

Abstract
The ABCF family protein Msr(A) confers high resistance to macrolides but only low resistance to ketolides in staphylococci. Mutations in conserved functional regions of ClpX as well as deletion of clpX significantly increased Msr(A)-mediated resistance to the ketolide antibiotic telithromycin. ClpX is the chaperone component of the ClpXP two-component proteolytic system. Nevertheless, no changes in resistance were observed in a clpP knockout strain expressing msr(A), demonstrating that ClpX affects Msr(A) independently of ClpP.
TEXT
Macrolides are important antibiotics used in clinical practice to treat infections caused by Staphylococcus aureus (1). One of the most frequent macrolide resistance determinants in staphylococci is msr(A), which confers high resistance to 14- and 15-membered macrolides as well as to streptogramin B but only low-level resistance to telithromycin (TEL), which is a member of the newest macrolide class—the ketolides (2–5). To examine the possibility of the development of TEL resistance as a result of changes in Msr(A) specificity, we selected and analyzed TEL-resistant mutants of S. aureus in which the resistance entirely depends on msr(A) expression. Unexpectedly, we found that TEL resistance was not associated with mutations in Msr(A) but rather was associated with mutations in the ClpX chaperone.
Strain S. aureus RN4220+MsrA, in which msr(A) expression is controlled by anhydrotetracycline (AnhTet), was constructed to eliminate TEL-resistant mutants whose resistance is Msr(A) independent. In brief, the msr(A) gene was PCR amplified, with a Shine-Dalgarno sequence added upstream of the gene and the sequence coding for a C-terminal Pro-(Gly)3-Ser-(His)6 tag added downstream. This construct was cloned into the pRMC2 plasmid and electroporated into S. aureus RN4220 (primers, plasmids, and strains used in the study are listed in Table S1 in the supplemental material). The resulting strain, RN4220+MsrA, was, as expected, highly resistant to erythromycin (ERY) and only moderately resistant to TEL in the presence of AnhTet (100 ng/ml). Highly TEL-resistant mutants derived from RN4220+MsrA were selected in the presence of 100 ng/ml of AnhTet on brain heart infusion (BHI) agar plates with 2, 4, 8, or 16 μg/ml of TEL following incubation at a subinhibitory concentration of TEL (0.5 MIC; 0.5 μg/ml) and AnhTet (100 ng/ml) for 12 h. Selection was performed in two independent parallel experiments.
MICs were determined using the standard broth microdilution method in Mueller-Hinton (MH) medium (6). All measurements were performed at least 3 times. For determinations in which MIC values differed between measurements, the range of MIC values is indicated in Table 1. By screening all 67 colonies that appeared on the plates, we identified 30 mutant strains in which TEL resistance was fully dependent on expression of msr(A). The MIC of TEL increased 4 to 16 times to 8 to 32 μg/ml in those mutants (Table 1). Surprisingly, sequencing of PCR-amplified msr(A) did not revealed any mutations in the msr(A) gene in any of the 30 strains (Table 1), suggesting that the mutations affecting Msr(A)-mediated resistance occurred on the chromosome. This interpretation is further supported by the observation that representative mutant strain 24 cured of the pRMC2+MsrA plasmid (strain 24C) was sensitive to TEL and that resistance was fully restored when the pRMC2+MsrA plasmid was reintroduced into the strain (strain 24R). To identify the chromosomal mutations, genomic DNA of mutant strain 24 was extracted using the