

Abstract
Artemisinin derivatives are used in combination with other antimalarial drugs for treatment of multidrug-resistant malaria worldwide. Clinical resistance to artemisinin recently emerged in southeast Asia, yet in vitro phenotypes for discerning mechanism(s) of resistance remain elusive. Here, we describe novel phenotypic resistance traits expressed by artemisinin-resistant Plasmodium falciparum. The resistant parasites exhibit altered patterns of development that result in reduced exposure to drug at the most susceptible stage of development in erythrocytes (trophozoites) and increased exposure in the most resistant stage (rings). In addition, a novel in vitro delayed clearance assay (DCA) that assesses drug effects on asexual stages was found to correlate with parasite clearance half-life in vivo as well as with mutations in the Kelch domain gene associated with resistance (Pf3D7_1343700). Importantly, all of the resistance phenotypes were stable in cloned parasites for more than 2 years without drug pressure. The results demonstrate artemisinin-resistant P. falciparum has evolved a novel mechanism of phenotypic resistance to artemisinin drugs linked to abnormal cell cycle regulation. These results offer insights into a novel mechanism of drug resistance in P. falciparum and new tools for monitoring the spread of artemisinin resistance.
INTRODUCTION
Artemisinin combination therapy (ACT) is the recommended treatment for multidrug-resistant malaria by the World Health Organization. These combinations rely on the fast-acting properties of the artemisinin derivative to rapidly clear parasites with the longer-acting partner drug clearing residual parasites, reducing the frequent observation of recrudescent infections when artemisinin drugs are used alone. Unfortunately, resistance to artemisinin derivatives has emerged in southeast Asia and now threatens the utility of the most important treatment options for Plasmodium falciparum malaria that is resistant to most antimalarial drugs. Clinical resistance to artemisinin is expressed as a reduced rate of parasite clearance from peripheral blood, resulting in a parasite clearance half-life of >5 h (1). Resistance to artemisinin is a heritable trait, linked to mutations in the Kelch propeller domains of Pf3D7_1343700, and it appears to be spreading in southeast Asia (1–3). Clearly, a public health disaster is looming if artemisinin resistance spreads globally, like the selective sweeps previously observed for chloroquine and pyrimethamine resistance (4–6).
Despite the well-characterized phenotype of clinical resistance to artemisinin, resistance phenotypes in classical in vitro drug susceptibility assays, used successfully for other antimalarial drugs, have not been useful for detecting artemisinin resistance (7). Two modified in vitro assays have been used to assess resistance with some success (8, 9), with both assessing reduced susceptibility in the ring stage of development in erythrocytes; however, stable in vitro phenotypes in culture-adapted parasites remain elusive. Given the apparent complexities of artemisinin resistance, we hypothesized that artemisinin-resistant parasites have evolved novel mechanisms of resistance that are not evident with previously reported phenotypes, and that possible fitness defects result in loss of resistance phenotypes and genotypes in vitro. In this study, we culture adapted and promptly cloned artemisinin-resistant P. falciparum. The clones then were used to demonstrate novel, stable resistance phenotypes that can be used to elucidate mechanisms of resistance to the most important class of antimalarial drugs. Remarkably, the artemisinin resistance phenotypes include dramatic alterations in intraerythrocytic development in the absence or presence of artemisinin drugs.
MATERIALS AND METHODS
Adapting patient samples to in vitro culture.
Cryopreserved infected erythrocytes were received on dry ice and immediately stored in liquid nitrogen. Modified thawing and culturing methods were used (10). Initially, patient samples were cultured with 15% heat-inactivated human AB+ plasma at 3% hematocrit. Once reaching 2% parasitemia, samples were immediately frozen in glycerolyte. In addition, isolates were cloned by limiting dilution to preserve the heterogeneity of the parasite population within a patient sample (11). Clones were immediately cryopreserved, and drug susceptibility was determined for clones and isolates from culture-adapted patient samples. Parasitized red blood cells of parental strains and clones were frozen for genomic DNA extraction, and filter spots were made for further analysis. Clones maintained in culture after an initial freeze-thaw were cryopreserved, and genomic DNA was extracted over the course of 2 years of the study. Additional details about patient samples adapted to in vitro culture can be found in Table S1 in the supplemental material.
Phenotype assays. (i) Time-zero (T0) [3H]hypoxanthine drug susceptibility assay.
Susceptibility of isolates and cloned progeny to artemisinin derivatives (artemisinin, dihydroartemisinin, artelinic acid, and artesunate) were analyzed by a modified [3H]hypoxanthine incorporation assay (8, 12). Artemisinin and artelinic acid (AL) were included in the artemisinin panel of drugs, in addition to clinically relevant dihydroartemisinin (DHA) and artesunate because of the large decrease in susceptibility observed with in vitro-selected artemisinin-resistant parasites (8). [3H]hypoxanthine and drugs were added to culture at the start of the assay and allowed to incubate for 48 h, and radiolabeled cells were harvested. The 50% inhibitory concentration (IC50) of mefloquine was determined for all parasites by adding [3H]hypoxanthine 24 h after drug exposure (standard [3H]hypoxanthine incorporation assay) (12).
(ii) Parasite clearance and recrudescence in vitro.
Late-stage parasites were synchronized using a magnetically activated cell sorting (MACS) column (Miltenyi Biotec). Once entering ring stage, synchronized parasites were exposed to 700 nM dihydroartemisinin (DHA) for 6 h at 1.5% parasitemia. DHA exposure was repeated 24 and 48 h after initial exposure. After 6 h of drug exposure, parasites were washed with RPMI medium three times. Giemsa smears were used to quantify parasitemia at hours 0, 6, 12, 18, 24, 36, and 48. These data were used to generate parasite clearance curves and parasite reduction ratios (PRRs). In a separate study, parasites were exposed to 30 nM DHA for 6 h, the drug was washed out, and parasite development was monitored by microscopy. Parasite maturation following drug exposure was measured by categorizing 100 parasites per sample at each of the above-mentioned time points. Parasites were categorized based on early- or late-stage normal morphology (ring or trophozoite/schizont). Parasites with dormant ring or pyknotic, dead forms were considered one category; therefore, parasite clearance rates and PRRs were based upon the prevalence of parasites with normal ring, trophozoite, or schizont morphology. Beginning at day 3, smears were made and stained with Giemsa every 24 h to observe recrudescence. Cultures were maintained under standard culturing conditions until parasitemia reached ≥1.5%. Three clones were randomly selected from each isolate, and clearance/recrudescence assays were run in duplicate. Clearance and recrudescence rates were compared to those of W2, an artemisinin-susceptible laboratory clone.
Parasite growth in the absence of drug pressure.
Two methods were used to monitor parasite growth: flow cytometry and microscopy of Giemsa-stained blood smears. A modified flow cytometry method was used to observe parasite growth under standard culturing conditions in the absence of drug pressure (13). Late-stage parasites were synchronized using a MACS magnetic column (Miltenyi Biotec, Auburn, CA). A 10-ml culture at 1.5% parasitemia was initiated at the start of the assay once parasites matured to ring stage. Every 2 h for approximately 60 h, a blood smear was made and 50 μl of culture, in triplicate, was fixed in 0.05% glutaraldehyde in a 96-well round-bottom plate for flow cytometry analysis. Flow cytometry samples were stained with ethidium bromide and analyzed as described by Balu et al. (13). The percentage of ring and schizont for 3 samples was averaged and plotted over time (standard deviations [SD], ≤0.025). Cell cycle progression of patient clones was compared to that of the W2 laboratory clone under standard culture conditions.
Stage-specific parasite viability following DHA exposure.
