).
The resulting library was plated on selective medium containing X-Gal (Fig. 1A). A total of ∼20,000 colonies were screened, and 14 isolates with a blue colony phenotype were obtained. An antibiotic susceptibility assay revealed that eight of these mutants were hyperresistant to AMP, a phenotype consistent with enhanced expression of the blaA gene (Fig. 1B). The remaining six, with modest increases in resistance, were unstable, reverting to white after subculturing. Four of the highly resistant mutants had transposon insertions that mapped to the coding region of the mrcA gene, which encodes PBP1a according to the genome annotation (Fig. 1C). Interestingly, insertion sites of three mutants were identified to be within the lppC gene (Fig. 1C), whose product is predicted to be an outer membrane lipoprotein homologous to E. coli LpoA. Given that inactivation of mrcA and lppC caused similar phenotypes against AMP, it seems probable that PBP1a and LppC participate in the same physiological processes by the same mechanism as that for E. coli PBP1a/LpoA. To be consistent, we renamed the S. oneidensis lppC gene lpoA.
The last of the highly resistant isolates had a transposon insertion that mapped to the initiation codon of the mdoH gene, from an operon responsible for the biosynthesis of membrane-derived oligosaccharides (MDOs) (49). Experiments designed to identify the role of MDOs in blaA regulation are under way.
Loss of PBP1a and LpoA increases blaA expression.
To further evaluate the effects of PBP1a and LpoA on the β-lactam resistance of S. oneidensis, we deleted their coding genes from the genome individually. Both the ΔmrcA and ΔlpoA strains displayed significant defects when grown in LB (Fig. 2A). The generation times of these two mutants increased 62% and 30%, respectively, relative to that of the wild type. Not surprisingly, the resistance of both the ΔmrcA and ΔlpoA strains to tested β-lactams was strongly increased (Table 2). Compared to the wild type, the ΔlpoA strain had 8-, 4-, and 2-fold increases in resistance to AMP, IPM, and CTX, respectively. The ΔmrcA strain had additional 2- to 4-fold increases in resistance to all tested β-lactams.
FIG 2.
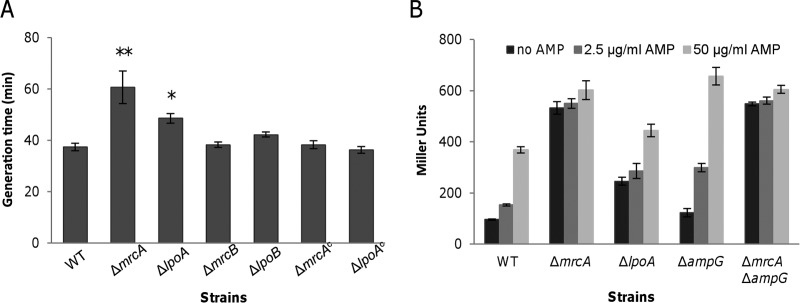
PBP1a and LpoA play important roles in cell growth and blaA expression. (A) PBP1a pathway mutants have impaired cell growth. The generation times for different strains were calculated from the OD600 values. ΔmrcAc and ΔlpoAc represent the ΔmrcA and ΔlpoA strains, respectively, complemented with pHG101 in trans. Results are averages for at least three replicates, and the error bars represent standard deviations (SD) (* and **, P < 0.05 and 0.01, respectively; two-tailed t test). (B) Enhanced β-lactam resistance in mrcA and lpoA mutants is dependent on blaA expression. PblaA promoter activities were determined by measuring β-galactosidase activity (in Miller units) by use of the PblaA-lacZ reporter system in different strains in the absence and presence of AMP at 50 μg/ml and 2.5 μg/ml. Results are averages for at least three replicates, and the error bars represent SD.
TABLE 2.
