

Background: Sodium-calcium exchanger (NCX1) regulates calcium in renal epithelial cells.
Results: Na,K-ATPase β-subunit regulates NCX1 membrane localization and reduced NCX1 expression or its functional inhibition increases cell migration.
Conclusion: NCX1 plays a pivotal role in activation of calcium dependent migration via calmodulin/PI3K/ERK.
Significance: Identifying regulators of epithelial cell motility is important in establishing novel therapeutic targets in fibrosis and cancer.
Keywords: Calcium, Cell Migration, Extracellular Signal-regulated Kinase (ERK), Na+/K+-ATPase, Sodium-Calcium Exchange, NCX1
Abstract
Na+/Ca2+ exchanger-1 (NCX1) is a major calcium extrusion mechanism in renal epithelial cells enabling the efflux of one Ca2+ ion and the influx of three Na+ ions. The gradient for this exchange activity is provided by Na,K-ATPase, a hetero-oligomer consisting of a catalytic α-subunit and a regulatory β-subunit (Na,K-β) that also functions as a motility and tumor suppressor. We showed earlier that mice with heart-specific ablation (KO) of Na,K-β had a specific reduction in NCX1 protein and were ouabain-insensitive. Here, we demonstrate that Na,K-β associates with NCX1 and regulates its localization to the cell surface. Madin-Darby canine kidney cells with Na,K-β knockdown have reduced NCX1 protein and function accompanied by 2.1-fold increase in free intracellular calcium and a corresponding increase in the rate of cell migration. Increased intracellular calcium up-regulated ERK1/2 via calmodulin-dependent activation of PI3K. Both myosin light chain kinase and Rho-associated kinase acted as mediators of ERK1/2-dependent migration. Restoring NCX1 expression in β-KD cells reduced migration rate and ERK1/2 activation, suggesting that NCX1 functions downstream of Na,K-β in regulating cell migration. In parallel, inhibition of NCX1 by KB-R7943 in Madin-Darby canine kidney cells, LLC-PK1, and human primary renal epithelial cells (HREpiC) increased ERK1/2 activation and cell migration. This increased migration was associated with high myosin light chain phosphorylation by PI3K/ERK-dependent mechanism in HREpiC cells. These data confirm the role of NCX1 activity in regulating renal epithelial cell migration.
Introduction
Enhanced cell migration is a prerequisite for tumor cell invasion and metastasis. Calcium-dependent signaling is essential for directional movement, reorganization of actin cytoskeleton, and cleaving of cell-cell and cell-substrate attachments in the regulation of cell migration (1–3). Thus, alteration in intracellular calcium levels can contribute to cell migration and invasion. Regulation of intracellular Ca2+ involves a balance between Ca2+ influx and efflux, which is governed by membrane-associated ion channels, ATPases, exchangers, and binding proteins. Indeed, modulation of specific calcium channels or pumps is associated with certain cancers. For example, transient receptor potential channel 8 (TRPM8) up-regulation in prostate cancer (4) and sarco/endoplasmic reticulum calcium transport ATPase 3 (SERCA3)2 down-regulation in colon cancer (5) has been reported.
In renal epithelial cells, the sodium-calcium exchanger 1 (NCX1), plasma membrane calcium ATPase (PMCA), and SERCA ATPases are the major regulators of intracellular Ca2+ ion concentration, with NCX1 being the protein responsible for Ca2+ extrusion (6, 7). The two other isoforms NCX2 and NCX3 are not expressed in the kidney (8). NCX1 consists of a 9 α-helical transmembrane domain and a large (550 residue) cytosolic domain that has two Ca2+ binding sites that mediate the extrusion of a Ca2+ and the influx of 3 Na+ in one exchange movement in the forward mode (9). NCX1 has also been shown to function in reverse mode, i.e. the exchanger can cause an influx of the Ca2+ ions into the cells depending on intracellular Na+, Ca2+, pH, ATP, and membrane potential (10).
Although there is no direct evidence linking NCX1 to cancer, there are isolated studies indicating that NCX1 is involved in cell adhesion. For example, cell adhesion in prostate epithelial cells induced by stromal cell co-culture caused an up-regulation of NCX1 transcript level among other genes involved in cell adhesion (11). Furthermore, inhibition of NCX1 activity by KB-R7943 down-regulated cell adhesion molecule ICAM1 and suppressed cell adhesion (12). NCX1 works in close partnership with Na,K-ATPase, by utilizing the sodium gradient generated by Na,K-ATPase to drive calcium efflux. Na,K-ATPase has also been shown to function as a motility and tumor suppressor (13, 14) and is involved in the maintenance of epithelial polarity (15) and cell adhesion (16, 17). Moreover, we previously reported that knockdown of Na,K-ATPase β1-subunit (Na,K-β) in MDCK cells led to mesenchymal phenotype accompanied by increased cell proliferation via activation of phosphoinositide-3 kinase (PI3K)/Akt and extracellular-signal-regulated kinase (ERK1/2) pathways (18).
In this study we demonstrate that MDCK cells with Na,K-β knockdown (β-KD) showed reduced NCX1 protein expression leading to an increase in intracellular calcium. Furthermore, we provide evidence that Na,K-β interacts with NCX1 and regulates NCX1 membrane localization. The activation of ERK1/2 and enhanced cell migration in β-KD cells was calcium-dependent and could be reversed when NCX1 was overexpressed in β-KD cells. Increased intracellular calcium activated calmodulin/PI3K/ERK signaling leading to myosin light chain kinase/Rho-associated protein kinase-dependent migration. Furthermore, inhibition of NCX1 in MDCK, LLC-PK1, and human primary renal epithelial cells (HREpiC) also led to activation of ERK1/2 and enhanced cell migration. Thus, our data reveal a novel role of forward mode NCX1 in regulation of cell migration in renal epithelial cells.
