

Abstract
The cell wall synthesis-inhibiting echinocandins, including caspofungin and micafungin, play important roles in the treatment of candidiasis and aspergillosis. Previous studies revealed that, in the haploid yeast Candida glabrata, sphingolipid biosynthesis pathway mutations confer caspofungin reduced susceptibility (CRS) but micafungin increased susceptibility (MIS). Here, we describe one Candida albicans strain (of 10 tested) that similarly yields CRS-MIS mutants at relatively high frequency. Mutants demonstrated increased levels of long-chain bases (sphingolipid pathway intermediates) and, unique to this strain, loss of His104/Pro104 heterozygosity in the TSC13-encoded enoyl reductase. CRS-MIS was similarly observed in a C. albicans homozygous fen1Δ fen12Δ laboratory strain and in diverse wild-type strains following exogenous long-chain-base treatment. Analogous to these results, CRS-MIS was demonstrated in an Aspergillus nidulans basA mutant encoding defective sphingolipid C4-hydroxylase and in its wild-type parent exposed to long-chain bases. Sphingolipids likely modulate echinocandin interaction with their Fks membrane target in all susceptible fungi, with potential implications for optimizing therapy with existing antifungals and the development of novel agents.
INTRODUCTION
Yeasts, including diploid Candida albicans and, to a lesser extent, haploid Candida glabrata, are commonly found at low levels among the normal mucosal flora. However, overgrowth of these opportunists due to reductions in the bacterial flora (e.g., following broad-spectrum antibiotic therapy) or deficiencies in cell-mediated immunity (e.g., associated with HIV/AIDS or cytotoxic chemotherapy) results in oropharyngeal or vaginal candidiasis and, furthermore, increases the risk of life-threatening systemic infection. Treatment of these and other fungal infections relies on a limited number of antifungals, specifically azoles, amphotericin B, and, increasingly, echinocandins. The latter include caspofungin, micafungin, and anidulafungin, which share a hexapeptide core but differ in their side chains, particularly their lipid moieties. In our current model for echinocandin mechanism of action, these lipid moieties target the drug to the external leaflet of the plasma membrane, where interaction with hot spot regions of Fks1 (and Fks2 in the case of C. glabrata) occurs, inhibiting the β-1,3-glucan synthase activity of this integral membrane protein (1, 2). Hot spot mutations are associated with treatment failure and, typically, cross-resistance to all three echinocandins (3).
We have previously described a novel, differential echinocandin susceptibility phenotype of C. glabrata mutants selected on low-level caspofungin (1, 4). These mutants, isolated at relatively high frequencies in 10 of 10 strains tested, exhibit caspofungin reduced susceptibility (CRS) but micafungin increased susceptibility (MIS). The responsible CRS-MIS mutations were mapped not to FKS genes but rather to FEN1, SUR4, SUR2, and IFA38, all four encoding enzymes involved in sphingolipid biosynthesis (Fig. 1). The mutations are loss of function, as evidenced by the equivalent phenotypes of gene disruptants, and result in the accumulation of sphingolipid pathway intermediates, in particular long-chain bases (LCBs), including phytosphingosine and dihydrosphingosine (Fig. 1).
FIG 1.

Sphingolipid biosynthesis pathway in yeast. Alternative names for certain enzymes are indicated in parentheses. The gray arrow indicates a minor alternative pathway yielding dihydroceramide in yeast. Loss-of-function mutation or absence (through gene disruption) of underlined proteins leads to accumulation of the LCBs phytosphingosine and dihydrosphingosine (or dihydrosphingosine alone in the case of Sur2/BasA mutation) and their phosphorylated derivatives (circled).
In light of these results with C. glabrata, it was important to examine other fungal pathogens for their CRS-MIS potential. We report here that 1 of 10 C. albicans strains tested yielded CRS-MIS mutants at relatively high frequency. This strain was uniquely heterozygous within the sphingolipid pathway gene TSC13, and its CRS-MIS mutants exhibited loss of this heterozygosity and apparent Tsc13 loss of function as evidenced by LCB accumulation. We also report that, in the model mold Aspergillus nidulans, mutation of the SUR2 ortholog basA conferred a CRS-MIS phenotype. Finally, exogenous LCBs conferred a CRS-MIS phenotype on wild-type strains of both C. albicans and A. nidulans, as well as Candida tropicalis and Candida krusei. Together, these data further support a general role for sphingolipids in modulating echinocandin susceptibility and have potential implications for antifungal therapy.
MATERIALS AND METHODS
Strains, media, and drugs.
Strains used in this study are listed in Table 1. Candida strains were cultured on or in YPD medium (1% yeast extract, 2% peptone, 2% dextrose) with or without 1.5% agar. An A. nidulans basA1 temperature-sensitive mutant and its parental strain GR5 were cultured on or in MAG medium (2% malt extract, 2% glucose, 0.2% peptone, trace elements, and vitamins) supplemented with uridine and uracil (5). Aspergillus fumigatus 293 was cultured on or in YPD or 2% dextrose-supplemented RPMI. All incubations were at 35°C (which yielded predominantly yeast rather than hyphal growth for C. albicans) except for routine propagation of A. nidulans strains, which was at 28°C. Sources and preparations of the echinocandins, myriocin, dihydrosphingosine, and phytosphingosine were described (1).