MICs of β-lactams and specific activities of BlaA in wild-type S. oneidensis and its derivative strains
| Straina | MIC (μg/ml)b |
Mean β-lactamase activityc ± SD |
|||
|---|---|---|---|---|---|
| AMP | CTX | IPM | No inducer | Inducer | |
| WT | 4 | 0.02 | 0.5 | 12 ± 0.8 | 125 ± 7 |
| ΔmrcA | 128 | 0.08 | 4 | 258 ± 22 | 228 ± 21 |
| ΔlpoA | 32 | 0.04 | 2 | 71 ± 11 | 241 ± 23 |
| ΔmrcB | 4 | 0.02 | 0.5 | 13 ± 0.8 | 116 ± 7.9 |
| ΔlpoB | 4 | 0.02 | 0.5 | 13 ± 1.2 | 128 ± 13 |
| ΔampG | 128 | 0.08 | 2 | 29 ± 4 | 238 ± 5.1 |
| ΔblaA | <0.5 | <0.005 | <0.06 | 2 ± 1 | 0 |
| ΔmrcA/mrcA | 128 | 0.08 | 4 | − | − |
| ΔmrcA/mrcA + Ara | 32 | 0.04 | 1 | 44 ± 2.3 | 227 ± 7.1 |
| ΔlpoA/lpoA | 32 | 0.04 | 2 | − | − |
| ΔlpoA/lpoA + Ara | 8 | 0.02 | 0.5 | 19 ± 0.1 | 174 ± 17 |
| ΔmrcA ΔblaA | <0.5 | <0.005 | <0.06 | 3 ± 0.5 | 0 |
| ΔlpoA ΔblaA | <0.5 | <0.005 | <0.06 | 3 ± 0.8 | 0 |
| ΔmrcA ΔampG | 128 | 0.08 | 2 | 216 ± 35 | 236 ± 18 |
ΔmrcA/mrcA and ΔlpoA/lpoA represent the ΔmrcA and ΔlpoA strains, respectively, complemented with pHGC02 harboring a copy of the corresponding S. oneidensis genes in trans. Ara, arabinose.
AMP, ampicillin; CTX, cefotaxime; IPM, imipenem.
Nanomoles of nitrocefin hydrolyzed per minute per milligram of protein. Induction was carried out with 200 μg/ml ampicillin for 2 h. −, not tested.
To confirm the observed phenotypes of the ΔmrcA and ΔlpoA strains described above, genetic complementation was carried out with the multicopy plasmid pHG101 harboring a copy of mrcA or lpoA under the control of its native promoter (43). The growth defect of these two mutants was fully corrected by expression of the corresponding genes in trans (Fig. 2A). However, although the arrangement reduced the β-lactam resistance of the ΔlpoA strain, it failed to do so with the ΔmrcA strain (see Fig. S2 in the supplemental material). Since genes within the vector are overexpressed (43, 50, 51), we supposed that this result was due to an excess of PBP1a, presumably by trapping a larger amount of β-lactams. To keep mrcA and lpoA expression at proper levels, the araBAD promoter (PBAD) along with araC was ligated into pHG101 to construct a tightly regulated plasmid, pHGC02. In the absence of arabinose, the resistance to β-lactams of both the ΔmrcA and ΔlpoA strains carrying the respective genes within pHGC02 for complementation was unaffected (Table 2). When arabinose was added to the cultures, the β-lactam MICs of these two mutants with proper complementation vectors were reduced 2- to 4-fold (Table 2). These results indicate that the phenotypes observed for the ΔmrcA and ΔlpoA strains were due to the intended mutations.
To validate that the elevated resistance to tested β-lactams of the ΔmrcA and ΔlpoA strains was due to enhanced expression of the blaA gene, a markerless integrative lacZ reporter system and nitrocefin hydrolysis were employed to determine the activity of the blaA promoter (PblaA) and the BlaA β-lactamase activity, respectively (41). The expression levels of β-galactosidase driven by the blaA promoter in the ΔmrcA and ΔlpoA strains were substantially higher than that in the wild type under all tested conditions (Fig. 2B). In particular, expression of lacZ in the ΔmrcA strain was constant at high levels, independently of AMP, suggesting that the blaA gene is constitutively expressed in the absence of PBP1a (Fig. 2B). Consistently, the basal levels of BlaA activity in the ΔmrcA strain were approximately equal to that induced by AMP at 200 μg/ml, which was ∼2-fold higher than the induced level in the wild type (Table 2). The ΔlpoA strain also had drastically increased basal levels of BlaA activity, i.e., approximately 6-fold higher than that of the wild type. Unlike that in the ΔmrcA strain, BlaA activity in the ΔlpoA strain could be induced by 200 μg/ml AMP, with an ∼3.4-fold increase compared to the level without induction (Table 2). Moreover, expression of mrcA and lpoA under the control of PBAD significantly decreased the basal levels of BlaA activity in the ΔmrcA and ΔlpoA strains, respectively (Table 2). Importantly, deletion of blaA from either the ΔmrcA or ΔlpoA strain completely disabled growth in the presence of β-lactams (Table 2). Collectively, these data indicate that inactivation of PBP1a or LpoA raises the level of blaA expression.
Loss of PBP1b has no effect on blaA expression.