EXPERIMENTAL PROCEDURES
Cell Lines and Maintenance
DMEM supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 25 units/ml penicillin, and 25 μg/ml streptomycin was used to grow MDCK and LLC-PK1 cells. Similarly, MDCK-Na,K-β1-KD and rescue cells (β-KD/R) as described in Barwe et al. (18) were also cultured in supplemented DMEM. β-KD cells were maintained in 10 μg/ml puromycin, and β-KD/R cells were maintained in 10 μg/ml puromycin and 500 μg/ml neomycin. Full-length canine NCX1, a kind gift from Dr. Kenneth Philipson, UCLA (19), was transfected in β-KD cells using the calcium phosphate transfection method, and NCX1 expressing β-KD cells were selected with 10 μg/ml puromycin and 100 μg/ml hygromycin post transfection. β-KD cells were also transfected with pWZL-neo Δ-p85 (Addgene Plasmid #10888) and selected with 10 μg/ml puromycin and 500 μg/ml neomycin post transfection. The cells that survived the selection media were confirmed to express the transfected constructs and were utilized for the experiments. HREpiC purchased from ScienCell™ (Carlsbad, CA) were maintained as per the supplier's recommendations and treated with inhibitors as indicated.
Antibodies and Chemicals
Monoclonal Na,K-β1 (M17-P5-F11) antibody from ThermoFisher Scientific Inc. (Waltham, MA) and monoclonal NCX1 antibody from Abcam® (Cambridge, MA) were used. Antibodies against PMCA4 and SERCA2 were purchased from BIOSS antibodies (Woburn, MA). Antibodies against phospho-p44/p42 MAPK (ERK1/2), total p44/p42 MAPK (ERK1/2), phospho-Akt (Ser-473), total Akt, phospho p70S6 kinase (Thr-389), phospho MLC2 (Ser-19), and horseradish peroxidase-conjugated secondary antibodies against mouse and rabbit IgG were obtained from Cell Signaling Technology® (Lexington, KY). Monoclonal β-actin antibody was purchased from Sigma.
KB-R7943, PD98059, U0126, and LY294002 were purchased from Tocris (Minneapolis, MN). MK-2206 and Y27632 were from Selleckchem (Houston, TX). W-13, KN-93, and ML-7 were from Cayman (Ann Harbor, MI).
Immunoblotting and Co-immunoprecipitation
Cells were lysed in a buffer containing 20 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerol phosphate, 1 mm sodium vanadate, 1 mm phenylmethylsulfonyl fluoride (PMSF), 5 mg/ml anti-papain, leupeptin, and pepstatin. For detection of NCX1 protein, cells were lysed in the lysis buffer supplemented with 2% sodium dodecyl sulfate (SDS). Based on protein estimation, 50 or 100 μg of cell lysates were resolved by SDS-PAGE and transferred onto nitrocellulose membrane. The immunoblots were blocked in 5% nonfat dried milk in Tris-buffered saline with 0.1% Tween 20 (TBST). Primary antibodies were diluted either in 5% bovine serum albumin (BSA) or nonfat dried milk in TBST and incubated overnight at 4 °C. Secondary antibodies were diluted in 5% nonfat dried milk in TBST. Immunoblots were developed with chemiluminescent lightning system ECL or ECL Prime (GE Healthcare) according to the manufacturer's recommendations. TINA 2.0 software (Open Source Image Analysis Environment) was utilized for immunoblot quantification and image analysis.
Cell lysate corresponding to 1 mg of protein was precleared with Protein A Mag-agarose beads (GE Healthcare) and incubated overnight with control IgG, Na,K-β, or NCX1 antibodies pre-coupled to Mag beads for 4 h. The beads were washed and separated by SDS-PAGE, and the proteins bound to the beads were immunoblotted as described above. For resolution of Na,K-β protein (55 kDa) from heavy chain IgG band (50 kDa), immunoprecipitates were treated with peptide N-glycosidase F (New England Biolabs, Ipswitch, MA) as described previously (13). The deglycosylated Na,K-β (32 kDa) appears as a single band.
Cell Surface Biotinylation
Cell surface biotinylation was performed as described previously (16). The sub-confluent monolayer of cells was labeled with 0.5 μg/ml membrane-impermeable EZ-Link Sulfo-NHS-LC-Biotin, Pierce in TEA buffer (10 mm triethanolamine, pH 9, 150 mm NaCl, 0.1 mm CaCl2, 1 mm MgCl2) on ice and quenched with buffer containing 50 mm NH4Cl in phosphate-buffered saline (PBS) with 0.1 mm CaCl2, and 1 mm MgCl2 and lysed in 1000 μl of lysis buffer (150 mm NaCl, 20 mm Tris, pH 8, 5 mm EDTA, 1% Triton X-100, 0.1% BSA, 1 mm PMSF, 5 μg/ml antipain, leupeptin, and pepstatin). The lysate was incubated with 30 μl of Ultralink streptavidin beads (Pierce) overnight at 4 °C. The beads were washed with lysis buffer, and the bound proteins were separated on SDS-PAGE and immunoblotted for NCX1 and Na,K-β.