method described in reference 7, followed by additional purification performed using a Wizard genomic DNA purification kit (Promega), and was sent to BGI Tech Solutions for sequencing. A total of 96 single nucleotide polymorphisms (SNPs) were identified when the S. aureus NCTC 8325 genome sequence (NC_007795.1) was used as the reference genome for SNP calling. However, a comparison of all SNPs to the published sequence of S. aureus RN4220 (8) identified only a single nucleotide mutation of C to A in the clpX gene, resulting in an amino acid change of cysteine to phenylalanine at position 13 of the ClpX protein. ClpX is the substrate-recognizing component of the ClpXP proteolytic system, which regulates protein quality and turnover through controlled proteolysis (9). ClpX consists of three domains: an N-terminal zinc-binding domain responsible for substrate recognition and an AAA+ module consisting of large and small AAA+ ATPase domains that assume a hexameric organization and unfold recognized proteins (10). The clpX gene was sequenced in 13 additional TEL-resistant mutant strains in which TEL resistance was dependent on the presence of Msr(A). In all cases, mutations distributed along the whole protein were identified (Table 1). Eleven of 14 sequenced clpX genes had deletions or stop codons (Table 1), suggesting that the increased TEL resistance is associated with complete loss of ClpX function. In contrast, the loss of ClpX function could be questionable in three strains in which only a single amino acid substitution, G150D or C13F, was detected. Nevertheless, both substitutions are located in conserved regions, suggesting that they may severely impair ClpX function. The G150D mutation detected in mutant 27 is within the conserved GYVG motif of the functionally important pore-1 loop (also called the Ar-Φ pore loop) that is responsible for ATP-dependent translocation of substrates through the pore formed by the ClpX hexamer. Mutations in the GYVG conserved motif of Escherichia coli ClpX affected the rate of (V154F) or even abolished (Y153A) substrate degradation by the ClpXP complex (11). The C13F mutation detected in mutants 19 and 24 changes one of the most conserved amino acid residues of the ClpX N-terminal zinc binding domain and disrupts the Zn-binding motif (12, 13). It has been observed that ClpX lacking the Zn-binding domain can assemble into a hexameric ring, can interact with ClpP, and can mediate protein degradation of some native substrates at rates similar to those seen with the wild type (14); however, the N-terminal domain of ClpX is conserved across all bacteria and plays an essential role in substrate recognition (15). It is therefore possible that elimination of the Zn-binding motif disrupts the ability of ClpX to recognize substrates and therefore affects ClpX activity. To confirm that loss of ClpX function is responsible for the increased Msr(A)-mediated TEL resistance, we compared the susceptibilities of the S. aureus 8325-4 wild-type strain and a ΔclpX mutant, both transduced (16) with plasmid pRMC2+MsrA. Indeed, the ΔclpX strain exhibited higher Msr(A)-mediated resistance to TEL than 8325-4+MsrA when expression of msr(A) was induced by AnhTet (Table 1). Moreover, the level of TEL resistance in the ΔclpX+MsrA strain (MIC = 16 to 32 μg/ml) was comparable to the resistance of RN4220+MsrA mutant strains.
TABLE 1.
MICs for telithromycin, erythromycin, and lincomycin in the presence or absence of AnhTet and mutations in amino acid sequences of Msr(A) and ClpX
| Strain | MIC (mg/liter) in presence or absence of AnhTet |