Like many other Gram-negative bacteria, S. oneidensis possesses three major class A PBPs: PBP1a, PBP1b, and PBP1c. The mrcB gene (encoding PBP1b) was not revealed in our screening, although transposon events in mrcA occurred multiple times. Given that these two genes are similar in length and that the number of total interruptions is sufficiently large to cover the entire genome, it is conceivable that PBP1b differs from PBP1a in its role in regulating blaA expression.
To test the involvement of PBP1b in the process, a strain devoid of the mrcB gene was constructed. In contrast to the ΔmrcA strain, neither growth nor the MICs for AMP, CTX, and IPM were affected in this strain (Fig. 2A and Table 2). Consistently, both basal and induced levels of BlaA activity in the ΔmrcB strain were similar to those in the wild type (Table 2). These results indicate that PBP1a and PBP1b differ from each other in mediating blaA expression in S. oneidensis.
SO1060 (LpoB) is likely the lipoprotein cofactor for PBP1b.
Similar to LpoA for PBP1a, the outer membrane lipoprotein LpoB is required for PBP1b function in E. coli (27, 28). Typas et al. (28) proposed that LpoA and PBP1a are evolutionarily limited to gammaproteobacteria but that LpoB and its docking domain (UB2H) in PBP1b are further restricted to enterobacteria. However, a recent study demonstrated that Vibrio cholerae contains both LpoB and the UB2H domain, indicating that LpoB is present in bacteria beyond enterobacteria (52). S. oneidensis, a gammaproteobacterium, possesses the UB2H domain in PBP1b (residues 73 to 157) (see Fig. S3 in the supplemental material), implying that a homologue of LpoB may have evolved in this bacterium.
To identify the lipoprotein cofactor for PBP1b, we performed a BLAST search using E. coli LpoB, revealing a single putative homologue, SO1060 (E value, 3e−13). While the protein sequence of SO1060 shares only 49% similarity and 28% identity with that of E. coli LpoB (59% coverage), the identity with V. cholerae LpoB (95% coverage) is 67%. In addition, SO1060 is predicted to be a lipoprotein, as it contains a lipobox motif with the consensus sequence L(A/S)(G/A)C (see Fig. S3) (27, 53).
To determine whether SO1060 is the outer membrane lipoprotein that works with PBP1b, we deleted the gene and characterized the resulting mutant. Neither growth nor the MICs for β-lactams were affected in the strain, a scenario observed with the ΔmrcB strain (Fig. 2A and Table 2). Subsequently, we made attempts to delete the SO1060 gene from the ΔmrcA, ΔlpoA, and ΔmrcB strains. As we expected, the ΔmrcB ΔSO1060 strain, but not the ΔmrcA ΔSO1060 or ΔlpoA ΔSO1060 strain, was successfully obtained. The phenotypes of the ΔmrcB ΔSO1060 strain with respect to growth and β-lactam resistance were identical to those of the wild-type, ΔmrcB, and ΔSO1060 strains (data not shown). These results indicate that the SO1060 gene likely encodes the counterpart of E. coli LpoB, as it could not be inactivated simultaneously with mrcA or lpoA.
To further confirm that SO1060 is essential for PBP1b function, we employed the pBAD inducible system to construct synthetically lethal mutants. The lpoA, mrcB, and SO1060 genes were deleted from a ΔmrcA strain which carried a copy of the gene within pHGC02 in trans. In parallel, mrcA, mrcB, and SO1060 were deleted from the ΔlpoA strain by the same strategy. In total, six strains were obtained, and all grew normally in LB supplemented with arabinose (Fig. 3). In the absence of arabinose, normal growth was observed with the ΔmrcA strain lacking LpoA and the ΔlpoA strain without PBP1a. In contrast, the loss of PBP1b or SO1060 upon mrcA depletion caused synthetic lethality. Similar results were obtained with the ΔlpoA strain. These results indicate that, similar to LpoA as the essential cofactor for PBP1a, SO1060 is required for the function of PBP1b. Overall, we concluded that SO1060 is the lipoprotein cofactor for PBP1b, and we thus designated its coding gene lpoB.
FIG 3.