Quantitative Real-time PCR Analysis
RNA was extracted by TRIzol, and cDNA was generated by iScript™ cDNA Synthesis kit (Bio-Rad) as per the manufacturer's instructions. Quantitative PCR was performed using the SYBR Green PCR Master Mix (Applied Biosystems) in a 384-well plate on a 7900HT Fast Real-time PCR system (Applied Biosystems). The following primers were used: Na,K-β (forward, TTACCCTTACTACGGCAAGCTCCT; reverse, TTCAGTGTCCATGGTGAGGTTGGT); NCX1 (forward, TTAGCCGTTGTGGCTCTCTT; reverse, TGTAGACCATGGCCACAAAA); GAPDH (forward, GCTGTCCAACCACATCTCCTC; reverse, TGGGGCCGAAGATCCTGTT). RNA levels were calculated by relative quantification (RQ) normalized to the endogenous control GAPDH. Samples were assayed in triplicate.
35S Metabolic Labeling
Cells (200,000) were grown on 60-mm dishes for 24 h in complete DMEM. Cells were incubated in methionine and cysteine-free DMEM containing 1% FBS 2 h before pulsing with 0.5 mCi/ml Tran35S-label (PerkinElmer Life Sciences) for 20 min. Cells were washed twice with PBS and lysed on ice in a lysis buffer. Lysates were incubated with protein A-agarose beads coated 1 μg/ml NCX1 antibody. Beads were washed and resolved by 10% SDS-PAGE and detected by fluorography using Typhoon Trio-phosphorimager (GE® Healthcare).
Immunofluorescence and Confocal Microscopy
Cells were cultured on glass coverslips and fixed with either ice-cold methanol (−20 °C) or 4% paraformaldehyde in PBS at room temperature. The coverslips were incubated overnight at 4 °C with primary antibodies diluted in 1% BSA in PBS with 100 μm calcium chloride and 1 mm magnesium chloride followed by Alexa-488™-, Alexa-546™-, or Alexa-633™-conjugated secondary antibodies and TO-PRO®-3-Iodide (Life Technologies) for nuclear staining. The coverslips were mounted on glass slides with ProLong gold antifade reagent (Life Technologies). The images were captured using Leica TCS SP5 Confocal Microscope (Leica Microsystems, Buffalo Grove, IL).
Wound-healing Migration Assay
Cells (200,000) were seeded in a 6-well tissue culture plate 24 h before a wound was made by scratching across the bottom of the well with a pipette tip. Wounded cell monolayers were washed three times with phosphate-buffered saline to remove the detached cells. Culture dishes were returned to the incubator for recovery of the wound in the presence or absence of the indicated inhibitors or reagents in serum-free media for 16 h. The scratches were photographed using an inverted microscope under the same configuration at the start and end of the experiment. The images were used to calculate the distance migrated by the cell sheet. The rate at which the wound was closed was calculated using the formula, speed = distance/time.
Electrical Cell-substrate Impedance Sensing (ECIS) Migration Assay
ECIS Model 1600R, Applied BioPhysics (Troy, NY) was utilized. 1 × 105 cells in 200 μl of media were plated on ECIS electrode arrays (8W1E), each array containing 8 individual wells, with an active gold electrode in the base, and allowed to incubate overnight. Cell impedance and resistance levels were continuously measured. The resistance gained steadily and plateaued when cell confluence was attained. At this point, the electrodes were supplied an AC signal of 1 μA, between a small measuring electrode (250-μm diameter) and a large counter electrode, resulting in a sharp drop in resistance. As the cells re-grew and spread on the small electrode, the resistance increased proportionally to the distance migrated by the cells. The rate of migration was calculated as a ratio of the area covered (49,062.5 μm2) in a given period of time (number of hours taken for the resistance to reach the plateau).
Calcium Measurements by Flow Cytometry
Cells (5 × 105) were plated in a 6-well dish for 48 h. On the day of the experiment cells were detached without the use of trypsin using 1× Hanks' buffer saline solution + 0.5 m EDTA at 37 °C for 10 min, centrifuged at 3000 rpm for 3 min, and resuspended in complete medium. 1 × 105 cells were resuspended in Tyrode's buffer without calcium containing 0.5 μm Fluo4-AM in pluronic acid (both from Life Technologies) and incubated at 37 °C for 20 min. Tyrode's buffer containing calcium was added to the cell suspension to make the final calcium concentration 0.5 mm before data acquisition using flow cytometer, BD Accuri C6 flow cytometer (BD Biosciences). The fluorescence intensity was continuously monitored for 8–10 min. 100 μm ouabain was added to the cells 2 min after start, and changes in intracellular calcium were calculated as described previously (20).
Calcium Imaging
Cells (10,000) were plated on glass-bottom dishes (MatTek Corp., Ashland, MA) and cultured until confluent. Cell cultures were then incubated with fluorescent calcium indicator, FURA-2, AM (Life Technologies) at a concentration of 10 μm in a Pluronic acid/DMSO mixture in a 37 °C incubator for 30 min protected from light. The MatTek plates were washed twice with HBSS containing 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 10 mm glucose, 10 mm 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid, pH 7.4. Cells were allowed to recover after loading in Hanks' buffer saline solution for 10 min before imaging. A xenon lamp equipped with quartz collector lenses was used to excite the cells, and the cells were imaged with a Nikon inverted microscope (Intracellular Imaging, Cincinnati, OH). The fluorescence intensity was measured continuously for 5 min to obtain baseline calcium values. The ratio of the fluorescence intensity emitted at 510 nm after excitation at 340 nm over excitation at 380 nm was used to calculate -fold change in intracellular calcium concentrations as described previously (21).