TABLE 1.
Strains used in this study
| Species | Strain | Source or reference |
|---|---|---|
| C. albicans | 34.80 | P. Nyirjesy (Drexel University) |
| 34.80-C1 | This study | |
| 34.80-C2 | This study | |
| 34.80-C3 | This study | |
| 34.80-C2r36 | This study | |
| 34.80-C2r37 | This study | |
| 20498.082 | 14 | |
| 20710-055 | 14 | |
| SC5314 | ATCC | |
| 90028 | ATCC | |
| 3356 | 8 | |
| 3359 | 8 | |
| 1489 | 8 | |
| 1490 | 8 | |
| 1491 | 8 | |
| BWP17 | 16 | |
| DAY286 | 16 | |
| BWP17-fen1Δ/FEN1 | This study | |
| BWP17-fen1Δ/fen1Δ | This study | |
| BWP17-fen12Δ/FEN12 | This study | |
| BWP17-fen12Δ/fen12Δ | This study | |
| SN95 | 10 | |
| SN95F1 (fen1Δ/fen1Δ) | 10 | |
| SN95F12 (fen12Δ/fen12Δ) | 10 | |
| SN95F1F12 (fen1Δ/fen1Δ fen12Δ/fen12Δ) | 10 | |
| SN95F1F12r11 (fen1Δ/fen1Δ fen12Δ/FEN12) | This study | |
| SN95F1F12r12 (fen1Δ/fen1Δ fen12Δ/FEN12) | This study | |
| C. tropicalis | 20491-032 | 14 |
| C. krusei | 6258 | ATCC |
| A. nidulans | GR5 | 5 |
| basA1 mutant | 5 | |
| A. fumigatus | 293 | Fungal Genetics Stock Center |
Sphingolipid analysis.
Cells were grown and prepared as described previously (1). Sphingosine species were extracted and analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) (6) at the Lipidomics Shared Resource of the Medical University of South Carolina (https://lipidomics.musc.edu/lipidomics-core).
Broth microdilution assays.
Yeast and conidia were assayed for echinocandin susceptibility as described previously (7). For Aspergillus species, conidia were harvested by suspension in phosphate-buffered saline supplemented with 0.01% Tween 20. Suspensions were diluted to a concentration of 1 × 104 conidia/ml. Combination studies with phytosphingosine (2.5 μg/ml) and myriocin (0.5 μg/ml) were performed as described previously (1) with 1 h of preincubation prior to the addition and serial dilution of echinocandins. MIC was defined as 80% inhibition relative to drug-free control growth following 24 h of incubation.
DNA purification, amplification, and sequencing.
DNA purification from yeast and conidia was performed as described previously (1). Very-long-chain fatty acid synthesis genes (FEN1, FEN12, IFA38, TPL1, and TSC13) and FKS1/GSC1 were PCR amplified and sequenced (for primers, see Table 2) in all C. albicans CRS-MIS mutants obtained. To examine the nucleotides encoding amino acid 104 in Tsc13, PCR products were sequenced in both directions. A. nidulans basA from the basA1 mutant and its parent strain GR5 were PCR amplified and sequenced (primers in Table 2) to confirm the Trp44Cys mutation previously described (5).
TABLE 2.
DNA primers used in this study and their application
| Primera | Application | Sequence (5′→3′)b |
|---|---|---|
| CaGSC1u229F | PCR | CCCATCATTGCAACAACAAAC |
| CaGSC1c1738R | PCR | TAAAGTGGCCACAGCAATG |
| CaGSC1c606R | Sequence | TGGTGACATACGGGAAGATC |
| CaGSC1c442F | Sequence | TATCCAGCTTGGTCTGCTGA |
| CaGSC1c1073F | Sequence | GTGTGATCACTCCACTTTACAG |
| CaGSC1c1554F | PCR | TTTCGTGCCTAGAGAATGGG |
| CaGSC1c3705R | PCR | CATCATGGCATTCATACCAGC |
| CaGSC1c1581F | Sequence | TCAACATTTGAGTCGTCGTATG |
| CaGSC1c2375F | Sequence | TGAGAGCTCCAACTTTCTTTG |
| CaGSC1c3061F | Sequence | GCTGAGTTCTTATTGCGTGC |
| CaGSC1c3504F | PCR | TTTGGGTGATGTTGCTGCTG |
| CaGSC1d41R | PCR | GAATAAACCACTTCCGCTCCC |