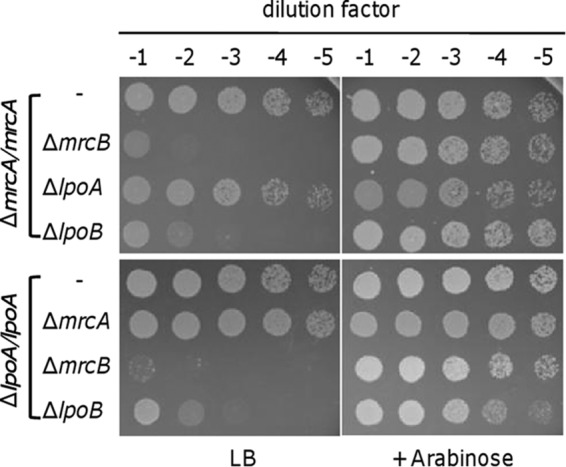
Synthetic lethality assay for PBP1a/LpoA and PBP1b/LpoB. ΔmrcA/mrcA and ΔlpoA/lpoA represent the ΔmrcA and ΔlpoA strains, respectively, complemented with pHGC02 harboring a copy of the corresponding S. oneidensis genes in trans. Cells from the late logarithmic phase (OD600 ≈ 0.6) were used to prepare a decimal dilution series. Three microliters of each dilution was placed on an LB plate supplemented with or without 0.002% arabinose.
PBP1a and AmpG mediate blaA expression via independent pathways.
It has been reported that inactivation of mrcA elevates the basal expression of L1 and L2 β-lactamases in Stenotrophomonas maltophilia, which is dependent on AmpR and AmpG (54). To assess whether PBP1a-mediated blaA expression is dependent on AmpG in S. oneidensis, we constructed a double mutant lacking mrcA and ampG for β-lactam susceptibility testing (Table 2). The ΔmrcA, ΔampG, and ΔmrcA ΔampG strains displayed similar levels of resistance to all tested β-lactams. However, differences between the ΔmrcA and ΔampG strains were observed by direct analyses of BlaA activity and blaA expression (Fig. 2B and Table 2). The basal levels of BlaA activity were constitutively robust in the ΔmrcA strain, contrasting with the relatively low but inducible levels in the ΔampG strain. Similar results were obtained in the PblaA activity assay. Importantly, the BlaA activities in the ΔmrcA ΔampG strain were comparable to those in the ΔmrcA strain (Table 2), indicating that PBP1a-mediated blaA expression is independent of AmpG.
DISCUSSION
Regulation of the ampC β-lactamase gene has been studied most intensively in the Enterobacteriaceae and P. aeruginosa (5–11), in which the LysR-type transcriptional regulator AmpR and peptidoglycan recycling are required. However, this paradigm is not applicable to the class D β-lactamase gene blaA in S. oneidensis, which lacks an AmpR homologue (40, 41). As a result, the loss of AmpG on expression of these two β-lactamase genes in the respective bacteria elicits contrasting effects. More importantly, the blaA gene in the S. oneidensis ampG mutant remains inducible, implicating the presence of a parallel regulatory pathway for blaA induction that is independent of AmpG (41). Here we present evidence to demonstrate that the targets of β-lactams play an important role in blaA expression. Using transposon mutagenesis, we found that inactivation of PBP1a or its lipoprotein cofactor LpoA substantially elevates blaA expression, conferring the bacterium with hyperresistance to many β-lactams. Although the complex of PBP1b and LpoB is functionally redundant, to some extent, with that of PBP1a/LpoA for peptidoglycan synthesis, the roles of the former in blaA expression are dispensable.
In E. coli, PBP1a and PBP1b are bifunctional enzymes containing both GTase and TPase domains, and the outer membrane lipoproteins LpoA and LpoB are essential cofactors for PBP1a and PBP1b function, respectively (27, 28). It has been shown that these cofactors improve both the GTase and TPase activities of their cognate PBPs (29). Although LpoB was initially predicted to be limited to enterobacteria (28), a homologue of LpoB was recently identified in V. cholerae (52), expanding the scope of bacteria that adopt this mechanism for regulation of PBP1s. In line with this, the present study demonstrated that LpoA and LpoB also exist in S. oneidensis. Like E. coli LpoA and LpoB, S. oneidensis counterparts are essential for their cognate PBP function. At least one of the PBP1/Lpo complexes is required for cell survival.
Although PBP1a/LpoA and PBP1b/LpoB are synthetically lethal and thus largely complement each other, S. oneidensis strains lacking each complex display somewhat different phenotypes. It is evident that the physiological roles of PBP1a/LpoA are more profound than those of PBP1b/LpoB, as growth is significantly affected by the loss of the former but not the latter. This difference coincides with those observed with the homologous protein pairs studied in other Gram-negative bacteria (24, 52). Additionally, the sensitivities of the E. coli PBP1b/LpoB and V. cholerae PBP1a/LpoA mutants to β-lactams are substantially increased, which may result from the high affinity between β-lactams and the remaining PBP1 protein (52, 55).