Alternatively, cells were incubated with 10 μm Fluo-4 AM in a Pluronic acid/DMSO mixture for 20 min at 37 °C. The plates were washed twice with Hanks' buffer saline solution with or without 2 mm CaCl2. After recovery for 15 min, the fluorescence intensity corresponding to cytosolic calcium was continuously measured by an LSM 710 confocal microscope system (Zeiss, Thornwood, NY). The amplitude of the response was calculated by subtracting basal fluorescence obtained during the initial 2 min from the maximum intensity of fluorescence after adding the 10 μm KB-R7943.
Statistical Analysis
Paired t test was used to evaluate the differences between average of two groups using data from at least three independent experiments, and a p of < 0.05 was considered statistically significant.
RESULTS
NCX1 Protein Level Is Reduced in β-KD Cells
MDCK cells with stable knockdown of Na,K-β using the RNA interference technique (β-KD) and with specific rescue of Na,K-β (β-KD/R), generated by introducing silent mutations within the shRNA recognition site in the Na,K-β cDNA, have been described previously (18). Immunoblot analysis confirmed 80% reduction in Na,K-β protein level in β-KD cells. These cells also showed 42% reduction in NCX1 protein level compared with MDCK cells when normalized to β-actin used as a loading control (Fig. 1A). β-KD/R cells with renewed Na,K-β expression had restored NCX1 protein, highlighting the specificity of Na,K-β in the regulation of NCX1. The reduction in NCX1 protein in β-KD cells was consistent with the reduction of NCX1 in β-KO hearts as we reported previously (22). Changes in the protein levels of other Ca2+ transport proteins such as PMCA and SERCA were not observed in β-KO hearts (22). Similarly, PMCA and SERCA protein levels in β-KD cells were comparable to parental MDCK cells (Fig. 1B), indicating that Na,K-β specifically regulates NCX1 protein.
FIGURE 1.
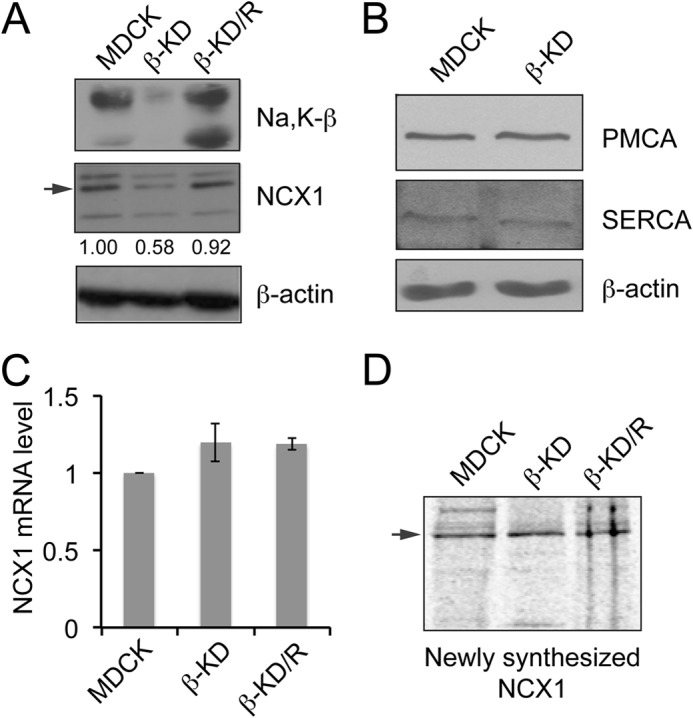
Knockdown of Na,K-β reduces NCX1 expression in MDCK cells. A, immunoblots showing Na,K-β, NCX1 and β-actin levels in the indicated cell lines. The arrow indicates 120-kDa NCX1 full-length protein. The bands at 160 and 70 kDa represent non-reduced exchanger and proteolytic fragment of NCX1, respectively. Quantification of NCX1 levels from three independent experiments expressed as -fold change normalized to β-actin loading control are indicated below the blot. The reduction in NCX1 protein level in β-KD cells is statistically significant (p < 0.05). B, immunoblots showing PMCA4, SERCA2, and β-actin levels in MDCK and β-KD cells. C, graph showing NCX1 mRNA level in MDCK, β-KD, and β-KD/R cells quantitated by quantitative real-time PCR. Mean values from three independent experiments are plotted. Error bars denote S.E. (p = 0.247). D, cells were metabolically labeled with [35S]methionine. Cell lysates were immunoprecipitated using anti-NCX1 antibody and resolved by SDS-PAGE. The newly synthesized NCX1 was detected by fluorography.
To determine whether Na,K-β alters NCX1 transcription, the mRNA level of NCX1 in β-KD cells was quantified. Compared with MDCK, β-KD cells did not show a significant change in NCX1 mRNA (Fig. 1C), suggesting that Na,K-β-mediated NCX1 regulation is post-transcriptional. It has been reported previously that Na,K-β regulates the rate of protein synthesis of its binding partner Na,K-α (23). Therefore, we tested NCX1 protein synthesis rate in β-KD cells by performing 35S metabolic labeling. The newly synthesized NCX1 protein detected by fluorography was comparable in MDCK, β-KD, and β-KD/R cells (Fig. 1D), indicating that Na,K-β does not alter the rate of synthesis of NCX1.