Late-stage parasites were synchronized using a MACS magnet column (Miltenyi Biotec). Once reaching ring stage, parasites were exposed to 700 nM DHA for 3 h, and the number of visible parasites was determined using a serial dilution method as described previously (14). Exposure was repeated with a new aliquot of parasite culture every 3 h for one complete life cycle (up to 48 h). The log of viable parasites was compared between resistant and sensitive parasites 14 days after exposure for each time point along the parasite life cycle.
Genetic determinants of resistance.
Genomic DNA was extracted using a phenol-chloroform method. Parasitized red blood cells were frozen at −80°C for at least 24 h and treated with 0.5% saponin to remove red blood cells from extract. Parasite pellet was frozen for 12 to 24 h in Tris-EDTA buffer. Thawed pellet was treated with 4% SDS and 20 mg/ml RNase (Life Technologies, Grand Island, NY). Equal volumes of phenol-chloroform-isoamylalcohol (25:24:1) were added and spun at 14,000 rpm for 5 min at room temperature. The upper aqueous phase was transferred to a new tube. This separation step was repeated with chloroform-isoamylalcohol (24:1) and then chloroform. The final aqueous transfer was treated with 3 M potassium acetate and 100% ethanol. Pellet was washed with 100% ethanol and suspended in Tris-EDTA buffer. Genomic DNA was quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA).
SNPs associated with artemisinin resistance.
Pyrosequencing was used to quantify allele frequency of known single-nucleotide polymorphisms (SNPs) associated with artemisinin resistance. PCR and sequencing primers were designed using Biotage Pyrosequencing Assay Design software (Charlottesville, VA) to amplify SNP-containing genes. Primers used in this study are listed in Table S2 in the supplemental material. A 5′ biotin label was added to the PCR primer binding to the opposite strand of the sequencing primer. Each 50-μl PCR mixture contained 31.5 μl sterile water, 10 μl 5× HF buffer, 1 μl 10 mM deoxynucleoside triphosphates (dNTPs), 2 μl each of forward and reverse primer, 0.5 μl Phusion hot start II DNA polymerase (Fisher Scientific, Pittsburg, PA), and 2 μl template DNA. Thermal cycling conditions were 98°C for 30 s (initial denaturation), followed by 40 cycles of denaturation at 98°C for 10 s, annealing at the temperature indicated in Table S2 for 30 s, and extension at 72°C for 50 s, followed by a final extension at 72°C for 7 min. PCR products (10 μl) were visualized using QiAxcel (Qiagen, Valencia, CA). Sequencing primer (0.6 μM) diluted in annealing buffer (Biotage, Charlottesville, VA) and 40 μl of mixture were aliquoted in a 96-well pyrosequencing plate (PSQ). Binding reaction mixture consisted of 4 μl of Streptavidin-coated Sepharose beads (GE Healthcare) and 51 μl of binding buffer. In each well of a 96-well plate, 55 μl of binding mixture was added to 25 μl of PCR product and mixed at 14,000 rpm for 10 min. Using a vacuum prep workstation (Biotage, Charlottesville, VA), the Sepharose-bound PCR products were captured with the vacuum prep tool immediately after being mixed and washed with 70% ethanol for 10 s, denaturing buffer (0.2 M NaOH) for 10 s, and washing buffer (10 mM Tris-acetate, pH 7.6) for 15 s. The vacuum was released and the Sepharose-bound PCR products were released into the PSQ plate containing sequencing primer. The plate was incubated at 80°C for 2 min and allowed to cool at room temperature. Plates were read on PyroMark ID (Biotage, Charlottesville, VA).
RESULTS
Cloning artemisinin-resistant isolates.
P. falciparum parasites isolated from patients often have multiple populations, and during growth in vitro, clones with fitness defects can be overgrown by more fit clones (15). Drug resistance may incur fitness costs, and recent evidence suggests that artemisinin-resistant phenotypes were lost during prolonged in vitro culture (15, 16). Therefore, we aimed to increase recovery of stably resistant parasites by cloning immediately upon establishment of growth in vitro. Cryopreserved isolates from clinical studies of artemisinin resistance in Cambodia and Thailand (7, 17, 18) were thawed, grown under standard culture conditions, and processed for cloning as soon as growth was established in vitro. Randomly selected clones (n = 3 per isolate) then were characterized for resistance phenotypes and genotypes.
Reduced sensitivity to artemisinin derivatives in vitro.
Ring-stage parasites of P. falciparum arrest development within the first 24 h following exposure to artemisinin drugs (8, 19). This is likely why standard in vitro assays have not been useful for confirmation of artemisinin resistance phenotypes. Therefore, we used a modified drug susceptibility assay to assess the susceptibility of isolates and clones to artemisinin derivatives in vitro (8). For these assays, the growth indicator ([3H]hypoxanthine) was added along with drug at the beginning of the assay (T = 0 h). By using this T0 assay, we observed significant shifts in dose-response curves, indicating reduced susceptibility of Cambodian P. falciparum isolates and clones to artemisinin, artesunate, dihydroartemisinin (DHA), and artelinic acid (AL) (Fig. 1A). Parasites displayed resistance to each artemisinin derivative tested, with the most pronounced changes occurring with artemisinin and AL. The level of resistance observed to each artemisinin derivative was similar to that of the artemisinin clones selected in the laboratory lines D6 and W2 (8). Interestingly, multiple clones from the same patient isolate displayed differing levels of resistance to multiple drugs (Fig. 1B). In some cases, drug-sensitive parasites were cloned from isolates with delayed clearance in vivo; however, cloning isolates very early during culture adaptation prevented loss of resistant phenotypes in long-term culture, as reported by others (16).
FIG 1.

Reduced susceptibility of artemisinin-resistant P. falciparum in a modified [3H]hypoxanthine assay (T0 assay). (A) Fifty percent inhibitory concentrations (IC50s) of isolates and clones from Cambodia and Thailand to artemisinin, artesunate, artelinic acid, and mefloquine were determined in the T0 assay, in which the label was added at the same time as drug. The largest shift in dose response was to artelinic acid and artemisinin. (B) Clones from isolates exhibited a range of responses to artemisinin drugs. For example, several clones of ARC08-88 (11H, 10G, and 9D) exhibited higher tolerance to artemisinin than the parent isolates. In addition, sensitive clones (5C and 7E) were isolated. Following initial drug susceptibility testing of clones, at least three clones from each isolate were selected for more detailed evaluation and repeat drug susceptibility testing (see Fig. S3 to S5 in the supplemental material). (C) Drug resistance phenotypes in the T0 assay were stable over 1 year of continuous culture, including several rounds of freeze and thaw. The results shown are averages from three independent replicates conducted at the start of culture and after a year in (mostly) continuous culture with multiple cryopreservation and thaw cycles. ARC08-88 clones are listed in descending order of IC50 to artemisinin for each graph.
The clinical phenotype for artemisinin resistance is a heritable trait (17, 18, 20), yet a common assumption has been that the resistance phenotypes are not stable in vitro or after cryopreservation. We cultivated P. falciparum clones continuously for more than 2 years and observed the stability of the artemisinin resistance phenotype in the T0 assay in vitro (Fig. 1C). Cryopreservation of resistant clones did not affect the resistance observed in subsequent T0 assays. In addition, parasites with decreased susceptibility to artemisinin derivatives exhibited decreased susceptibility to other antimalarial drugs. Our results demonstrate the T0 assay is a stable, reliable method for assessing reduced sensitivity to artemisinin derivatives in artemisinin-resistant parasites selected in vitro or from patient isolates and clones thereof (8). It is interesting that AL has never been used clinically, yet decreased susceptibility to this derivative was observed, suggesting a common mechanism of resistance that may affect many endoperoxide-containing antimalarial drugs.