In S. oneidensis, the removal of PBP1a/LpoA leads to enhanced resistance to β-lactams, a scenario also observed in S. maltophilia (54, 56, 57). However, the regulatory mechanisms of PBP1a on β-lactamase expression utilized by these two microorganisms are certainly different. The regulation in S. maltophilia is dependent on AmpG and AmpR (54). Although both the S. oneidensis ΔampG and ΔmrcA strains have increased resistance to β-lactams, the blaA gene in the former remains inducible by AMP, but not that in the latter (41). Moreover, it appears that PBP1a functionally overwhelms AmpG in basal expression of blaA and that the pathways involving these two proteins have no additive effect, because the blaA expression in the ΔmrcA ΔampG strain is comparable to that in the mrcA single mutant strain. Given that S. oneidensis lacks AmpR and its PBP1a/LpoA-mediated blaA expression is independent of AmpG, it is undoubted that the mechanisms by which PBP1a and LpoA regulate β-lactamase expression are novel.
Generally, strains lacking PBP1 or its cognate Lpo display identical phenotypes (27, 28, 52). Nevertheless, our data showed a significant difference between S. oneidensis PBP1a- and LpoA-deficient mutants with respect to blaA expression. Compared to the strain lacking PBP1a, the ΔlpoA strain has a lower basal level of blaA expression and remains inducible by AMP. Given that LpoA increases the GTase and TPase activities of PBP1a (29), it is reasonable to assume that the enzymatic activities of PBP1a account for the elevated BlaA production. In the absence of LpoA, PBP1a retains a share of the GTase and TPase activities, thus leading to an unsaturated basal level of blaA expression.
Except for PBP1, LMW PBPs are also involved in the regulation of β-lactamase expression in Gram-negative bacteria. Compared to the mysterious effects of PBP1a, loss of PBP4 (DacB) triggers overexpression of the β-lactamase AmpC mediated by the CreBC (BlrAB) two-component system in P. aeruginosa and Aeromonas spp. (58, 59). In S. oneidensis, our previous results demonstrate that inactivation of PBP5 (DacA) decreases the induced level of blaA expression via an unknown mechanism (39). Although the homologues of AmpR and CreBC are absent in S. oneidensis, it cannot be ruled out that other regulators are involved in the PBP1a-mediated blaA expression pathway.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Natural Science Foundation of China (grants 31270097 and 41476105), the Major State Basic Research Development Program (973 Program; grant 2010CB833803), and the Doctoral Fund of the Ministry of Education of China (grant 20130101110142).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04669-14.
REFERENCES
- 1.Typas A, Banzhaf M, Gross CA, Vollmer W. 2012. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zapun A, Contreras-Martel C, Vernet T. 2008. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 32:361–385. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 3.Poole K. 2004. Resistance to beta-lactam antibiotics. Cell Mol Life Sci 61:2200–2223. doi: 10.1007/s00018-004-4060-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilke MS, Lovering AL, Strynadka NC. 2005. Beta-lactam antibiotic resistance: a current structural perspective. Curr Opin Microbiol 8:525–533. doi: 10.1016/j.mib.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 5.Dietz H, Pfeifle D, Wiedemann B. 1997. The signal molecule for beta-lactamase induction in Enterobacter cloacae is the anhydromuramyl-pentapeptide. Antimicrob Agents Chemother 41:2113–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanson N, Sanders C. 1999. Regulation of inducible AmpC beta-lactamase expression among Enterobacteriaceae. Curr Pharm Des 5:881–894. [PubMed] [Google Scholar]
- 7.Jacobs C, Frère J-M, Normark S. 1997. Cytosolic intermediates for cell wall biosynthesis and degradation control inducible beta-lactam resistance in Gram-negative bacteria. Cell 88:823–832. doi: 10.1016/S0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs C, Huang L, Bartowsky E, Normark S, Park J. 1994. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for beta-lactamase induction. EMBO J 13:4684–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs C, Joris B, Jamin M, Klarsov K, Beeumen J, Mengin-Lecreulx D, Heijenoort J, Park JT, Normark S, Frère JM. 1995. AmpD, essential for both beta-lactamase regulation and cell wall recycling, is a novel cytosolic N-acetylmuramyl-l-alanine amidase. Mol Microbiol 15:553–559. doi: 10.1111/j.1365-2958.1995.tb02268.x. [DOI] [PubMed] [Google Scholar]