| CaGSC1c3553F | Sequence | GCAAGAACTTTGGCACAAATTGG |
| CaGSC1c4264F | Sequence | ACCGTTGGTGGTGCTAGATA |
| CaGSC1c4947F | Sequence | GACTGGTGCTGTTATTGCTG |
| CaFEN1u159F | PCR and screens | TTCACCACCAACTGAGTTCC |
| CaFEN1d54R | PCR | ACTAGCGACAACAACCGTCA |
| CaFEN1c523R | Sequence and screens | AAGCAGTAGCTCCATGATGAT |
| CaFEN1c321F | Sequence | GTGTGAACAAATCATCCCCAT |
| CaFEN1-URA3F | fen1Δ/FEN1 | ATTAATTATCACATTTTTTATTCTGGGTTTACCATTATCATGACAGTCAACACTAAGACC |
| CaFEN1-URA3R | fen1Δ/FEN1 | TCATACCTCTCTAAATACAACTCTTCTGCCTGACTATTTATAATTGGCCAGTTTTTTTCA |
| CaFEN1-ARG4F | fen1Δ/fen1Δ | TACACATTCGCTTAATTAATTATCACATTTTTTATTCTGGGTTTACCATTATCATGTCACAACAACAAGATAAAC |
| CaFEN1-ARG4R | fen1Δ/fen1Δ | CTAGCGACAACAACCGTCATACCTCTCTAAATACAACTCTTCTGCCTGACTATTTAACTTAAAATTGATTTTAAATTTC |
| CaFEN12-URA3F | fen12Δ/FEN12 | TCCTTCCTTCTTTATATTTTAATATCTTAACCACTGAACATGACAGTCAACACTAAGACC |
| CaFEN12-URA3R | fen12Δ/FEN12 | ACTCTCATCTTGACATGGTTGGGTTTACTTTTCCTTTTTATAATTGGCCAGTTTTTTTCA |
| CaFEN12-ARG4F | fen12Δ/fen12Δ | CCCACTTGTTCATTTCCTTCCTTCTTTATATTTTAATATCTTAACCACTGAACATGTCACAACAACAAGATAAAC |
| CaFEN12-ARG4R | fen12Δ/fen12Δ | CTGTGGTAACCAATTCACTCTCATCTTGACATGGTTGGGTTTACTTTTCCTTTTTAACTTAAAATTGATTTTAAATTTC |
| CaURA3c364R | Δ screens | TAGTAATATCTGCCCAACTAC |
| CaARG4c232R | Δ screens | GTCTGATTTGTTCTAATCCTTG |
| CaFEN12u300F | PCR and Δ screens | AATGGAAGAGGGAAGGCATAT |
| CaFEN12d110R | PCR | CCAATCGGGGATTGATCTAG |
| CaFEN12c558R | Sequence and Δ screens | TGGCACCCATTGAACTGAAG |
| CaFEN12c329F | Sequence | TCCATTCTGGTGGTTCATATC |
| CaFEN12u251F | Reconstitution | TAGATATTGCTGGAAGAGGAG |
| CaFEN12d215R | Reconstitution | TGCCCTTCAGATATCGTATCT |
| CaFEN12u403F | PCR screen | GGAACATTCGGAACCCTTCA |
| CaIFA38u351F | PCR | TCACCAACGACAACACGTCA |
| CaIFA38d170R | PCR | GGAAGAGACGTTAACTATGGG |
| CaIFA38c612F | Sequence | TAATGTCGGGCAATCCCACT |
| CaIFA38c700R | Sequence | GTGGGTTTTCAACGGTAGAG |
| CaTPL1u217F | PCR | CCCTGGATTCTTGTACCTTTA |
| CaTPL1d255R | PCR | GTTGGAATGTGCGATGGTATC |
| CaTPL1u183F | Sequence | GGCCAACGAACTAAAGTGTTG |
| CaTSC13u164F | PCR | GCTCTGTTGGTAAAGTGTCG |
| CaTSC13d120R | PCR | CTCGAACTGTGACGGACATT |
| CaTSC13c14F | Sequence | AAGTCAAATCACGTTCCAGATC |
| CaTSC13c612R | Sequence | ACCATATACAACTGAGCTTGG |
| CaTSC13c449F | Sequence | CTAACGCAACAATGCCAGCT |
| CaTSC13c81F | Reconstitution | TCAAGAATTAATCAACGAATTGAGTTCcGATAATCATATTc |
| CaTSC13c605R | Reconstitution | ACAACTGAGCTTGGTAATTCATTAACATGaAAAAAGTATTc |
| AnBasAc3F | PCR, sequence | GGCTACAAACACAACTTTGCT |
| AnBasAc660R | PCR | GTGAAGGAAATACTGCCATGT |
Numbers in primer names correspond to nucleotide location upstream (u) or within the coding region (c) relative to the start codon or downstream (d) relative to the stop codon.
Underlined regions of disruption primers correspond to selection marker sequences.
Lowercase nucleotides represent synonymous mutations used to confirm a transformant's origins.
Multilocus sequence typing.
Portions of 7 housekeeping genes (AAT1, AAC, ADP1, PMI1, SYA1, VPS13, and ZWF1) were PCR amplified (using the recommended primers), sequenced, and compared to the C. albicans MLST database sequences (http://calbicans.mlst.net) (8). Five strains with equivalent sequence types were obtained through MLST curator M.-E. Bougnoux (Fungal Biology and Pathogenicity Unit, Institut Pasteur, France).