DCA in vitro.
Clinical artemisinin resistance is defined as a reduced parasite clearance rate following treatment with artesunate alone or in combination with other antimalarial drugs (1, 7). We hypothesized that a similar phenotype could be expressed in vitro in resistant parasites exposed to clinically relevant concentrations of drug. We used highly synchronous ring-stage clones exposed to 700 nM DHA for 6 h every 24 h for 3 days to approximate a three-day dosing regimen in vivo. At the end of each 6-h drug exposure, drug was washed out with media (3×) and parasites placed back into culture. Parasite clearance in the delayed clearance assay (DCA) was determined by quantifying the proportion of asexual-stage parasites remaining over time following exposure to DHA. For all artemisinin-resistant clones from Cambodia, we observed slower clearance of morphologically normal rings, trophozoites, and schizonts compared to the control parasite clone W2 (Fig. 2). In the control clone (W2), only dormant ring-stage parasites were detected by 18 h, whereas clones from artemisinin-resistant isolates remained positive for morphologically normal asexual blood-stage parasites for up to 48 h (Fig. 2). Even though normal rings, trophozoites, and schizonts were cleared by 48 h, there was a large proportion of parasites with the previously characterized dormant morphology of ring-stage-arrested growth following the second and third exposures to artemisinin drugs (Fig. 3; also see Fig. S1 in the supplemental material) (8).
FIG 2.

Delayed parasite clearance of P. falciparum clones following dihydroartemisinin (DHA) exposure. Three clones per isolate plus the control W2 clone were tested in duplicate. Highly synchronous ring-stage parasites were exposed to three 6-h pulses of 700 nM DHA (indicated in gray) in the delayed clearance assay (DCA). Delayed parasite clearance of morphologically normal rings, trophozoites, or schizonts was observed for all artemisinin-resistant clones compared to artemisinin-susceptible W2. In contrast, parasite clearance of W2 was observed by hour 18 of the experiment, whereas resistant clones took as long as 48 h to clear parasites. Dead and dormant parasites were excluded from this analysis.
FIG 3.

Parasite development was monitored following exposure to DHA. Parasites (n = 100) were categorized as ring, trophozoite/schizont, or dormant/dead for each time point. In the delayed parasite clearance assay (DCA), P. falciparum clones were exposed to three 6-h pulses of 700 nM DHA. (A, C, E, and G) Only ring-stage parasites were observed in W2, whereas mature-stage parasites were observed in artemisinin-resistant clones following exposure to DHA. DCA results for additional clones are shown in Fig. S1 in the supplemental material. (B, D, F, and G) In similar studies, the clones were exposed to one 6-h pulse of 30 nM DHA (10× IC50 for W2) and monitored for parasite development. W2 progressed to mature stages, yet after one asexual cycle all parasites remaining were either dead or in a ring-arrested dormant state. In contrast, the artemisinin clones progressed through two cycles at an accelerated rate, especially compared to 4G in the absence of drug (see Fig. 5).
We estimated in vitro parasite clearance by calculating the parasite reduction ratio (PRR) from the first 12 or 24 h of the DCA and compared this phenotype to parasite clearance half-lives from the clinical data (Fig. 4A). Interestingly, in vivo clearance half-life significantly correlated with in vitro PRR12 h or PRR24 h (24-h data not shown) following exposure to DHA (Fig. 4A). For example, positive associations between in vitro and in vivo parasite clearance were observed with isolate ARC08-88 and clones thereof. ARC08-88 clones 5A and 8A were more resistant to artemisinin derivatives than ARC08-88 clone 11G and took longer to clear 50% of the parasite in vitro (Fig. 2; also see Fig. S1 in the supplemental material).
FIG 4.
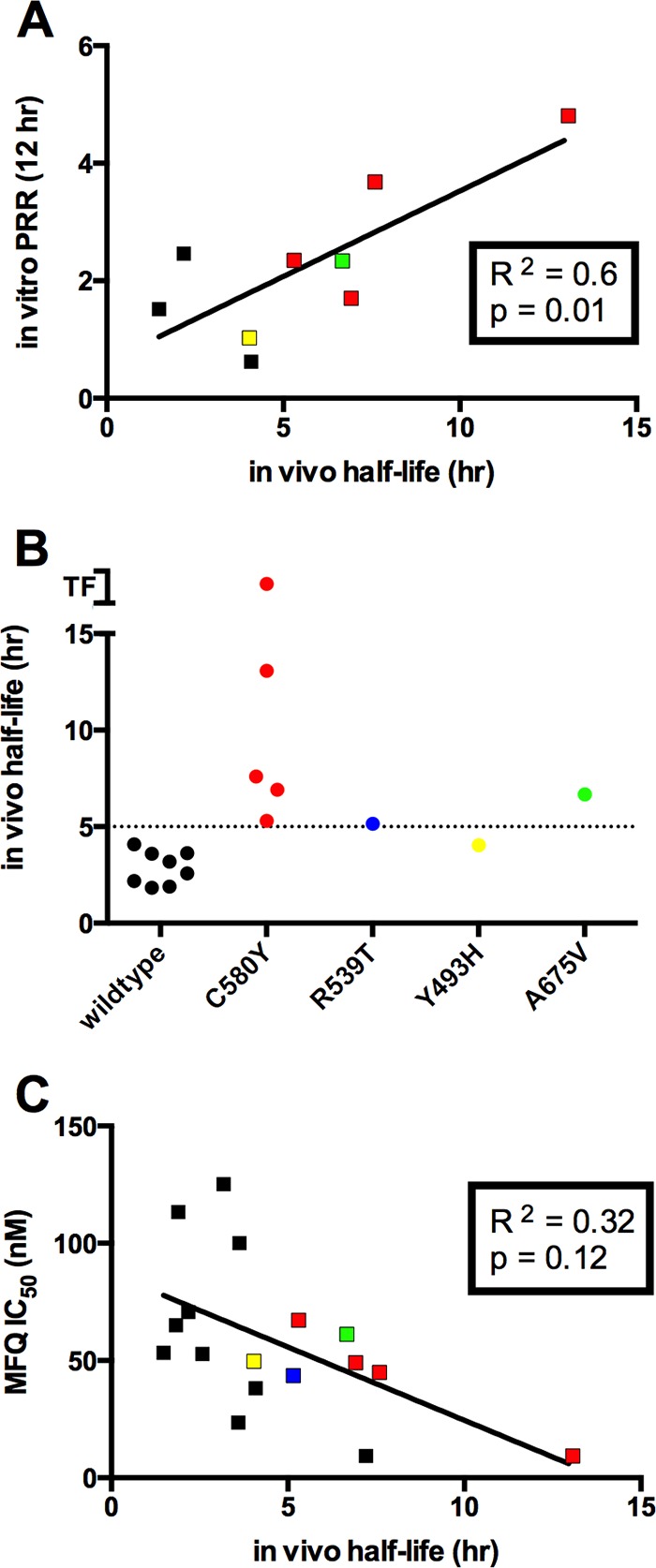
Correlation of in vitro artemisinin resistance phenotypes with in vivo parasite clearance half-lives and mutations in the Kelch (K13) propeller domains of Pf3D7_1343700. (A) The results from the delayed clearance assay (DCA) were used to determine parasite reduction ratios (PRR) during the first 12 h following exposure to 700 nM DHA. There was a significant correlation between the in vitro phenotype (PRR1–12 h) and in vivo parasite clearance half-life. (B) The delayed clearance phenotype in vitro also was positively correlated with mutations in Pf3D7_1343700 that were previously shown to be markers for artemisinin resistance (TF, treatment failure). (C) Although mefloquine-resistant clones were prevalent (see Fig. 1), there was an inverse correlation between mefloquine resistance and the delayed clearance phenotype in vitro (PRR1–12 h). Color symbols in each panel correspond to the absence or presence of Pf3D7_1343700 (K13) mutations as annotated in panel B.