- 10.Juan C, Moyá B, Pérez JL, Oliver A. 2006. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level beta-lactam resistance involves three AmpD homologues. Antimicrob Agents Chemother 50:1780–1787. doi: 10.1128/AAC.50.5.1780-1787.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindberg F, Westman L, Normark S. 1985. Regulatory components in Citrobacter freundii ampC beta-lactamase induction. Proc Natl Acad Sci U S A 82:4620–4624. doi: 10.1073/pnas.82.14.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindquist S, Weston-Hafer K, Schmidt H, Pul C, Korfmann G, Erickson J, Sanders C, Martin HH, Normark S. 1993. AmpG, a signal transducer in chromosomal beta-lactamase induction. Mol Microbiol 9:703–715. doi: 10.1111/j.1365-2958.1993.tb01731.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Bao Q, Gagnon LA, Huletsky A, Oliver A, Jin S, Langaee T. 2010. ampG gene of Pseudomonas aeruginosa and its role in beta-lactamase expression. Antimicrob Agents Chemother 54:4772–4779. doi: 10.1128/AAC.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chahboune A, Decaffmeyer M, Brasseur R, Joris B. 2005. Membrane topology of the Escherichia coli AmpG permease required for recycling of cell wall anhydromuropeptides and AmpC beta-lactamase induction. Antimicrob Agents Chemother 49:1145–1149. doi: 10.1128/AAC.49.3.1145-1149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korfmann G, Sanders CC. 1989. ampG is essential for high-level expression of AmpC beta-lactamase in Enterobacter cloacae. Antimicrob Agents Chemother 33:1946–1951. doi: 10.1128/AAC.33.11.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zamorano L, Reeve TM, Juan C, Moyá B, Cabot G, Vocadlo DJ, Mark BL, Oliver A. 2011. AmpG inactivation restores susceptibility of pan-beta-lactam-resistant Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother 55:1990–1996. doi: 10.1128/AAC.01688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balcewich MD, Reeve TM, Orlikow EA, Donald LJ, Vocadlo DJ, Mark BL. 2010. Crystal structure of the AmpR effector binding domain provides insight into the molecular regulation of inducible AmpC beta-lactamase. J Mol Biol 400:998–1010. doi: 10.1016/j.jmb.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 18.Bartowsky E, Normark S. 1991. Purification and mutant analysis of Citrobacter freundii AmpR, the regulator for chromosomal AmpC beta-lactamase. Mol Microbiol 5:1715–1725. doi: 10.1111/j.1365-2958.1991.tb01920.x. [DOI] [PubMed] [Google Scholar]
- 19.Bartowsky E, Normark S. 1993. Interactions of wild-type and mutant AmpR of Citrobacter freundii with target DNA. Mol Microbiol 10:555–565. doi: 10.1111/j.1365-2958.1993.tb00927.x. [DOI] [PubMed] [Google Scholar]
- 20.Kuga A, Okamoto R, Inoue M. 2000. ampR gene mutations that greatly increase class C beta-lactamase activity in Enterobacter cloacae. Antimicrob Agents Chemother 44:561–567. doi: 10.1128/AAC.44.3.561-567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindquist S, Lindberg F, Normark S. 1989. Binding of the Citrobacter freundii AmpR regulator to a single DNA site provides both autoregulation and activation of the inducible ampC beta-lactamase gene. J Bacteriol 171:3746–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. 2006. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol Rev 30:673–691. doi: 10.1111/j.1574-6976.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- 23.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 24.Banzhaf M, van den Berg van Saparoea B, Terrak M, Fraipont C, Egan A, Philippe J, Zapun A, Breukink E, Nguyen-Distèche M, den Blaauwen T. 2012. Cooperativity of peptidoglycan synthases active in bacterial cell elongation. Mol Microbiol 85:179–194. doi: 10.1111/j.1365-2958.2012.08103.x. [DOI] [PubMed] [Google Scholar]
- 25.Bertsche U, Kast T, Wolf B, Fraipont C, Aarsman ME, Kannenberg K, Von Rechenberg M, Nguyen-Distèche M, Den Blaauwen T, Höltje JV. 2006. Interaction between two murein (peptidoglycan) synthases, PBP3 and PBP1B, in Escherichia coli. Mol Microbiol 61:675–690. doi: 10.1111/j.1365-2958.2006.05280.x. [DOI] [PubMed] [Google Scholar]