Change(s) in amino acid sequencea |
||||||
|---|---|---|---|---|---|---|---|---|
| TEL |
ERY |
LIN |
Msr(A) | ClpX | ||||
| − | + | − | + | − | + | |||
| RN4220 | 0.25 | 0.25 | 1 | 1 | 1 | 0.5–1 | ||
| RN4220+MsrA | 1 | 2 | 2 | 64 | 1 | 1 | None | |
| 1 | 0.5–1 | 16 | 4 | 128 | None | Δ104–154 | ||
| 4 | 0.5–1 | 16 | 4 | 128 | None | Stop at 26 | ||
| 7 | 0.5–1 | 16 | 4 | 128 | None | Stop at 327 | ||
| 8 | 2 | 16–32 | None | |||||
| 11 | 0.5–1 | 16 | 4 | 128 | None | Δ52–128 | ||
| 14 | 0.5–1 | 16 | None | Stop at 337 | ||||
| 15 | 1 | 16 | None | |||||
| 16 | 2 | 16–32 | None | |||||
| 17 | 2 | 16 | None | |||||
| 18 | 2 | 16–32 | None | Stop at 280 | ||||
| 19 | 0.5–1 | 16 | 4 | 128 | None | C13F | ||
| 20 | 1 | 16 | None | Stop at 41 | ||||
| 21 | 1 | 16 | None | |||||
| 23 | 1 | 16 | None | |||||
| 24 | 0.5–1 | 16 | 4 | 128 | 1 | 1 | None | C13F |
| 25 | 1 | 16 | None | |||||
| 26 | 1 | 16 | None | |||||
| 27 | 0.5–1 | 16 | 4 | 128 | 1 | 1 | None | G150D |
| 28 | 4 | 16 | None | Stop at 230 | ||||
| 30 | 1 | 16 | None | |||||
| 37 | 2 | 16–32 | None | |||||
| 38 | 1 | 16 | None | |||||
| 39 | 1 | 16 | None | |||||
| 40 | 1 | 16 | None | Stop at 26 | ||||
| 43 | 0.5 | 8 | None | |||||
| 54 | 1 | 16 | None | |||||
| 58 | 0.5–1 | 16 | 4 | 128 | 1 | 1 | None | Stop at 200 |
| 62 | 1 | 16 | None | |||||
| 64 | 0.5–1 | 16 | 4 | 128 | 1 | 1 | None | Δ0–67/382–420 |
| 70 | 1 | 16 | None | |||||
| 24C | <1 | <1 | ||||||
| 24C+pRMC2 | <1 | <1 | ||||||
| 24R | <1 | 16–32 | ||||||
| 8325-4 | 0.25–0.5 | 0.25 | 1 | 1 | 1 | 0.5 | ||
| 8325-4+MsrA | 1 | 2 | 2 | 16 | 1 | 1 | ||
| ΔclpX | 0.25 | 0.5 | 1 | 1 | 1 | 1 | ||
| ΔclpX+MsrA | 1 | 16–32 | 4 | 128 | 1 | 1 | ||
| ΔclpC | 0.5 | 0.5 | 1 | 2 | 1 | 1 | ||
| ΔclpC+MsrA | 1 | 2 | 2 | 16 | 1 | 1 | ||
| ΔclpP | 0.5 | 0.5 | 1 | 1 | 1 | 1 | ||
| ΔclpP+MsrA | 0.5 | 2 | 2 | 16 | 1 | 1 | ||
The presence or absence of mutations in Msr(A) and ClpX is shown only for the strains in which the respective genes were sequenced.
In addition to TEL, resistance to ERY was also increased in the ClpX mutant strains (Table 1). However, susceptibility to lincomycin, which is not affected by Msr(A), did not change. This shows that loss of ClpX function results in a change in the overall efficiency of Msr(A) rather than in a change of antibiotic specificity. Increased efficiency of Msr(A) in ClpX mutant strains could be explained by increased amounts of the protein. We tested Msr(A)-6His levels using Western blot analysis in three independent experiments using a monoclonal anti-His antibody (Novagen). However, we did not detect any differences in Msr(A) protein levels between strain RN4220+MsrA and its mutants (mutants 1, 4, and 24) or between mutant 8325-4+MsrA and mutant ΔclpX+MsrA (Fig. 1). Notably, the level of Msr(A) in the 8325-4-derived strains was lower than in the RN4220-derived strains despite the use of the same total protein load (2.5 μg/lane). Nevertheless, the MICs were comparable (Table 1), suggesting that the observed differences in Msr(A) levels have little effect on the level of resistance.
FIG 1.
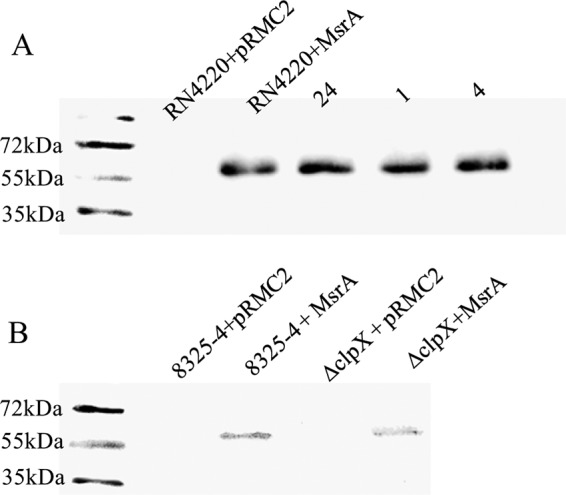
Western blot analysis of Msr(A). (A) Western immunoblots of Msr(A)-6His in wild-type S. aureus RN4220, expressing msr(A) (RN4220+MsrA) and in telithromycin-resistant mutant strains 24, 1, and 4. (B) Western immunoblots of Msr(A)-6His in wild-type S. aureus 8325-4 and in the isogenic ΔclpX mutant, both expressing msr(A) (strains 8325-4+MsrA and ΔclpX+MsrA, respectively). Strains with empty pRMC2 plasmids were used as negative controls.