Na,K-β Interacts with NCX1 and Targets NCX1 to the Cell Surface
Next, we determined the membrane localization of NCX1 using cell surface biotinylation assay. Na,K-β on the cell surface was reduced by 85% in β-KD cells. The NCX1 level on the cell membrane was drastically diminished (by 89%) (Fig. 2A), although the NCX1 total protein level was reduced only by 42% (Fig. 1A). NCX1 membrane expression was restored in β-KD/R cells, highlighting the specificity of Na,K-β in targeting NCX1 to the membrane. The cell surface level of occludin, a well known tight junction protein, remained the same and was used as a control for successful biotinylation and equal loading. Immunofluorescence analysis confirmed that NCX1 staining was diminished in β-KD cells. Moreover, whereas NCX1 was localized to the membrane in MDCK and β-KD/R cells, such localization was not prominent in β-KD cells (Fig. 2B). Immunofluorescence analysis also showed that Na,K-β and NCX1 co-localize on the cell surface in MDCK and β-KD/R cells. To test the possibility that Na,K-β interacts with NCX1, a co-immunoprecipitation analysis was performed. Na,K-β was captured by anti-NCX1 immunoprecipitation, and NCX1 was detected in Na,K-β antibody immunoprecipitates (Fig. 2C), indicating that Na,K-β associates with NCX1.
FIGURE 2.
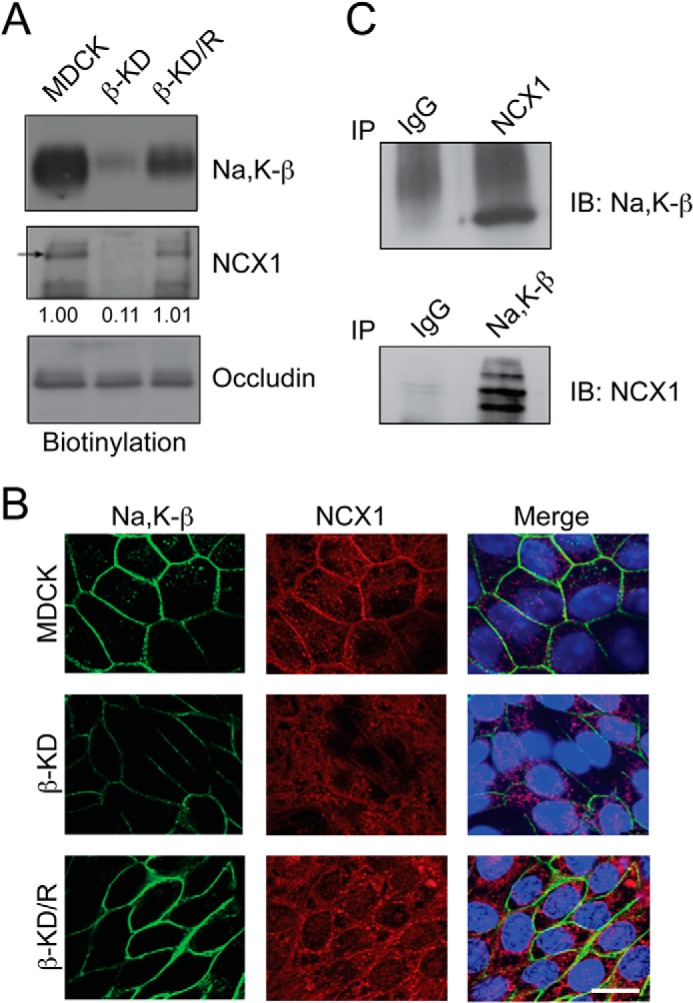
Na,K-β associates with NCX1 and its knockdown reduces NCX1 cell surface localization. A, cell surface proteins were pulled down with streptavidin beads and immunoblotted for indicated proteins. Immunoblots show cell surface levels of Na,K-β, NCX1, and occludin in MDCK, β-KD, and β-KD/R cells. NCX1 cell surface levels expressed, as -fold change with respect to MDCK cells, are shown below the blot. The reduction in NCX1 cell surface levels in β-KD cells is statistically significant (p = 0.003). B, representative images showing immunofluorescence staining of Na,K-β (green) and NCX1 (red) in MDCK, β-KD, and β-KD/R cells. The TOPRO-3 stained nuclei are shown in blue. Scale bar = 25 mm. C, immunoblots demonstrating association of NCX1 and Na,K-β by co-immunoprecipitation (IP) analysis in MDCK cells. Representative blots from three independent experiments are shown.
β-KD Cells Have Reduced NCX1 Activity and High Baseline Level of Intracellular Calcium
NCX1 activity is required for ouabain-induced increase in intracellular calcium (24). Therefore, the effect of ouabain on intracellular calcium was tested to determine whether NCX1 function is altered in β-KD cells. Treatment of MDCK and β-KD/R cells with 100 μm ouabain elicited an immediate increase in intracellular calcium shown by the spike in fluorescence intensity above the threshold, i.e. the percentage of response to ouabain treatment above the baseline calcium level (Fig. 3A). However, the percentage of response to ouabain treatment was 50% lower in β-KD cells compared with MDCK cells, suggesting that β-KD cells have reduced NCX1 activity, consistent with reduced NCX1 protein.
FIGURE 3.