- 26.Müller P, Ewers C, Bertsche U, Anstett M, Kallis T, Breukink E, Fraipont C, Terrak M, Nguyen-Distèche M, Vollmer W. 2007. The essential cell division protein FtsN interacts with the murein (peptidoglycan) synthase PBP1B in Escherichia coli. J Biol Chem 282:36394–36402. doi: 10.1074/jbc.M706390200. [DOI] [PubMed] [Google Scholar]
- 27.Paradis-Bleau C, Markovski M, Uehara T, Lupoli TJ, Walker S, Kahne DE, Bernhardt TG. 2010. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143:1110–1120. doi: 10.1016/j.cell.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Typas A, Banzhaf M, van den Berg van Saparoea B, Verheul J, Biboy J, Nichols RJ, Zietek M, Beilharz K, Kannenberg K, von Rechenberg M. 2010. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 143:1097–1109. doi: 10.1016/j.cell.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lupoli TJ, Lebar MD, Markovski M, Bernhardt T, Kahne D, Walker S. 2014. Lipoprotein activators stimulate Escherichia coli penicillin-binding proteins by different mechanisms. J Am Chem Soc 136:52–55. doi: 10.1021/ja410813j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fredrickson JK, Romine MF, Beliaev AS, Auchtung JM, Driscoll ME, Gardner TS, Nealson KH, Osterman AL, Pinchuk G, Reed JL. 2008. Towards environmental systems biology of Shewanella. Nat Rev Microbiol 6:592–603. doi: 10.1038/nrmicro1947. [DOI] [PubMed] [Google Scholar]
- 31.Konstantinidis KT, Serres MH, Romine MF, Rodrigues JL, Auchtung J, McCue L-A, Lipton MS, Obraztsova A, Giometti CS, Nealson KH. 2009. Comparative systems biology across an evolutionary gradient within the Shewanella genus. Proc Natl Acad Sci U S A 106:15909–15914. doi: 10.1073/pnas.0902000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Héritier C, Poirel L, Nordmann P. 2004. Genetic and biochemical characterization of a chromosome-encoded carbapenem-hydrolyzing Ambler class D beta-lactamase from Shewanella algae. Antimicrob Agents Chemother 48:1670–1675. doi: 10.1128/AAC.48.5.1670-1675.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janda JM, Abbott SL. 2014. The genus Shewanella: from the briny depths below to human pathogen. Crit Rev Microbiol 40:293–312. doi: 10.3109/1040841X.2012.726209. [DOI] [PubMed] [Google Scholar]
- 34.Poirel L, Héritier C, Nordmann P. 2004. Chromosome-encoded Ambler class D beta-lactamase of Shewanella oneidensis as a progenitor of carbapenem-hydrolyzing oxacillinase. Antimicrob Agents Chemother 48:348–351. doi: 10.1128/AAC.48.1.348-351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poirel L, Héritier C, Nordmann P. 2005. Genetic and biochemical characterization of the chromosome-encoded class B beta-lactamases from Shewanella livingstonensis (SLB-1) and Shewanella frigidimarina (SFB-1). J Antimicrob Chemother 55:680–685. doi: 10.1093/jac/dki065. [DOI] [PubMed] [Google Scholar]
- 36.Poirel L, Rodriguez-Martinez J-M, Mammeri H, Liard A, Nordmann P. 2005. Origin of plasmid-mediated quinolone resistance determinant QnrA. Antimicrob Agents Chemother 49:3523–3525. doi: 10.1128/AAC.49.8.3523-3525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potron A, Poirel L, Nordmann P. 2011. Origin of OXA-181, an emerging carbapenem-hydrolyzing oxacillinase, as a chromosomal gene in Shewanella xiamenensis. Antimicrob Agents Chemother 55:4405–4407. doi: 10.1128/AAC.00681-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heidelberg JF, Paulsen IT, Nelson KE, Gaidos EJ, Nelson WC, Read TD, Eisen JA, Seshadri R, Ward N, Methe B. 2002. Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat Biotechnol 20:1118–1123. doi: 10.1038/nbt749. [DOI] [PubMed] [Google Scholar]
- 39.Yin J, Sun L, Dong Y, Chi X, Zhu W, Qi S-h, Gao H. 2013. Expression of blaA underlies unexpected ampicillin-induced cell lysis of Shewanella oneidensis. PLoS One 8:e60460. doi: 10.1371/journal.pone.0060460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walther-Rasmussen J, Hoiby N. 2006. OXA-type carbapenemases. J Antimicrob Chemother 57:373–383. doi: 10.1093/jac/dki482. [DOI] [PubMed] [Google Scholar]