Differential parasite maturation during drug exposure.
We next evaluated the maturation of intraerythrocytic, asexual-stage parasites following exposure to artemisinin drugs in vitro and observed that artemisinin-sensitive and artemisinin-resistant parasite clones had different responses to drug exposure (Fig. 3; also see Fig. S1 in the supplemental material). In assays with three 6-h pulse exposures to 700 nM DHA, only ring-stage parasites were observed in the control clone W2, and all morphologically normal rings were cleared by 18 h (Fig. 3A). Conversely, with artemisinin-resistant clones the rate of ring-stage clearance was significantly reduced (Fig. 3B to D; also see Fig. S1). For example, clone 5A of ARC08-88 exhibited a prolonged rate of ring-stage parasite clearance with morphologically normal rings present for up to 48 h despite two 6-h exposures to DHA. Interestingly, a small proportion of mature erythrocyte-stage parasites (trophozoites and schizonts) were observed up to 36 h following DHA exposure only in artemisinin-resistant clones (see Fig. S1). Despite the increased resistance exhibited by delayed clearance of both rings and trophozoites, all artemisinin-resistant clones eventually arrested as dormant ring-stage parasites following the second pulse exposure to 700 nM DHA (Fig. 3; also see Fig. S1). The induction of ring-stage dormancy by DHA is consistent with our previous studies with both drug-susceptible and laboratory-induced artemisinin-resistant parasites (8, 19).
Altered patterns of intraerythrocytic development.
Morphological observations of artemisinin-resistant isolates and clones during continuous culture demonstrated an unusually high proportion of ring-stage parasites compared to artemisinin-susceptible parasites. Therefore, we assessed the intraerythrocytic development of synchronous parasites over one to two asexual life cycles in vitro in the absence of drug pressure. By using highly synchronous ring-stage cultures, we observed a prolonged phase of ring-stage development in the artemisinin-resistant clones (Fig. 5) compared with that of the control clone (W2). For example, in the artemisinin-resistant clone 4G, the ring-stage parasites were smaller and were morphologically consistent with ring-stage parasites that persisted up to 30 h; in comparison, W2 completed ring-stage development in ∼16 h (Fig. 5). Following the prolonged ring phase, the trophozoite stage of resistant clones was temporally compressed into a remarkably fast period of approximately 8 h, followed by normal schizont development (Fig. 5 and 6). The prolonged ring phase of development in artemisinin-resistant clones was stable during long-term culture, and importantly, this phenotype was observed in multiple artemisinin-resistant clones in the absence of any drug pressure.
FIG 5.

Altered patterns of intraerythrocytic development of artemisinin-resistant clones in the absence of drug pressure. Tightly synchronized parasites were prepared for artemisinin-resistant clones, and the timing of asexual stage development was compared with that of the control clone W2. (A) ARC08-22 (4G) exhibited a prolonged ring-phase phenotype with rings observed up to 30 h of development. (B) The trophozoite stages of 4G were temporally compressed, and the schizogony of 4G was completed simultaneously with that of W2. (C) Microscopic examination of Giemsa-stained blood smears revealed the rings of 4G were smaller and persisted ∼14 h longer than those of W2. This prolonged ring phase then was followed by rapid progression through the trophozoite stage. Although most artemisinin-resistant clones expressed the prolonged ring-phase phenotype, one clone (5C) from PL08-009 completed it full asexual life cycle in only 36 h.
FIG 6.
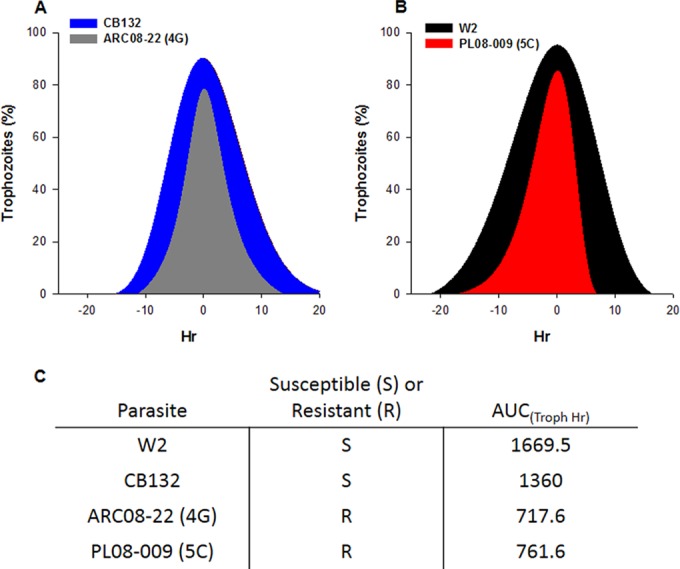
Rapid progression through trophozoite stage in artemisinin-resistant P. falciparum. The unusually rapid progression of artemisinin-resistant clones through the trophozoite stage was examined by quantifying the percentage of rings, trophozoites, and schizonts present throughout the asexual life cycle. The trophozoite prevalence data from two independent experiments then were fit to a polynomial function and, for comparison purposes, were plotted with time at 0 h as the peak of trophozoite prevalence. (A and B) Both of the artemisinin-resistant clones obviously progressed through the trophozoite stage at an accelerated rate compared to sensitive parasites (W2 and CB132). (C) Trophozoite prevalence data then were used to estimate the AUC(Troph h); both artemisinin-resistant clones had similar AUCs and spent approximately half the time that W2 and CB132 did in the trophozoite stage per asexual life cycle. CB132 is an artemisinin-sensitive isolate from Pailin, Cambodia, that was adapted to culture in 1992 (21).
The remarkably reduced phase of trophozoite development was further examined in multiple artemisinin-resistant parasites by assessing the proportion of trophozoites in the population during one intraerythrocytic asexual life cycle (Fig. 6). For comparison between different clones, we estimated the area under the curve (AUC) for trophozoite development in hours [termed AUC(troph h)] and observed that resistant clones had a much reduced AUC(troph h) compared to that of artemisinin-susceptible parasites (Fig. 6). Although most artemisinin-resistant clones exhibited the prolonged ring phase of development followed by accelerated development through the trophozoite stage, the 5C clone from PL08-09 was found to have highly accelerated asexual development of all stages and completed its intraerythrocytic life cycle within 36 h. Compared with other artemisinin-resistant clones, the 5C clone also exhibited a significantly shorter period of development in trophozoite-stage parasites than in artemisinin-susceptible parasites (Fig. 6).
The unusual alterations of the cell cycle in artemisinin-resistant clones of P. falciparum prompted additional evaluation of parasite development following drug exposure. In the 700 nM DHA pulse experiments, a small proportion of clones progressed to trophozoites earlier than would be expected from the development patterns observed in the absence of drug (Fig. 3; also see Fig. S1 in the supplemental material). Therefore, we repeated the pulse experiments but used a single 6-h pulse of 30 nM DHA, followed by media washes and evaluation of parasite development by microscopy. This concentration was chosen because it was 10× the W2 IC50 for DHA. The control clone W2 progressed from rings into trophozoites and schizonts, yet after 30 h only ring-stage dormant parasites were observed (Fig. 3). In contrast, the artemisinin-resistant clones 4G and 5C progressed from rings to trophozoites to schizonts following the pulse exposure to 30 nM DHA (Fig. 3). Interestingly, the ring-to-trophozoite progression was accelerated in the 4G resistant clone. In fact, both artemisinin-resistant clones completed the asexual cycle in ∼36 h following a low-dose pulse of 30 nM DHA (Fig. 3). These results suggest the low-dose pulse of drug actually stimulated accelerated asexual development in the 4G resistant clone to a rate similar to that of 5C in the absence of drug pressure.