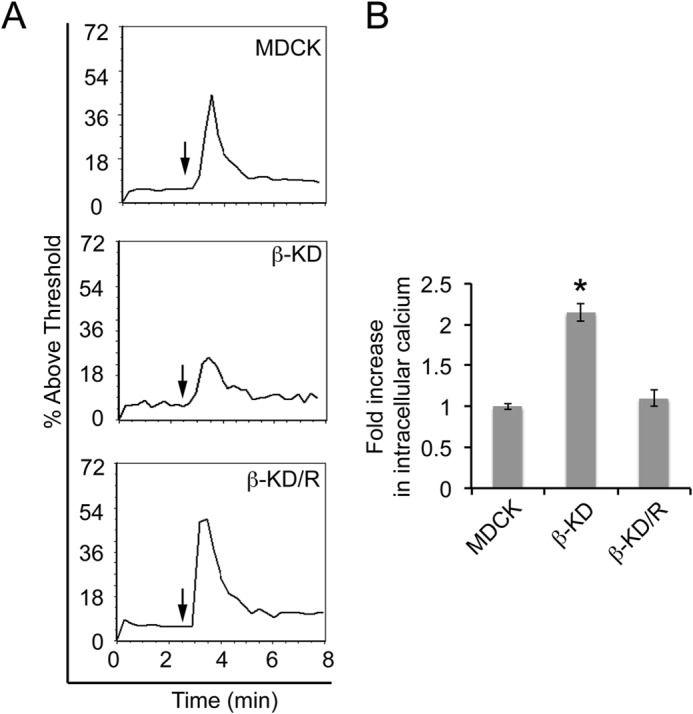
β-KD cells are ouabain-insensitive and have elevated baseline intracellular calcium due to reduced NCX1 levels. A, the plots show flow cytometric analysis of intracellular calcium in MDCK, β-KD, and β-KD/R cells. The data are represented as the percentage of fluorescence intensity above the threshold after the addition of 100 μm ouabain (arrow). Representative plots from three independent experiments in triplicate are shown. B, the graph shows -fold change in baseline intracellular calcium concentrations compared with MDCK cells. Average values from three independent experiments are plotted. Error bars denote S.E. The asterisk indicates statistical significance (*, p < 0.0001).
NCX1 is the major Ca2+ extrusion mechanism in kidney. To test whether reduced NCX1 affects calcium concentration in β-KD cells, the baseline intracellular free calcium levels were measured by Fura-2AM ratiometric imaging. The baseline intracellular calcium was 2.1-fold higher in β-KD cells compared with MDCK cells (Fig. 3B). The baseline calcium level in β-KD/R cells was similar to MDCK cells. Thus, the baseline intracellular calcium levels were inversely proportional to NCX1 protein levels.
β-KD Cells Migrate Faster and Require Calcium for Increased Migration Rate
Because enhanced intracellular calcium can increase cell migration and Na,K-β is a motility suppressor (13, 15), the rate of cell migration in β-KD cells was quantified by two different techniques. In a scratch wound healing assay, β-KD cells (13.6 ± 1.5 μm/h) showed a 1.8-fold higher migration rate than MDCK (8.2 ± 0.5 μm/h) and β-KD/R (7.8 ± 0.7 μm/h) cells (Fig. 4, A and B). A wound-healing assay was performed in the presence of calcium chelator, BAPTA-AM, to determine if the enhanced migration rate in β-KD cells was calcium-dependent. Treatment of β-KD cells with BAPTA-AM reduced the migration rate (5.6 ± 0.3 μm/h) (Fig. 4, A and B), suggesting that migration in β-KD cells is calcium-dependent.
FIGURE 4.
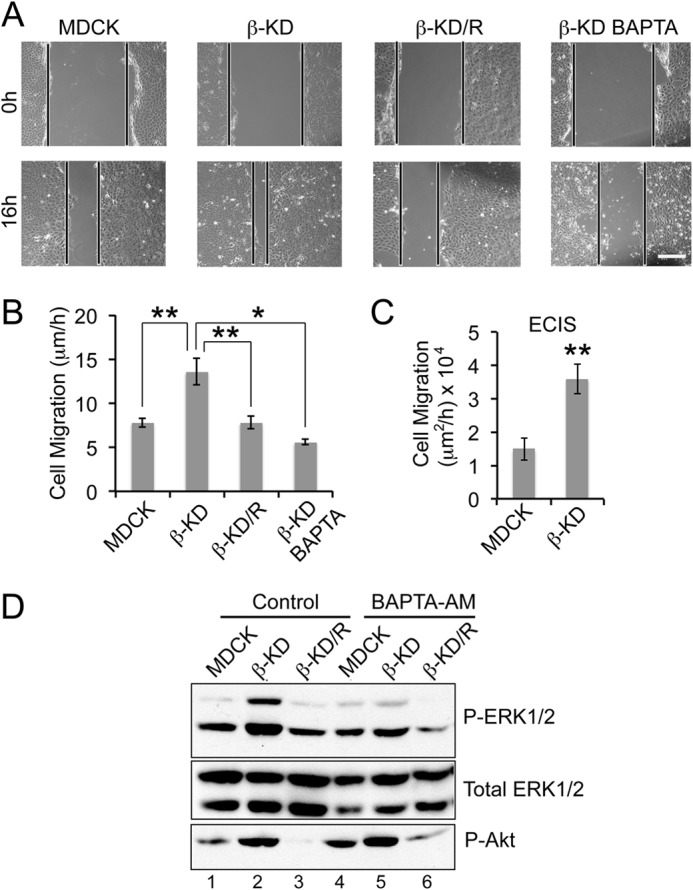
The role of Ca2+ in inducing cell migration and ERK1/2 phosphorylation in β-KD cells. A, representative images of the wound at 0 h and after 16 h in wound healing assay. Similar images were used to calculate the distance of migration over 16 h. Scale bar = 100 μm. B, the graph represents the average rate of migration calculated from three independent experiments in triplicate. Error bars denote S.E., and the asterisks indicate statistical significance (*, p < 0.05; **, p < 0.005). C, the graph represents the rate of migration obtained by ECIS wound healing assay from three independent experiments. Error bars denote S.E., and the asterisks indicate statistical significance (**, p < 0.005). D, immunoblots show the phosphorylation status of ERK1/2 and Akt (Ser-473) and total ERK1/2 (loading control) in the indicated cell lines under control conditions or with 10 μm BAPTA-AM treatment. Representative blots from four independent experiments are shown.