C. albicans gene deletion and mutant complementation.
Deletion of FEN1 and FEN12 in strain BWP17 was performed with two rounds of transformation. In the first round, wild-type BWP17 (ura3/ura3 his1/his1 arg4/arg4) was transformed with a PCR-amplified CaURA3 PRODIGE cassette (for primers, see Table 2) and selection on SD-Ura as previously described (9). Transformants were screened by PCR with the indicated primers (Table 2) to identify heterozygous disruptants. Homozygous disruptants were obtained by transforming a heterozygous disruptant with a PCR-amplified CaARG4 PRODIGE cassette and selection on SD-Arg. PCR screens and sequencing (Table 2) confirmed disruption of both alleles.
Revertants of CRS-MIS mutant 34.80-C2 were constructed by transformation with a PCR product generated with wild-type SC5314 DNA and primers CaTSC13c81F and CaTSC13c605R (Table 2; both include a synonymous mutation to confirm the transformant's origins). Transformants were selected at 42°C, a temperature which permits growth of wild-type C. albicans but restricts growth of CRS-MIS mutants. Transformants were subsequently screened to confirm loss of temperature sensitivity at 42°C, further screened by sequencing their CaTSC13u164F-CaTSC13d120R PCR products in both directions, and tested by broth microdilution for echinocandin susceptibility.
Revertants of SN95F1F12 (fen1Δ/fen1Δ fen12Δ/fen12Δ) were constructed by transformation with a PCR product generated with wild-type SC5314 DNA and primers CaFEN12u251F and CaFEN12d215R (Table 2). Transformants were selected at 42°C, screened to confirm loss of temperature sensitivity, further screened by PCR with two pairs of primers (CaFEN12c329F-CaFEN12d110R and CaFEN12u403F-CaFEN12c558R) to confirm FEN12 restoration, and tested by broth microdilution.
RESULTS
C. albicans strain 34.80 readily yields CRS-MIS mutants.
C. albicans CRS mutants were selected by spreading 1 × 106 cells of 10 diverse strains on YPD plates containing caspofungin ranging in concentration from 0.12 to 0.35 μg/ml, followed by incubation at 35°C for 3 to 4 days. Colonies (10 to 48 per strain) were screened for micafungin hypersusceptibility by spot assay on 0.01 μg/ml micafungin-containing YPD plates, and candidate CRS-MIS mutants were subsequently assayed by broth microdilution. Only one of these strains, the vaginal isolate 34.80, yielded CRS-MIS mutants (specifically, 5 mutants from 30 colonies screened). Compared to their parent, these mutants exhibited 8-fold elevated caspofungin MIC and 8- to 16-fold reduced micafungin MIC (64- to 128-fold differential) (Table 3).
TABLE 3.
Echinocandin MICs of C. albicans mutants relative to their parent strains and revertants
| Strain | MIC (μg/ml) |
Fold differential | |
|---|---|---|---|
| Caspofungin | Micafungin | ||
| 34.80 | 0.03 | 0.03 | |
| 34.80-C1 | 0.25 | 0.002 | 128 |
| 34.80-C2 | 0.25 | 0.002 | 128 |
| 34.80-C3 | 0.25 | 0.004 | 64 |
| 34.80-C2r36 | 0.03 | 0.03 | 1 |
| 34.80-C2r37 | 0.03 | 0.03 | 1 |
| BWP17 | 0.03 | 0.03 | |
| DAY286 | 0.03 | 0.03 | 1 |
| BWP17-fen1Δ/FEN1 | 0.06 | 0.03 | 2 |
| BWP17-fen1Δ/fen1Δ | 0.06 | 0.016 | 4 |
| BWP17-fen12Δ/FEN12 | 0.03 | 0.03 | 1 |
| BWP17-fen12Δ/fen12Δ | 0.06 | 0.03 | 2 |
| SN95 | 0.03 | 0.016 | |
| SN95F1 | 0.06 | 0.016 | 2 |
| SN95F12 | 0.06 | 0.016 | 2 |
| SN95F1F12 | 0.25 | 0.002 | 64 |
| SN95F1F12r11 | 0.06 | 0.016 | 2 |
| SN95F1F12r12 | 0.06 | 0.016 | 2 |
CRS-MIS mutants of strain 34.80 have wild-type FKS1 but accumulate LCBs.
Sequencing of the FKS1 (GSC1; orf19.2929) complete coding region from three 34.80 CRS-MIS mutants revealed no changes compared to the parental strain (data not shown). Therefore, a representative mutant (34.80-C2) and its parent were subjected to sphingolipid analysis. This mutant exhibited a 5.5-fold increase in dihydrosphingosine (600 pmol per sample) and 12-fold increase in phytosphingosine (5,300 pmol) compared to wild-type 34.80 (110 and 440 pmol, respectively). Additionally, phosphorylated forms of these LCBs (Fig. 1) increased 10-fold and 68-fold, respectively, in the mutant (40 and 270 pmol compared to 4 and 4 pmol, respectively).
CRS-MIS in strain 34.80 is associated with loss of TSC13 heterozygosity.