Stage-specific resistance to artemisinin drugs.
The current hypothesis is that artemisinin-resistant parasites have reduced susceptibility to drug in the ring stage of development. As noted above, artemisinin-resistant parasites express a prolonged ring phase of development followed by a shortened trophozoite phase; therefore, we next examined the stage-specific resistance of the clones to DHA following 3-h pulse exposures throughout the asexual life cycle of highly synchronized clones. Following synchronization, parasite aliquots were exposed to 3-h pulses of DHA (700 nM) followed by three washes with drug-free media and then serial dilution into 96-well plates, and the cultures were allowed to grow out for 14 days to determine the number of viable parasites remaining following drug exposure (14). For comparison, we used W2 and CB132, an artemisinin-susceptible isolate collected from Pailin, Cambodia, in 1991 (21). In the first few hours of ring-stage development, there were no differences in viability of artemisinin-resistant or -susceptible parasites following exposure to drug, and between hours 3 and 9 of development we observed hypersusceptibility to drug in both artemisinin-resistant and -susceptible clones, as previously demonstrated (22), whereas from hours 9 through 20 there was a log increase in viability of artemisinin-resistant ring-stage parasites compared to the level for CB132 (Fig. 7). As noted above, in the presence of 700 nM DHA, a proportion of the resistant clones accelerated asexual development from ring to trophozoite (Fig. 7B). Interestingly, when artemisinin-resistant parasites were in the trophozoite stage, they were equally susceptible to drug, yet during schizogony resistance again was observed with an approximately 1-log increased viability in the resistant parasite compared to the level for CB132 (Fig. 7). These data support the hypothesis that ring stages are less susceptible to drug yet also for the first time demonstrate accelerated ring-to-trophozoite transition during drug exposure as well as resistance in the later asexual stages of development.
FIG 7.
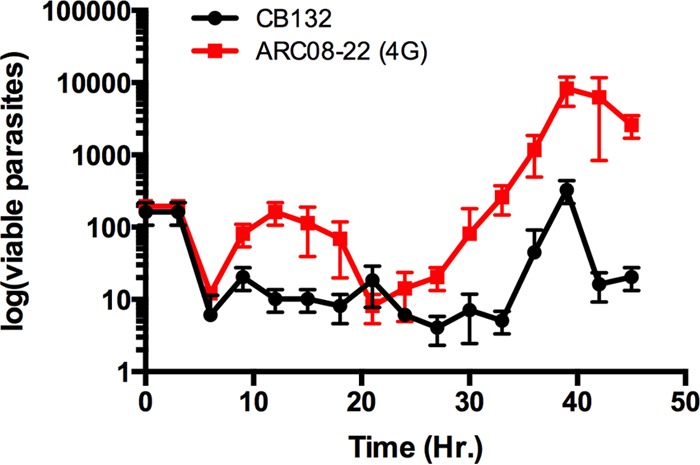
Stage specificity of artemisinin resistance in P. falciparum. The relative susceptibility of each stage of asexual parasite development was assessed by exposing tightly synchronous parasites to 3-h pulses of 700 nM DHA at 3-h increments throughout one cycle (duplicate experiments). Resistance then was assessed by parasite viability of each stage of development, with viability defined as the ability to grow logarithmically within 14 days of drug exposure. Initially there were no differences in viability of 4G and CB132, and a brief window of hypersusceptibility was observed for both clones. In the presence of drug, the ring-to-trophozoite transition for 4G was accelerated, and in this brief trophozoite window both clones were equally susceptible. Resistance to artemisinin was observed in both ring stages and throughout schizogony in the artemisinin-resistant 4G clone, and this was expressed as an ∼1 log increase in viability.
Recrudescence following DHA exposure.
Previous studies demonstrated a ring-stage arrest following exposure to artemisinin derivatives (8, 19). These ring-stage-arrested parasites are known as dormant rings due to their ability to persist in culture for days to weeks prior to recrudescence of normal growth. Since all artemisinin-resistant clones eventually arrested as dormant ring-stage parasites, we next determined the recrudescence patterns for the dormant rings following three 6-h pulse exposures to DHA (700 nM). As shown in Fig. 8, the control clone (W2) remained dormant through day 11, followed by recrudescent growth evident on day 12 with subsequent recovery of the parasites with greater than initial parasitemia by day 14. Interestingly, artemisinin-resistant clones expressed divergent dormancy recrudescence patterns. Some resistant clones recrudesced 2 days earlier than the control W2 clone (e.g., 8A clone of ARCO8-88), whereas other clones were significantly delayed in their recrudescence patterns. Some clones from artemisinin-resistant isolates did not recover from dormancy until well after 20 days (Fig. 8; also see Fig. S3 to S5 in the supplemental material). These different recrudescence patterns from dormancy were consistent between duplicate assays (n ≥ 2), suggesting inherited differences in recrudescence following dormancy in different cloned artemisinin-resistant P. falciparum strains.
FIG 8.
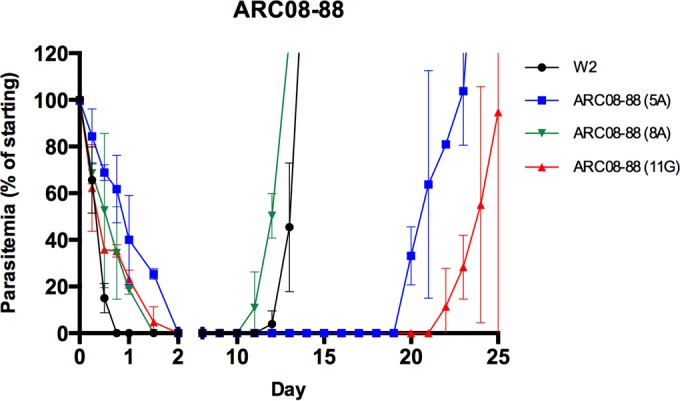
Recrudescence from DHA-induced ring-stage dormancy in P. falciparum. Three 6-h pulses of 700 nM DHA induced dormant ring stages in both susceptible (W2) and artemisinin-resistant clones (also see Fig. S3 to S5 in the supplemental material). There was a divergent pattern of recrudescence observed, with some clones recovering up to 2 days prior to W2, whereas other resistant clones remained dormant and only recovered later than 20 days (n = 2; bars represent ±1 SD).
Association of in vitro and in vivo drug resistance phenotypes.
Previous studies have suggested a strong association of the clinical resistance phenotype with mutations in the propeller domains of the Kelch gene (K13) on chromosome 13 (Pf3D7_1343700) and ring-stage survival in a modified in vitro assay (9). The presence or absence of K13 mutations was determined by pyrosequence analysis for all isolates and clones used in this study (see Table S3 in the supplemental material for a complete list). We compared the phenotypes observed in this study to in vivo parasite clearance half-life and found the strongest association was with the in vitro DCA (Fig. 4). Interestingly, the reduced rate of parasite clearance in vitro also was observed in parasites with C580Y, R539T, Y493H, or A675V mutations in K13 (Fig. 4B). In contrast, susceptibility to mefloquine was inversely correlated with in vitro clearance half-life (PRR12 h) and with the presence of K13 mutations (Fig. 4C). Despite the reduced susceptibility to multiple artemisinin derivatives in the T0 hypoxanthine assays, there was no significant correlation between hypoxanthine assay results and in vivo parasite clearance rates or K13 mutations (see Fig. S2 to S5).