In ECIS wound-healing assay, the resistance of cells grown on electrodes is continuously monitored. A wound is created by the application of high current to the cell-covered electrode resulting in cell death and causing a subsequent drop in the resistance. The rate of migration is calculated by determining how quickly the resistance reverts to the levels of the cell-covered electrode due to the migration of cells on to the electrode (25). β-KD cells showed a 2.4-fold higher migration rate when compared with parental MDCK cells (Fig. 4C). The differences in fold increase in the rate of migration could be due to the variances in the sensitivity of the equipment employed for migration analysis and the changes in local cell density as described previously (26). Thus, β-KD cells migrated faster in both assays, supporting the reliability of the ECIS wound-healing assay.
Cell Migration in β-KD Cells Requires Calcium-dependent ERK1/2 Activation
We previously demonstrated that β-KD cells have high ERK1/2 and Akt activation mediated by PI3K (18). These signaling pathways are known to play significant roles in inducing cell migration (27–29). Therefore, to determine which of these signaling molecules are regulated by intracellular calcium, MDCK, β-KD, and β-KD/R cells were treated with BAPTA-AM, and the phosphorylation status of ERK1/2 and Akt was determined by immunoblot analysis. BAPTA-AM treatment of β-KD cells reduced phosphorylated ERK1/2 to levels similar to MDCK cells (compare lanes 2 and 5), but the Akt phosphorylation remained high (Fig. 4D), indicating that ERK1/2 activation was calcium-dependent and was likely involved in the regulation of cell migration.
To determine the role of ERK1/2 and Akt activation in cell migration, β-KD cells were treated with specific inhibitors of these signaling pathways. Treatment with inhibitors of MEK1/2 (upstream regulator of ERK1/2), PD98059 and U0126, reduced the migration rate of β-KD cells from 9.5 ± 0.9 μm/h to 4.7 ± 0.7 μm/h and 5.7 ± 1.1 μm/h respectively, suggesting a significant role for ERK1/2 in migration (Fig. 5A).
FIGURE 5.
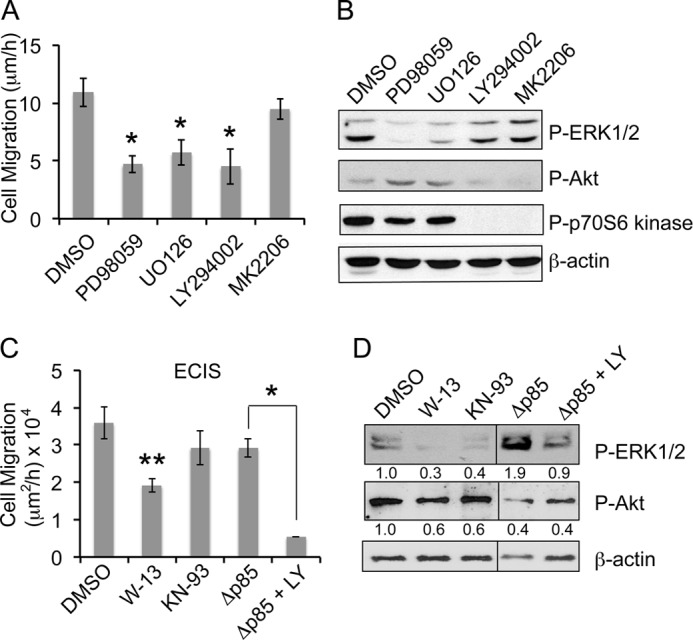
Increased migration in β-KD cells is dependent on calmodulin/PI3K-mediated ERK1/2 activation. A, the graph shows the average rate of migration of β-KD cells treated with the indicated inhibitors (LY294002, U0126, PD98059, MK2206 at 10 μm each) from three independent experiments performed in triplicate. Error bars denote S.E. Treatment with all inhibitors except MK2206 attained statistical significance (*, p < 0.05). B, the immunoblots show the levels of phospho-ERK1/2, phospho-Akt, phospho p70S6 kinase and β-actin (loading control) after inhibitor treatment for 16 h in β-KD cells. Representative blots from three independent experiments are shown. C, the graph shows the average rate of migration by ECIS wound healing assay in β-KD cells treated with DMSO, 30 μm W-13 or 30 μm KN-93, and β-KD/ΔP85 cells after treatment with DMSO or 10 μm LY294002 from three independent experiments. Error bars denote S.E. Asterisk(s) indicate statistical significance (*, p < 0.05; **, p < 0.005). D, the immunoblots show the levels of phospho-ERK1/2, phospho-Akt, and β-actin after inhibitor treatment for 16 h. Quantification from three independent experiments expressed as -fold change normalized to β-actin loading control are indicated below the blot.