The accumulation of both dihydrosphingosine and phytosphingosine and their phosphorylated derivatives suggests a loss-of-function mutation in one of the enzymes involved in very-long-chain fatty acid elongation (Fig. 1). We therefore amplified and sequenced the C. albicans FEN1 (orf19.6343), FEN12 (SUR4; orf19.908), IFA38 (orf19.3859), TPL1 (PHS1; orf19.5156), and TSC13 (orf19.3293) coding regions from CRS-MIS mutants of strain 34.80. For all but one gene, these sequences were identical to those of the wild-type C. albicans strains SC5314 and WO-1, whose genomes have been sequenced (www.candidagenome.org). The only difference was observed in TSC13, where a homozygous mutation was discovered in all 34.80 CRS-MIS mutants. This mutation would result in substitution of the wild-type histidine at amino acid position 104 (His104/His104) with proline (Pro104/Pro104). The corresponding position in TSC13 of the 34.80 parental strain was clearly heterozygous in both the forward (Fig. 2) and reverse (not shown) sequences, encoding His104/Pro104. Thus, CRS-MIS mutants of strain 34.80 exhibited a loss-of-heterozygosity event. TSC13 was sequenced from all 10 C. albicans strains including SC5314 that failed to yield CRS-MIS mutants, and all encode His104/His104 (Fig. 2 and data not shown).
FIG 2.

CRS-MIS mutants of C. albicans strain 34.80 exhibit loss of TSC13/tsc13 heterozygosity. DNA sequence chromatograms showing TSC13/tsc13 heterozygosity in the 34.80 parent, loss of heterozygosity in representative CRS-MIS mutant 34.80-C2, and heterozygosity again in the 34.80-C2 revertant (confirmed as originating from the transforming DNA by the synonymous T-to-C mutation incorporated into primer CaTSC13c81F). For comparison, the equivalent TSC13 region from laboratory strain SC5314 is also shown.
To determine if the identified tsc13/tsc13 mutation was solely responsible for CRS-MIS, representative mutant 34.80-C2 was transformed with a TSC13 PCR product generated from wild-type SC5314 DNA and primers (Table 2) containing synonymous mutations to confirm the transformant's origins. Transformants were selected at 42°C, since the growth of C. albicans CRS-MIS mutants is restricted at this temperature (Fig. 3A). Two of 13 transformants (r36 and r37) exhibited 42°C tolerance, equivalent to parent 34.80 (Fig. 3A), and both were confirmed by sequencing (Fig. 2) and microdilution assay (Table 3) to represent TSC13/tsc13 revertants.
FIG 3.
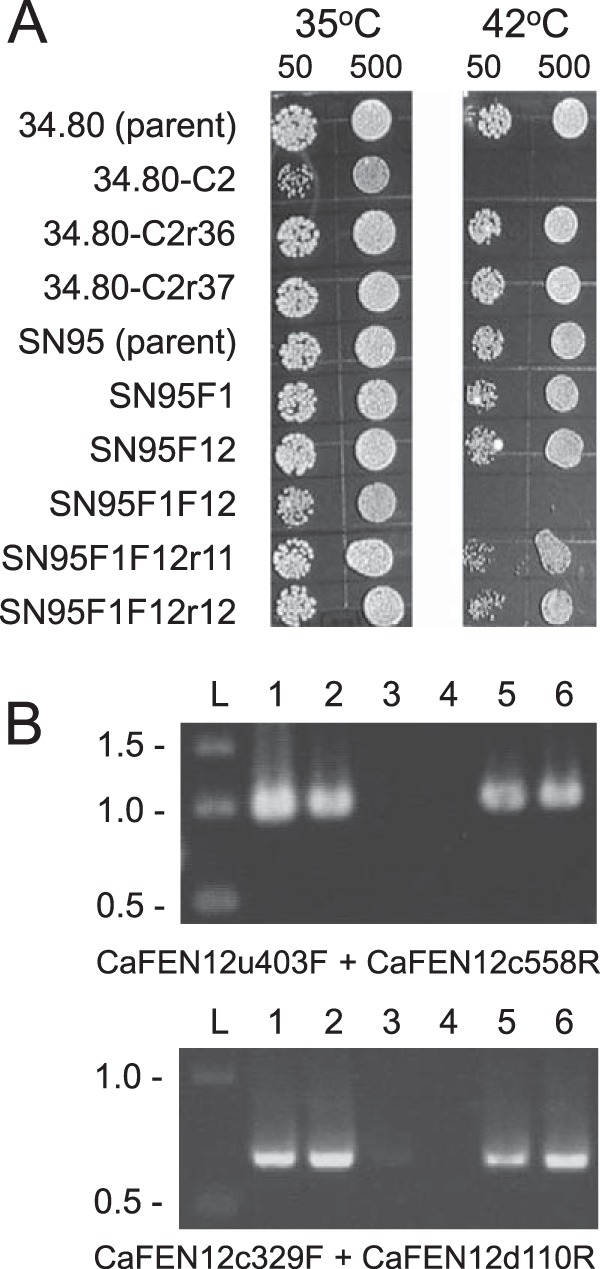
(A) Temperature sensitivity of C. albicans mutants exhibiting CRS-MIS (34.80-C2 and SN95F1F12) at 42°C and its reversion following reconstitution of wild-type TSC13 (34.80-C2r36 and 34.80-C2r37) or FEN12 (SN95F1F12r11 and SN95F1F12r12). Approximately 50 or 500 cells were spotted in duplicate on YPD agar plates and incubated at 35°C or 42°C for 2 days. (B) PCR confirmation of FEN12 restoration in SN95F1F12r11 and SN95F1F12r12. Genomic DNA was amplified with the indicated primer pairs and analyzed by gel electrophoresis. Lane L, DNA ladder (kbp); lane 1, SN95 parent; lane 2, SN95F1; lane 3, SN95F12; lane 4, SN95F1F12; lane 5; SN95F1F12r11; lane 6, SN95F1F12r12. Expected size of CaFEN12u403uF-CaFEN12c558R product = 961 bp; expected size of CaFEN12c329F-CaFEN12d110R product = 671 bp.