DISCUSSION
Our current understanding of mechanisms of antimalarial drug resistance have emerged from the ability to continuously culture drug-resistant parasites from patients and for these parasites to express phenotypes that can be used for phenotype-genotype association studies to identify genetic markers of resistance. Classic examples of this approach are the identification of pfcrt mutations that confer chloroquine resistance and mutations in dihydrofolate reductase and dihydropteroate synthase that confer resistance to antifolate drugs (4–6). Conversely, for artemisinin resistance, the typical in vitro drug assay phenotypes used for previous antimalarial drug-resistant studies have not been useful (7). Since these standard in vitro phenotypes have not yielded success, genome-wide association studies had to be undertaken to identify heritable traits associated with clinical resistance (3, 17, 18).
In this study, we demonstrate for the first time multiple new artemisinin resistance-associated phenotypes that are expressed stably in parasite clones in vitro. These include decreased susceptibility to artemisinin drugs in a T0 [3H]hypoxanthine assay, altered patterns of intraerythrocytic development with prolonged ring-stage and temporally compressed trophozoite development, a slow-clearance in vitro phenotype in the presence of drug, and association of slow clearance in vitro with K13 mutations as well as with reduced rate of clearance phenotypes in vivo. These novel in vitro phenotypes offer new opportunities for elucidating the role of K13 mutations in artemisinin resistance and for identifying additional molecular markers associated with clinical resistance to artemisinin.
Perhaps the most remarkable phenotype expressed by artemisinin-resistant clones was the altered patterns of intraerythrocytic development in the absence or presence of artemisinin drugs. In the absence of drug pressure, we observed a prolonged phase of ring-stage development in artemisinin-resistant clones that lasted up to 30 h of the life cycle; this was approximately 14 h longer than that of the control (W2). The prolonged ring phase then was followed by a temporally compressed trophozoite stage (Fig. 6) prior to normal schizont development. Despite the dramatic differences, the overall intraerythrocytic life cycle completion of these resistant clones was the same as that for W2. Interestingly, we also discovered a resistant clone (5C) that had a significantly abbreviated intraerythrocytic life cycle that was completed in only 36 h. Since we observed no differences in intraerythrocytic stage development in drug-sensitive clones or isolates from southeast Asia (e.g., CB132), the prolonged ring and temporally compressed trophozoite development phenotype is strongly associated with artemisinin resistance. This result is consistent with a recent study of the ex vivo transcriptome of artemisinin-resistant parasites (23) that suggested an extended phase of ring-stage development coincident with upregulation of unfolded protein response (UPR) pathways. Our results show, for the first time, this phenotype in stable clones and that some resistant isolates have a significantly abbreviated asexual life cycle.
Intriguing questions are what advantage does the altered intraerythrocytic development pattern provide and how might this phenotype confer resistance? Mok et al. (23) suggest the prolonged ring phase of development allows the upregulated UPR pathways to mitigate protein damage caused by drug exposure. Although it is plausible that UPR pathways are involved, this does not explain why we observed the prolonged ring and compressed trophozoite development phenotypes stably expressed in the absence of drug pressure. In addition, the accelerated development of the intraerythrocytic life cycle of some artemisinin clones (5C) in the absence of drug pressure, as well as the accelerated ring-to-trophozoite transition in response to drug exposure, appear inconsistent with the hypothesis of delayed ring development and UPR damage mitigation.
Our data from cloned, stably artemisinin-resistant parasites suggest two advantages of the altered intraerythrocytic development patterns observed; these are increased viability of ring stages and reduced exposure to drug in the most susceptible stage of development (trophozoites). Although most of the resistant clones had a prolonged ring phase of development, one resistant clone also had an accelerated 36-h life cycle in the red cell. One consistency between these phenotypes is the dramatically shortened period of development in the trophozoite stage (Fig. 6). Previous studies suggest that the trophozoite stage of development is the most sensitive in the life cycle (22), and our studies confirmed that trophozoites are the most susceptible stage, even in artemisinin-resistant clones (Fig. 7). Therefore, we hypothesize that the intraerythrocytic development patterns evolved as a mechanism to reduce exposure of the parasite to drug in the most susceptible stage of development. The similar AUC(troph h) for the prolonged-ring-phase phenotype in the absence of drug, the accelerated ring-to-trophozoite development in the presence of drug, and the fully abbreviated life cycle (clone 5C) are consistent with this hypothesis. Second, the prolonged ring phase of development offers another survival advantage to the parasite. In previous studies, we demonstrated that ring-stage parasites are less susceptible to drug due to the ability of ring stages to arrest development following drug exposure; these dormant rings can survive for days to weeks before recovering and growing normally. We hypothesize the prolonged ring phase of development is a resistance advantage by extending the period in which the ring-stage parasites can be stably arrested in growth with subsequent recovery from dormancy and survival. Evidence that supports this hypothesis is shown in Fig. 7, where we observed significantly increased viability of resistant parasites that were exposed to drug in the ring stages of development. Since ring stages were not able to grow continuously and complete the life cycle in the presence of 700 nM DHA (Fig. 2 and 3), recovery from dormancy was a major component of the ring-stage resistance and long-term viability differences we observed in artemisinin-resistant clones (Fig. 7 and 8). Given that drug-induced dormancy is a phenotype expressed in sensitive and resistant isolates, this trait appears to be independent of drug resistance selection; however, it is plausible the survival advantage conferred by drug-induced dormant ring stages factored into the ultimate selection of the prolonged ring-phase phenotype in artemisinin-resistant P. falciparum.
An intriguing correlate of the altered patterns of intraerythrocytic development is why this phenotype has not been observed previously. Most antimalarial drugs are most effective against trophozoite-stage parasites; thus, why other antimalarial drugs have not selected for this artemisinin resistance phenotype is not known. We hypothesize it is related to the novel resistance of ring stages to artemisinin drugs and that enhanced resistance is due to altered cell cycle regulation that conveys the ability of artemisinin-resistant parasites to lengthen the ring-stage development and temporally compress trophozoite development. Further studies are needed to assess the potential effect of the altered cell cycle resistance phenotype on resistance to other antimalarial drugs, since it could accelerate resistance to other drugs acting on trophozoite stages.