- 41.Yin J, Mao Y, Ju L, Jin M, Sun Y, Jin S, Gao H. 2014. Distinct roles of major peptidoglycan recycling enzymes in beta-lactamase production in Shewanella oneidensis. Antimicrob Agents Chemother 58:6536–6543. doi: 10.1128/AAC.03238-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin M, Jiang Y, Sun L, Yin J, Fu H, Wu G, Gao H. 2013. Unique organizational and functional features of the cytochrome c maturation system in Shewanella oneidensis. PLoS One 8:e75610. doi: 10.1371/journal.pone.0075610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu L, Wang J, Tang P, Chen H, Gao H. 2011. Genetic and molecular characterization of flagellar assembly in Shewanella oneidensis. PLoS One 6:e21479. doi: 10.1371/journal.pone.0021479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong SM, Mekalanos JJ. 2000. Genetic footprinting with mariner-based transposition in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 97:10191–10196. doi: 10.1073/pnas.97.18.10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das S, Noe JC, Paik S, Kitten T. 2005. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J Microbiol Methods 63:89–94. doi: 10.1016/j.mimet.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 46.Clinical and Laboratory Standards Institute. 2014. Performance standards for antimicrobial susceptibility testing; 24th informational supplement. CLSI document M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 47.Fu H, Chen H, Wang J, Zhou G, Zhang H, Zhang L, Gao H. 2013. Crp-dependent cytochrome bd oxidase confers nitrite resistance to Shewanella oneidensis. Environ Microbiol 15:2198–2212. doi: 10.1111/1462-2920.12091. [DOI] [PubMed] [Google Scholar]
- 48.Fu H, Jin M, Ju L, Mao Y, Gao H. 2014. Evidence for function overlapping of CymA and the cytochrome bc1 complex in the Shewanella oneidensis nitrate and nitrite respiration. Environ Microbiol 16:3181–3195. doi: 10.1111/1462-2920.12457. [DOI] [PubMed] [Google Scholar]
- 49.Ebel W, Vaughn GJ, Peters HK III, Trempy JE. 1997. Inactivation of mdoH leads to increased expression of colanic acid capsular polysaccharide in Escherichia coli. J Bacteriol 179:6858–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong Y, Wang J, Fu H, Zhou G, Shi M, Gao H. 2012. A Crp-dependent two-component system regulates nitrate and nitrite respiration in Shewanella oneidensis. PLoS One 7:e51643. doi: 10.1371/journal.pone.0051643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun L, Dong Y, Shi M, Jin M, Zhou Q, Luo Z-Q, Gao H. 2014. Two residues predominantly dictate functional difference in motility between Shewanella oneidensis flagellins FlaA and FlaB. J Biol Chem 289:14547–14559. doi: 10.1074/jbc.M114.552000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorr T, Moll A, Chao MC, Cava F, Lam H, Davis BM, Waldor MK. 2014. Differential requirement for PBP1a and PBP1b in in vivo and in vitro fitness of Vibrio cholerae. Infect Immun 82:2115–2124. doi: 10.1128/IAI.00012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tokuda H, Matsuyama S. 2004. Sorting of lipoproteins to the outer membrane in E. coli. Biochim Biophys Acta 1693:5–13. doi: 10.1016/j.bbamcr.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 54.Lin C-W, Lin H-C, Huang Y-W, Chung T-C, Yang T-C. 2011. Inactivation of mrcA gene derepresses the basal-level expression of L1 and L2 beta-lactamases in Stenotrophomonas maltophilia. J Antimicrob Chemother 66:2033–2037. doi: 10.1093/jac/dkr276. [DOI] [PubMed] [Google Scholar]
- 55.Sarkar SK, Dutta M, Kumar A, Mallik D, Ghosh AS. 2012. Sub-inhibitory cefsulodin sensitization of E. coli to beta-lactams is mediated by PBP1b inhibition. PLoS One 7:e48598. doi: 10.1371/journal.pone.0048598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang Y-W, Hu R-M, Lin C-W, Chung T-C, Yang T-C. 2012. NagZ-dependent and NagZ-independent mechanisms for beta-lactamase expression in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 56:1936–1941. doi: 10.1128/AAC.05645-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Talfan A, Mounsey O, Charman M, Townsend E, Avison MB. 2013. Involvement of mutation in ampD I, mrcA, and at least one additional gene in beta-lactamase hyperproduction in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 57:5486–5491. doi: 10.1128/AAC.01446-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moya B, Dötsch A, Juan C, Blázquez J, Zamorano L, Haussler S, Oliver A. 2009. β-Lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog 5:e1000353. doi: 10.1371/journal.ppat.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tayler AE, Ayala JA, Niumsup P, Westphal K, Baker JA, Zhang L, Walsh TR, Wiedemann B, Bennett PM, Avison MB. 2010. Induction of beta-lactamase production in Aeromonas hydrophila is responsive to beta-lactam-mediated changes in peptidoglycan composition. Microbiology 156:2327–2335. doi: 10.1099/mic.0.035220-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.