Double disruption of FEN1 and FEN12 confers CRS-MIS in a C. albicans laboratory strain.
To confirm that a mutational defect in sphingolipid biosynthesis confers CRS-MIS in C. albicans, gene disruption studies were undertaken in laboratory strains. TSC13 is likely an essential gene (candidagenome.org); consequently, the nonessential genes FEN1 (orf19.6343) and FEN12 (SUR4; orf19.908) were targeted. Compared to both parent BWP17 and marker-matched strain DAY286, heterozygous disruptants of either gene exhibited echinocandin susceptibilities that were essentially unchanged (≤2-fold differentials), and their homozygous disruptants exhibited only minimal CRS-MIS (4-fold differentials; Table 3). Similarly, homozygous disruptants in these individual genes constructed by Sharma et al. (10) in strain SN95 by an alternative mechanism exhibited little or no change in echinocandin susceptibilities. In contrast, their double homozygous disruptant SN95F1F12 (fen1Δ/fen1Δ fen12Δ/fen12Δ) exhibited a 128-fold CRS-MIS differential (Table 3). This result was confirmed by transforming SN95F1F12 with a wild-type FEN12 PCR product followed by selection at 42°C; as noted above, C. albicans CRS-MIS mutants exhibit temperature sensitivity at 42°C. Two of 20 transformants (SN95F1F12 r11 and SN95F1F12 r12) were temperature tolerant, equivalent to parent SN95 (Fig. 3A), demonstrated reconstitution of one FEN12 allele in PCR screens (Fig. 3B), and exhibited reversion of their CRS-MIS phenotype (Table 3). The requirement for double disruption implies that FEN1 and FEN12 are largely redundant, at least with respect to the CRS-MIS phenotype.
Strain 34.80 does not represent a high-risk CRS-MIS clade.
The relatively high frequency of CRS-MIS mutation through loss of TSC13/tsc13 heterozygosity in C. albicans strain 34.80 is analogous to the frequent development of flucytosine resistance due to loss of FUR1/fur1 heterozygosity in clade I strains (11). We hypothesized that strain 34.80 may similarly represent a clade of “high-risk” CRS-MIS strains. To test this, strain 34.80 was subjected to multilocus sequence typing (http://calbicans.mlst.net) (8), which identified it as sequence type 124. Five strains reported to have the equivalent sequence type were obtained from the typing database curators, and their TSC13 genes were amplified and sequenced. None, however, demonstrated heterozygosity, and consistent with this, none yielded CRS-MIS mutants (data not shown).
Exogenous LCB induces CRS-MIS in wild-type C. albicans.
Although mutations yielding the CRS-MIS phenotype may be rare in C. albicans, we knew from our studies with C. glabrata that this phenotype could be induced in wild-type strains upon exogenous addition of phytosphingosine or dihydrosphingosine LCBs (1). Indeed, CRS-MIS was similarly induced in all four wild-type C. albicans strains tested after preincubation with phytosphingosine (Fig. 4A). Subinhibitory levels of myriocin, an inhibitor of the initial step (serine palmitoyltransferase) of LCB synthesis, did not have this effect (Fig. 4B), consistent with the mechanism previously proposed in which LCB accumulation is responsible for CRS-MIS. Indeed, myriocin partially reversed the CRS-MIS phenotype of mutant 34.80-C2, while exogenous phytosphingosine exaggerated the phenotype (Fig. 4C). As expected, phytosphingosine supplementation could overcome the effects of myriocin (Fig. 4B).
FIG 4.

Effects of exogenous LCB and myriocin on wild-type C. albicans and CRS mutants. (A) Cells were pretreated with 2.5 μg/ml phytosphingosine (PHS) for 1 h and then assayed for caspofungin (CSF) and micafungin (MCF) susceptibilities. Results are shown as fold changes in MIC compared to the same strains without phytosphingosine. (B) Cells were pretreated as indicated with 2.5 μg/ml phytosphingosine, 0.5 μg/ml myriocin, or both for 1 h and then assayed for echinocandin susceptibilities. Results are shown as fold changes in MIC compared to the same strains without pretreatment. (C) CRS-MIS mutant 34.80-C2 treated and assayed as described above. MICs are representative of three independent experiments.
Exogenous LCB induces CRS-MIS in other susceptible fungi.
To further examine the CRS-MIS spectrum, echinocandin susceptibilities were determined in additional Candida and Aspergillus species in the presence and absence of exogenous phytosphingosine. Similar to C. glabrata and C. albicans, a CRS-MIS phenotype was induced in wild-type C. krusei strain 6258 (4-fold CRS, 4-fold MIS), C. tropicalis strain 20491-032 (4-fold CRS, 4-fold MIS), and A. nidulans strain GR5 (4-fold CRS, 8-fold MIS). With A. fumigatus strain 293, there was no detectable CRS with LCB supplementation, but 4- to 8-fold MIS was observed.