As noted by others, some standard in vitro drug susceptibility assays do not correlate with the clinical phenotype of a reduced rate of parasite clearance (7). Results from two modified in vitro assays have been shown to correlate to some degree with artemisinin resistance in vitro; these are the T0 [3H]hypoxanthine assay (8) and a ring-stage survival assay (RSA) (9). The RSA is reported to assess the survival of ring-stage parasites exposed to a single pulse of 700 nM DHA. The endpoint is the percentage of normal parasites counted in Giemsa-stained blood smears at 72 h. The RSA was used successfully for association studies with clinical resistance and were found to have statistically significant correlation with in vivo clearance and mutations in the propeller domains of K13 (1, 2). Although the RSA results correlate with the clinical resistance phenotype, our data suggest the underlying mechanisms measured in the RSA differ from what has been described previously. For example, in our studies, 700 nM DHA was sufficient to induce dormancy even in resistant parasites, yet the timing of dormancy induction was delayed significantly from sensitive parasites. The pyknotic parasites that were present at the 72-h time point in the RSA were thought to be dead (9, 24), yet clearly the results of our study demonstrate parasites remain viable in the dormant stage; thus, the RSA underestimates viability of resistant parasites. This viability of dormant rings was despite exposure to three 6-h pulse exposures to 700 nM DHA. Conversely, one underlying mechanism for the higher survival rates in the RSA could be the prolonged ring phase of development observed in artemisinin-resistant parasites. Therefore, we believe the prolonged ring phenotype was incorrectly interpreted as quiescent rings in the previous study (24), given that we observed the prolonged ring phase in resistant clones in the absence of drug. Interestingly, the clinical phenotype of reduced parasite clearance is due primarily to the presence of ring-stage parasites in the peripheral blood of patients following treatment with artemisinin drugs. The basis of the in vivo slow-clearance phenotype likely is due to the prolonged-ring-phase phenotype that we have observed. Since ring stages do not sequester in deep vasculature, the prolonged ring phase of development probably contributes to reduced parasite clearance rates observed in vivo. Regardless of the mechanism(s) involved, these results suggest the common thread between the in vivo slow-clearance phenotype, reduced rate of parasite clearance in vitro, and K13 mutations is the prolonged-ring-phase phenotype. Therefore, the other resistance-associated phenotypes described here may be additional traits that contribute to an artemisinin resistance mechanism(s) that appears to be more complex than resistance to other antimalarial drugs.
The increased prevalence and apparent spread of artemisinin resistance increases the urgency for fielding effective tools for epidemiological monitoring of resistance. Currently, there are two tools that can be used: presence of K13 mutations and the RSA phenotype. In this study, we demonstrate that the reduced rate of parasite clearance in vivo correlates well with the results of a new delayed clearance assay in vitro. The main advantages over the RSA is the DCA can be conducted in as little as 12 or 24 h, it is less reliant upon tight synchronization of cultures, it more accurately assesses the prolonged ring phenotype, and the microscopy endpoint could be rapidly adapted to other quantitative measures (e.g., flow cytometry). The DCA also should be useful with ex vivo parasite cultures (directly from patients) and needs to be assessed for potential utility in the field.
The resistance phenotypes described here offer great potential for studies of drug resistance mechanisms for artemisinin and related compounds in development. Recent advances in rapid, selective integration of mutations in the parasite genome have accelerated the confirmation of drug resistance mechanisms. Several groups have found that insertion of specific K13 mutations into susceptible parasites confer an RSA resistance phenotype in vitro (25, 26). These studies did not describe any resistance phenotypes other than RSA; thus, it would be interesting to know if these transformed resistant lines display any of the new resistant phenotypes we observed in artemisinin-resistant parasite clones from southeast Asia or if there are yet-to-be-identified mechanisms that control the unusual cell cycle regulation in artemisinin-resistant parasites.
Artemisinin-induced dormancy in ring stages has been observed in both sensitive and resistant parasites in vitro (8, 19). One of the interesting phenotypes we observed in this study was the divergent pattern of recrudescence following dormancy in artemisinin-resistant clones. As shown in Fig. 8, some resistant clones recrudesced earlier than the W2 control parasite, whereas others had a significantly prolonged period of dormancy followed by recovery after 20 days. At present, there is no obvious explanation for these differences. The prolonged recovery could be due to differences in the proportion of dormant parasites that recover, given the possibility that increased numbers of viable dormant parasites might recover at a higher rate, as observed for a laboratory-selected resistant line (8). Conversely, the ability to remain dormant for longer periods of time may be an advantageous response to combination drug therapy where the partner drug has prolonged systemic exposure; these artemisinin-resistant clones could persist in the dormant ring state until the partner drug exposure is alleviated. Given that dormant parasites remain in drug-exposed cultures even after viable parasites start to recover, elucidation of the dormancy recovery mechanisms and the true proportion of parasites that recover from dormancy will require further studies.
Previous results suggest that artemisinin resistance is confined to the ring stages of parasite development. Our data confirm this hypothesis, yet we also observed, for the first time, resistance to artemisinin drugs in mature stages. The mechanisms involved in late-stage resistance are not known, but this could be a sign that increased levels of resistance are developing; thus, the phenotype is expressed more broadly in the intraerythrocytic development of the parasite. Another interesting observation from these studies is the stable expression of reduced susceptibility to multiple artemisinin drugs in vitro, as measured by the T0 assay. This assay captures parasite development within the first 24 h following exposure to drug before ring stages become arrested, and growth differences are not able to be measured (as in other in vitro susceptibility assays). In our previous studies, the laboratory-derived artemisinin-resistant parasites were most resistant to AL and artemisinin, with reduced levels of susceptibility to DHA being observed (8). Surprisingly, we observed the same resistance pattern in the artemisinin-resistant clones from Cambodia, despite the fact that AL has never been used in the field. We hypothesize that the resistance mechanism recognizes related compounds; therefore, these results highlight a significant concern for artemisinin derivatives in development. The importance of resistance to artemisinin drugs in the T0 assay has yet to be determined. In our studies, the IC50s obtained did not correlate with slow parasite clearance in vivo, yet there was a significant difference in IC50s for many resistant isolates, and this phenotype was stable in culture (Fig. 1). The facts that others found similar reduced susceptibility in Cambodia when adding [3H]hypoxanthane at the beginning of the assay (27) and that we found many clones with reduced susceptibility in the T0 assay suggest this phenotype is due to a resistance mechanism unrelated to the slow-clearance phenotype.
Resistance to artemisinin drugs and the potential for a selective sweep of these parasites in regions outside southeast Asia is one of the most important threats to malaria control and elimination efforts. The novel resistance phenotypes described here offer potential for epidemiological monitoring with the DCA and highlight a previously unrecognized plasticity of intraerythrocytic development of asexual stages of P. falciparum that confers a selective advantage to parasites exposed to artemisinin derivatives. The regulatory mechanism(s) involved in altering the cell cycle and intraerythrocytic development is unknown, yet it seems likely to be due to more than mutations in K13. We hypothesize that altered regulation of exit from G1 in the cell cycle is the basis for the novel phenotypes observed. The artemisinin-resistant clones that stably express these artemisinin resistance-associated phenotypes offer an opportunity to better understand both the mechanisms of resistance and the mechanisms of cell cycle regulation in P. falciparum.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Institute of Allergy and Infectious Diseases (R01 AI058973) and the Bill and Melinda Gates Foundation (via a grant to the World Health Organization).
We thank Francois Nosten, Arjen Dorndorp, Chris Plowe, Delia Bethell, and Mark Fukuda for provision of cryopreserved parasites and/or parasite clearance time information.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00197-15.