Because ClpX mainly acts in complex with ClpP, the effect of ClpX inactivation on resistance could be interpreted to be a dysfunction of the ClpXP protein degradation system. Thus, inactivation of ClpP could have an effect on Msr(A)-mediated resistance to TEL similar to that of the inactivation of ClpX. In addition to ClpX, ClpP may interact with ClpC (17), which is a homologue of ClpX and is an alternative means of recognizing and unfolding some cellular proteins targeted for degradation by ClpP. To clarify how inactivation of each component of the protein degradation machinery affects Msr(A)-mediated resistance, plasmid pRMC2+MsrA was transduced into mutant strains ΔclpC and ΔclpP. The resulting strains exhibited susceptibility to TEL similar to that seen with strain 8325-4+MsrA (Table 1), showing that only ClpX and not ClpC or ClpP interferes with Msr(A)-mediated TEL resistance. It has been shown that the ClpP-independent activity of ClpX plays a role in destabilization of protein-DNA and protein-protein complexes (18, 19). Considering two hypotheses of Msr(A) function (20), we can speculate that ClpX may destabilize the interaction of Msr(A) with a membrane protein that assists in antibiotic efflux or that it may destabilize the interaction of Msr(A) with a ribosome, thereby interfering with displacement of an antibiotic from the ribosome.
Although we observed that S. aureus expressing Msr(A) might develop TEL resistance through “loss-of-function” mutations in ClpX, it is unlikely that it causes a problem in clinical practice. It has been shown that ClpX affects the expression of virulence genes and is required for S. aureus virulence (21–23); therefore, clpX mutants resistant to TEL should be quickly eliminated unless the immune system of the host is compromised.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Free for the gift of the S. aureus knockout strains. We thank T. Foster for the gift of the pRMC2 plasmid. We thank M. Horsburgh and J. Alorabi for providing help transducing S. aureus strains.
This work was supported by Grant Agency of the Czech Republic project P302-12-P632 and Ministry of Education, Youth and Sports of the Czech Republic projects CZ.1.07/2.3.00/20.0055 and CZ.1.07/2.3.00/30.0003 and BIOCEV—Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University no. CZ.1.05/1.1.00/02.0109, from the European Regional Development Fund in the Czech Republic.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04367-14.
REFERENCES
- 1.Adriaenssens N, Coenen S, Versporten A, Muller A, Minalu G, Faes C, Vankerckhoven V, Aerts M, Hens N, Molenberghs G, Goossens H, ESAC Project Group . 2011. European Surveillance of Antimicrobial Consumption (ESAC): outpatient antibiotic use in Europe (1997–2009). J Antimicrob Chemother 66(Suppl 6):vi3–vi12. doi: 10.1093/jac/dkr453. [DOI] [PubMed] [Google Scholar]
- 2.Zmantar T, Kouidhi B, Miladi H, Bakhrouf A. 27 October 2011, posting date. Detection of macrolide and disinfectant resistance genes in clinical Staphylococcus aureus and coagulase-negative staphylococci. BMC Res Notes doi: 10.1186/1756-0500-4-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gatermann SG, Koschinski T, Friedrich S. 2007. Distribution and expression of macrolide resistance genes in coagulase-negative staphylococci. Clin Microbiol Infect 13:777–781. doi: 10.1111/j.1469-0691.2007.01749.x. [DOI] [PubMed] [Google Scholar]
- 4.Novotná G, Spízek J, Janata J. 2007. In vitro activity of telithromycin and quinupristin/dalfopristin against methicillin-resistant coagulase-negative staphylococci with defined resistance genotypes. Folia Microbiol (Praha) 52:593–599. doi: 10.1007/BF02932188. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds ED, Cove JH. 2005. Resistance to telithromycin is conferred by msr(A), msrC and msr(D) in Staphylococcus aureus. J Antimicrob Chemother 56:1179–1180. doi: 10.1093/jac/dki378. [DOI] [PubMed] [Google Scholar]