An A. nidulans sphingolipid pathway mutant exhibits CRS-MIS.
The Aspergillus basA gene is orthologous to yeast SUR2, encoding sphingolipid C4-hydroxylase, responsible for converting dihydrosphingosine to phytosphingosine. Unlike yeast SUR2, Aspergillus basA is essential, but a temperature-sensitive basA1 mutant has been isolated and characterized (5, 12). In C. glabrata, sur2 mutations lead to dihydrosphingosine accumulation and a CRS-MIS phenotype (1). Similarly, the A. nidulans basA1 mutant was shown to accumulate dihydrosphingosine (5) and indeed exhibits a CRS-MIS phenotype (64-fold differential in caspofungin compared to micafungin MIC relative to parent strain GR5) (Fig. 5). As in C. glabrata and C. albicans CRS-MIS mutants, myriocin treatment reversed the CRS-MIS phenotype of the basA1 mutant (Fig. 5).
FIG 5.

A. nidulans basA mutation yields CRS-MIS. A. nidulans basA1 mutant (Trp44Cys substitution) and its parent strain GR5 were assayed for caspofungin (CSF) and micafungin (MCF) susceptibilities with or without myriocin pretreatment (4 μg/ml, 1 h). Results are shown as fold changes in MIC compared to the parent strain. MICs are representative of three independent experiments.
DISCUSSION
Based on our previous studies with C. glabrata, we examined the potential for CRS-MIS-conferring mutations in C. albicans, the dominant yeast among the normal mucosal flora and a major agent of life-threatening fungal infection in the immunocompromised. Initial studies with a small number of strains that included the recent vaginal isolate 34.80 suggested that CRS-MIS mutations might be similarly common in C. albicans. Furthermore, sphingolipid analysis showing LCB accumulation, and sequence analysis identifying a mutation in TSC13-encoded enoyl reductase, confirmed that CRS-MIS in this diploid yeast employed the same mechanism as in haploid C. glabrata. However, further studies with additional strains revealed 34.80 to be the exception in terms of CRS-MIS potential and relatedly in terms of TSC13 heterozygosity. While 9 other strains examined here, and both strains whose genomes have been sequenced (www.candidagenome.org), encode His104/His104, strain 34.80 encodes His104/Pro104, which undergoes loss of heterozygosity to Pro104/Pro104 in all of its CRS-MIS mutants. In addition to Pro being a nonconservative change, His at this position is well conserved in orthologs throughout the fungal kingdom (although notable exceptions include most Aspergillus species, where Pro is at this position). We surmise that Pro104 substitution reduces but does not eliminate C. albicans enoyl reductase activity, since the latter would likely have deleterious effects on growth, which were not apparent in any of the CRS-MIS mutants examined.
The relatively high frequency of CRS-MIS mutant recovery in haploid C. glabrata can be explained by the many possible loss-of-function mutations in any one of at least four different sphingolipid pathway enzymes. On the other hand, in the absence of heterozygosity analogous to the TSC13/tsc13 of strain 34.80, diploid C. albicans would be expected to yield CRS-MIS mutants at a much lower frequency. Nonetheless, strain 34.80 established the potential for CRS-MIS in this yeast, and indeed this phenotype was demonstrated in both a homozygous fen1Δ fen12Δ laboratory strain and in wild-type strains treated with exogenous LCB.
Evolutionarily, C. glabrata and C. albicans are relatively distant. We anticipated, therefore, that other echinocandin-susceptible fungal species would possess the potential for CRS-MIS, and this was confirmed for C. tropicalis, C. krusei, and A. nidulans in susceptibility assays with and without exogenous LCB. As final confirmation, an A. nidulans basA (SUR2 ortholog) mutant was tested and shown to exhibit CRS-MIS.
CRS-MIS has clear relevance for understanding the echinocandin mechanism of action, i.e., the differential modulation by LCBs of caspofungin compared to micafungin interaction with Fks hot spots, which we have recently shown to be adjacent to or embedded within the outer leaflet of the plasma membrane (2). Phytosphingosine is known to alter cell signaling, and consequently cell wall integrity, in vitro (13); however, we would expect cell signaling effects to impact susceptibility to caspofungin and micafungin equally. Therefore, the differential effects associated with CRS-MIS are more likely a result of the direct interaction of sphingolipids, echinocandins, and Fks proteins within the membrane environment.
For C. glabrata, there is indirect evidence based on the characterization of several clinical isolates that CRS-MIS mutations occur in patients, potentially in response to caspofungin treatment (1, 4). If so, then therapeutic strategies for C. glabrata infection involving sequential treatment with caspofungin followed by micafungin might be preferable to monotherapy. The same rationale may be applicable to Aspergillus infections, in light of our demonstration here of CRS-MIS in the A. nidulans genetic model. As with C. glabrata, Aspergillus species have haploid genomes and multiple mutational targets within the sphingolipid pathway. To date, however, no Aspergillus CRS-MIS mutants have been reported; this may be attributed to reduced fitness, as suggested by the observation that basA disruption is lethal, while a comparable sur2 mutation is tolerated in yeast (1).