REFERENCES
- 1.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale JC, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takala-Harrison S, Jacob CG, Arze C, Cummings MP, Silva JC, Dondorp AM, Fukuda MM, Hien TT, Mayxay M, Noedl H, Nosten F, Kyaw MP, Nhien NT, Imwong M, Bethell D, Se Y, Lon C, Tyner SD, Saunders DL, Ariey F, Mercereau-Puijalon O, Menard D, Newton PN, Khanthavong M, Hongvanthong B, Starzengruber P, Fuehrer HP, Swoboda P, Khan WA, Phyo AP, Nyunt MM, Nyunt MH, Brown TS, Adams M, Pepin CS, Bailey J, Tan JC, Ferdig MT, Clark TG, Miotto O, MacInnis B, Kwiatkowski DP, White NJ, Ringwald P, Plowe CV. 2014. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in southeast Asia. J Infect Dis doi: 10.1093/infdis/jiu491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ, Su XZ. 2002. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 418:320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- 5.Roper C, Pearce R, Bredenkamp B, Gumede J, Drakeley C, Mosha F, Chandramohan D, Sharp B. 2003. Antifolate antimalarial resistance in southeast Africa: a population-based analysis. Lancet 361:1174–1181. doi: 10.1016/S0140-6736(03)12951-0. [DOI] [PubMed] [Google Scholar]
- 6.Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, Guthmann JP, Smithuis FM, Hien TT, White NJ, Nosten F, Anderson TJ. 2003. A selective sweep driven by pyrimethamine treatment in southeast Asian malaria parasites. Mol Biol Evol 20:1526–1536. doi: 10.1093/molbev/msg162. [DOI] [PubMed] [Google Scholar]
- 7.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucker MS, Mutka T, Sparks K, Patel J, Kyle DE. 2012. Phenotypic and genotypic analysis of in vitro-selected artemisinin-resistant progeny of Plasmodium falciparum. Antimicrob Agents Chemother 56:302–314. doi: 10.1128/AAC.05540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Witkowski B, Khim N, Chim P, Kim S, Ke S, Kloeung N, Chy S, Duong S, Leang R, Ringwald P, Dondorp AM, Tripura R, Benoit-Vical F, Berry A, Gorgette O, Ariey F, Barale JC, Mercereau-Puijalon O, Menard D. 2013. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob Agents Chemother 57:914–923. doi: 10.1128/AAC.01868-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 11.Rosario V. 1981. Cloning of naturally occurring mixed infections of malaria parasites. Science 212:1037–1038. doi: 10.1126/science.7015505. [DOI] [PubMed] [Google Scholar]
- 12.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. 1979. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 16:710–718. doi: 10.1128/AAC.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balu B, Singh N, Maher SP, Adams JH. 2010. A genetic screen for attenuated growth identifies genes crucial for intraerythrocytic development of Plasmodium falciparum. PLoS One 5:e13282. doi: 10.1371/journal.pone.0013282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanz LM, Crespo B, De-Cozar C, Ding XC, Llergo JL, Burrows JN, Garcia-Bustos JF, Gamo FJ. 2012. P. falciparum in vitro killing rates allow to discriminate between different antimalarial mode-of-action. PLoS One 7:e30949. doi: 10.1371/journal.pone.0030949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenthal PJ. 2013. The interplay between drug resistance and fitness in malaria parasites. Mol Microbiol 89:1025–1038. doi: 10.1111/mmi.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chotivanich K, Tripura R, Das D, Yi P, Day NP, Pukrittayakamee S, Chuor CM, Socheat D, Dondorp AM, White NJ. 2014. Laboratory detection of artemisinin-resistant Plasmodium falciparum. Antimicrob Agents Chemother 58:3157–3161. doi: 10.1128/AAC.01924-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheeseman IH, Miller BA, Nair S, Nkhoma S, Tan A, Tan JC, Al Saai S, Phyo AP, Moo CL, Lwin KM, McGready R, Ashley E, Imwong M, Stepniewska K, Yi P, Dondorp AM, Mayxay M, Newton PN, White NJ, Nosten F, Ferdig MT, Anderson TJ. 2012. A major genome region underlying artemisinin resistance in malaria. Science 336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takala-Harrison S, Clark TG, Jacob CG, Cummings MP, Miotto O, Dondorp AM, Fukuda MM, Nosten F, Noedl H, Imwong M, Bethell D, Se Y, Lon C, Tyner SD, Saunders DL, Socheat D, Ariey F, Phyo AP, Starzengruber P, Fuehrer HP, Swoboda P, Stepniewska K, Flegg J, Arze C, Cerqueira GC, Silva JC, Ricklefs SM, Porcella SF, Stephens RM, Adams M, Kenefic LJ, Campino S, Auburn S, MacInnis B, Kwiatkowski DP, Su XZ, White NJ, Ringwald P, Plowe CV. 2013. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in southeast Asia. Proc Natl Acad Sci U S A 110:240–245. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teuscher F, Chen N, Kyle DE, Gatton ML, Cheng Q. 2012. Phenotypic changes in artemisinin-resistant plasmodium falciparum lines in vitro: evidence for decreased sensitivity to dormancy and growth inhibition. Antimicrob Agents Chemother 56:428–431. doi: 10.1128/AAC.05456-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miotto O, Almagro-Garcia J, Manske M, Macinnis B, Campino S, Rockett KA, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Duong S, Nguon C, Chuor CM, Saunders D, Se Y, Lon C, Fukuda MM, Amenga-Etego L, Hodgson AV, Asoala V, Imwong M, Takala-Harrison S, Nosten F, Su XZ, Ringwald P, Ariey F, Dolecek C, Hien TT, Boni MF, Thai CQ, Amambua-Ngwa A, Conway DJ, Djimde AA, Doumbo OK, Zongo I, Ouedraogo JB, Alcock D, Drury E, Auburn S, Koch O, Sanders M, Hubbart C, Maslen G, Ruano-Rubio V, Jyothi D, Miles A, O'Brien J, Gamble C, Oyola SO, Rayner JC, Newbold CI, Berriman M, Spencer CC, McVean G, Day NP, White NJ, Bethell D, Dondorp AM, Plowe CV, Fairhurst RM, Kwiatkowski DP. 2013. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat Genet 45:648–655. doi: 10.1038/ng.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hopperus Buma AP, van Thiel PP, Lobel HO, Ohrt C, van Ameijden EJ, Veltink RL, Tendeloo DC, van Gool T, Green MD, Todd GD, Kyle DE, Kager PA. 1996. Long-term malaria chemoprophylaxis with mefloquine in Dutch marines in Cambodia. J Infect Dis 173:1506–1509. doi: 10.1093/infdis/173.6.1506. [DOI] [PubMed] [Google Scholar]
- 22.Klonis N, Xie SC, McCaw JM, Crespo-Ortiz MP, Zaloumis SG, Simpson JA, Tilley L. 2013. Altered temporal response of malaria parasites determines differential sensitivity to artemisinin. Proc Natl Acad Sci U S A 110:5157–5162. doi: 10.1073/pnas.1217452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mok S, Ashley EA, Ferreira PE, Zhu L, Lin Z, Yeo T, Chotivanich K, Imwong M, Pukrittayakamee S, Dhorda M, Nguon C, Lim P, Amaratunga C, Suon S, Hien TT, Htut Y, Faiz MA, Onyamboko MA, Mayxay M, Newton PN, Tripura R, Woodrow CJ, Miotto O, Kwiatkowski DP, Nosten F, Day NP, Preiser PR, White NJ, Dondorp AM, Fairhurst RM, Bozdech Z. 2014. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 347:431–435. doi: 10.1126/science.1260403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, Kim S, Lim P, Mao S, Sopha C, Sam B, Anderson JM, Duong S, Chuor CM, Taylor WR, Suon S, Mercereau-Puijalon O, Fairhurst RM, Menard D. 2013. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 13:1043–1049. doi: 10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Menard D, Fidock DA. 2014. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. 2014. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 32:819–821. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- 27.Lim P, Wongsrichanalai C, Chim P, Khim N, Kim S, Chy S, Sem R, Nhem S, Yi P, Duong S, Bouth DM, Genton B, Beck HP, Gobert JG, Rogers WO, Coppee JY, Fandeur T, Mercereau-Puijalon O, Ringwald P, Le Bras J, Ariey F. 2010. Decreased in vitro susceptibility of Plasmodium falciparum isolates to artesunate, mefloquine, chloroquine, and quinine in Cambodia from 2001 to 2007. Antimicrob Agents Chemother 54:2135–2142. doi: 10.1128/AAC.01304-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.