- 6.Urbaskova P. 1998. Resistance of bacteria to antibiotics. Selected methods [Rezistence bakterií k antibiotikům. Vybrané metody] TRIOS, Praha. [Google Scholar]
- 7.Novotna G, Janata J. 2006. A new evolutionary variant of the streptogramin A resistance protein, Vga(A)LC, from Staphylococcus haemolyticus with shifted substrate specificity towards lincosamides. Antimicrob Agents Chemother 50:4070–4076. doi: 10.1128/AAC.00799-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nair D, Memmi G, Hernandez D, Bard J, Beaume M, Gill S, Francois P, Cheung AL. 2011. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J Bacteriol 193:2332–2335. doi: 10.1128/JB.00027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. 2009. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 139:744–756. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stinson BM, Nager AR, Glynn SE, Schmitz KR, Baker TA, Sauer RT. 2013. Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell 153:628–639. doi: 10.1016/j.cell.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin A, Baker TA, Sauer RT. 2008. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol 15:1147–1151. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donaldson LW, Wojtyra U, Houry WA. 2003. Solution structure of the dimeric zinc binding domain of the chaperone ClpX. J Biol Chem 278:48991–48996. doi: 10.1074/jbc.M307826200. [DOI] [PubMed] [Google Scholar]
- 13.Thibault G, Yudin J, Wong P, Tsitrin V, Sprangers R, Zhao R, Houry WA. 2006. Specificity in substrate and cofactor recognition by the N-terminal domain of the chaperone ClpX. Proc Natl Acad Sci U S A 103:17724–17729. doi: 10.1073/pnas.0601505103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker TA, Sauer RT. 27 June 2011, posting date. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojtyra UA, Thibault G, Tuite A, Houry WA. 2003. The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. J Biol Chem 278:48981–48990. doi: 10.1074/jbc.M307825200. [DOI] [PubMed] [Google Scholar]
- 16.Cohen S, Sweeney HM. 1970. Transduction of methicillin resistance in Staphylococcus aureus dependent on an unusual specificity of the recipient strain. J Bacteriol 104:1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng J, Michalik S, Varming AN, Andersen JH, Albrecht D, Jelsbak L, Krieger S, Ohlsen K, Hecker M, Gerth U, Ingmer H, Frees D. 2013. Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J Proteome Res 12:547–558. doi: 10.1021/pr300394r. [DOI] [PubMed] [Google Scholar]
- 18.Haeusser DP, Lee AH, Weart RB, Levin PA. 2009. ClpX inhibits FtsZ assembly in a manner that does not require its ATP hydrolysis-dependent chaperone activity. J Bacteriol 191:1986–1991. doi: 10.1128/JB.01606-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burton BM, Williams TL, Baker TA. 2001. ClpX-mediated remodeling of mu transpososomes: selective unfolding of subunits destabilizes the entire complex. Mol Cell 8:449–454. doi: 10.1016/S1097-2765(01)00307-0. [DOI] [PubMed] [Google Scholar]
- 20.Kerr ID, Reynolds ED, Cove JH. 2005. ABC proteins and antibiotic drug resistance: is it all about transport? Biochem Soc Trans 33:1000–1002. doi: 10.1042/BST20051000. [DOI] [PubMed] [Google Scholar]
- 21.Frees D, Qazi SNA, Hill PJ, Ingmer H. 2003. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol Microbiol 48:1565–1578. doi: 10.1046/j.1365-2958.2003.03524.x. [DOI] [PubMed] [Google Scholar]
- 22.Frees D, Chastanet A, Qazi S, Sørensen K, Hill P, Msadek T, Ingmer H. 2004. Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol Microbiol 54:1445–1462. doi: 10.1111/j.1365-2958.2004.04368.x. [DOI] [PubMed] [Google Scholar]
- 23.Farrand AJ, Reniere ML, Ingmer H, Frees D, Skaar EP. 2013. Regulation of host hemoglobin binding by the Staphylococcus aureus Clp proteolytic system. J Bacteriol 195:5041–5050. doi: 10.1128/JB.00505-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.