With respect to C. albicans, the data presented here suggest that CRS-MIS mutants are likely to be rare, and consistent with this we were unable to identify such mutants in a large collection of clinical isolates for which caspofungin and micafungin susceptibility data were available (14) (data not shown). Interestingly, however, caspofungin treatment of C. albicans cells has been reported to downregulate expression of FEN12 (15), which could lead to a transient increase in LCBs that would enhance micafungin susceptibility. Whether or not this mechanism operates and can be exploited to improve echinocandin therapy of C. albicans infection warrants further study.
ACKNOWLEDGMENTS
We thank D. Diekema, M. Pfaller, M. Castanheira, P. Nyirjesy, and M. E. Bougnoux for Candida strains, S. Harris for A. nidulans GR5 and basA1 strains, and K. Ganesan for C. albicans SN95 and its fen1Δ fen12Δ disruptants.
These studies were supported by National Institutes of Health grants R56AI100580 and AI075272 and Astellas Investigator-Initiated Research grant 251117.
REFERENCES
- 1.Healey KR, Katiyar SK, Raj S, Edlind TD. 2012. CRS-MIS in Candida glabrata: sphingolipids modulate echinocandin-Fks interaction. Mol Microbiol 86:303–313. doi: 10.1111/j.1365-2958.2012.08194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson ME, Katiyar SK, Edlind TD. 2011. New Fks hot spot for acquired echinocandin resistance in Saccharomyces cerevisiae and its contribution to intrinsic resistance of Scedosporium species. Antimicrob Agents Chemother 55:3774–3781. doi: 10.1128/AAC.01811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arendrup MC, Perlin DS. 2014. Echinocandin resistance: an emerging clinical problem? Curr Opin Infect Dis 27:484–492. doi: 10.1097/QCO.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Healey KR, Katiyar SK, Castanheira M, Pfaller MA, Edlind TD. 2011. Candida glabrata mutants demonstrating paradoxical reduced caspofungin susceptibility but increased micafungin susceptibility. Antimicrob Agents Chemother 55:3947–3949. doi: 10.1128/AAC.00044-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S, Bao D, Yuen G, Harris SD, Calvo AM. 2007. basA regulates cell wall organization and asexual/sexual sporulation ratio in Aspergillus nidulans. Genetics 176:243–253. doi: 10.1534/genetics.106.068239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bielawski J, Pierce JS, Snider J, Rembiesa B, Szulc ZM, Bielawska A. 2010. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). Adv Exp Med Biol 688:46–59. doi: 10.1007/978-1-4419-6741-1_3. [DOI] [PubMed] [Google Scholar]
- 7.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61:704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 8.Bougnoux ME, Tavanti A, Bouchier C, Gow NA, Magnier A, Davidson AD, Maiden MC, D'Enfert C, Odds FC. 2003. Collaborative consensus for optimized multilocus sequence typing of Candida albicans. J Clin Microbiol 41:5265–5266. doi: 10.1128/JCM.41.11.5265-5266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edlind TD, Henry KW, Vermitsky JP, Edlind MP, Raj S, Katiyar SK. 2005. Promoter-dependent disruption of genes: simple, rapid, and specific PCR-based method with application to three different yeast. Curr Genet 48:117–125. doi: 10.1007/s00294-005-0008-3. [DOI] [PubMed] [Google Scholar]
- 10.Sharma S, Alfatah M, Bari VK, Rawal Y, Paul S, Ganesan K. 2014. Sphingolipid biosynthetic pathway genes FEN1 and SUR4 modulate amphotericin B resistance. Antimicrob Agents Chemother 58:2409–2414. doi: 10.1128/AAC.02130-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dodgson AR, Dodgson KJ, Pujol C, Pfaller MA, Soll DR. 2004. Clade-specific flucytosine resistance is due to a single nucleotide change in the FUR1 gene of Candida albicans. Antimicrob Agents Chemother 48:2223–2227. doi: 10.1128/AAC.48.6.2223-2227.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li S, Du L, Yuen G, Harris SD. 2006. Distinct ceramide synthases regulate polarized growth in the filamentous fungus Aspergillus nidulans. Mol Biol Cell 17:1218–1227. doi: 10.1091/mbc.E05-06-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dickson RC. 2010. Roles for sphingolipids in Saccharomyces cerevisiae. Adv Exp Med Biol 688:217–231. doi: 10.1007/978-1-4419-6741-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfaller MA, Boyken L, Hollis RJ, Messer SA, Tendolkar S, Diekema DJ. 2005. In vitro activities of anidulafungin against more than 2,500 clinical isolates of Candida spp., including 315 isolates resistant to fluconazole. J Clin Microbiol 43:5425–5427. doi: 10.1128/JCM.43.11.5425-5427.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu TT, Lee RE, Barker KS, Wei L, Homayouni R, Rogers PD. 2005. Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrob Agents Chemother 49:2226–2236. doi: 10.1128/AAC.49.6.2226-2236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson RB, Davis D, Mitchell AP. 1999. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriol 181:1868–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]