ERK1/2 Activation and Cell Migration in β-KD Cells Is Calmodulin- and PI3K-dependent but Akt-independent
As we showed previously that ERK1/2 phosphorylation in β-KD cells is PI3K-dependent (18), we tested the role of PI3K in β-KD cell migration. PI3K inhibition by LY294002 reduced phosphorylated ERK1/2 (47.9 ± 1.3% of control) and also reduced the rate of migration (4.5 ± 1.5 μm/h) showing that PI3K is involved in inducing cell migration via its effect on ERK1/2. Because Akt is a downstream target of PI3K and is activated in β-KD cells, we tested whether Akt was also involved in the regulation of cell migration. Treatment with MK2206, a specific Akt inhibitor (10.9 ± 1.2 μm/h), did not alter the migration rate of β-KD cells (9.5 ± 0.9 μm/h) (p = 0.355), indicating that migration in β-KD cells is Akt-independent. Immunoblot analysis of inhibitor-treated cells confirmed the reduction in phosphorylated ERK1/2 by PD98059, U0126, and LY294002 (Fig. 5B). MK2206 did not change phosphorylated ERK1/2 levels significantly (83.9 ± 1.9%, p = 0.780). As expected, phosphorylation of Akt and p70S6 kinase, a downstream target of Akt, was reduced after LY294002 and MK2206 treatments (Fig. 5B).
To confirm the involvement of PI3K in cell migration, β-KD cells were transfected with a dominant negative mutant of PI3K p85 subunit lacking the binding site for the catalytic subunit p110 (β-KD/Δp85) (30). β-KD/Δp85 cells had reduced Akt activation, consistent with suppression of PI3K signaling (Fig. 5D). Surprisingly, these cells migrated at rates similar to β-KD cells (Fig. 5C) and showed no reduction in ERK1/2 phosphorylation (Fig. 5D). This apparent discrepancy could be resolved by considering the fact that in addition to its well known lipid kinase activity, PI3K is also a protein kinase (31, 32) that directly phosphorylates MEK resulting in ERK1/2 activation (33). The PI3K inhibitor LY294002 inhibits both activities of PI3K (34). β-KD/Δp85 cells treated with LY294002 showed reduced migration and ERK1/2 phosphorylation (Fig. 5, C and D), indicating that the protein kinase activity of PI3K is involved in ERK1/2 phosphorylation and migration in β-KD cells.
Enhanced intracellular calcium can activate PI3K via Ca2+ sensor calmodulin or calmodulin-dependent kinase II, which are key players in calcium-mediated cell migration (35–37). β-KD cells were treated with W-13 or KN-93, which inhibit calmodulin and calmodulin-dependent kinase II, respectively. W-13 reduced β-KD cell migration by 47% (p = 0.001), whereas KN-93 did not significantly alter cell migration rate (p = 0.173) (Fig. 5C). W-13 also reduced ERK1/2 phosphorylation by 70% (Fig. 5D). Taken together, these results indicate that calmodulin is involved in inducing ERK1/2-dependent cell migration in β-KD cells.
Pharmacological Inhibition of NCX1 Induces PI3K-dependent ERK1/2 Activation and Promotes Cell Migration in Renal Epithelial Cells
To determine if the functional inhibition of NCX1 is sufficient to activate ERK1/2 and induce migration, we blocked the Na+/Ca2+ exchange current with KB-R7943 that has been shown to inhibit both inward and outward calcium flux (38, 39). Treatment of MDCK cells with 10 μm KB-R7943 increased intracellular calcium in the presence or absence of calcium in the culture medium (Fig. 6, A and B). KB-R7943-treated MDCK cells also showed a 2.8-fold increase in ERK1/2 phosphorylation, but the phosphorylated Akt level remained comparable with MDCK cells (Fig. 6C). Furthermore, KB-R7943 treatment in MDCK cells increased the migration rate by 1.5-fold (Fig. 6D), highlighting the role of NCX1 in the regulation of cell migration.
FIGURE 6.
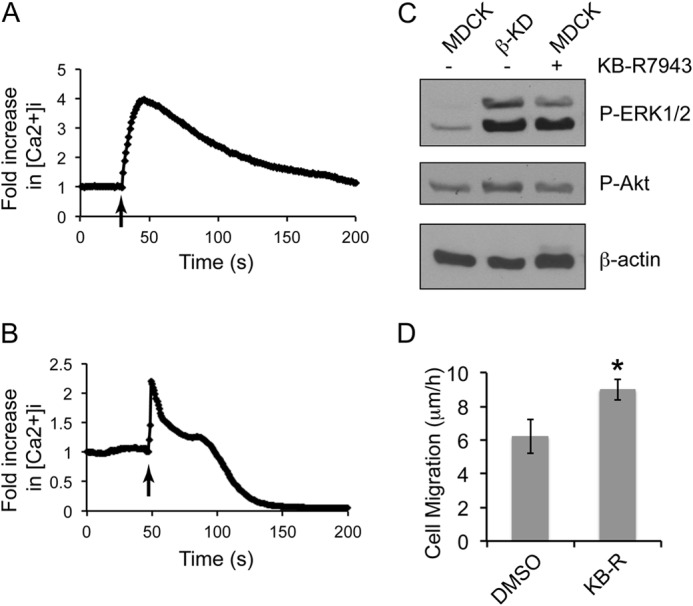
Inhibition of NCX1 activity increases intracellular calcium, activates ERK1/2, and increases rate of migration in MDCK cells. Plots depicting the intracellular calcium response of MDCK cells loaded with 10 μm Fluo-4 AM to 10 μm KB-R7943 with (A) or without (B) calcium in the medium. The arrow indicates the time of KB-R7943 addition. Representative plots from three independent experiments (each with greater than 25 cells) are shown. Note the increase in intracellular calcium concentration upon the addition of 10 μm KB-R7943 (arrow). C, immunoblots show the phosphorylation status of ERK1/2 and Akt with β-actin as loading control in MDCK, β-KD, and MDCK cells treated with 10 μm KB-R7943. Representative blots from three independent experiments are shown. D, the graph denotes the average rate of migration of MDCK cells with or without 10 μm KB-R7943 treatment as measured by wound healing assay from three independent experiments performed in triplicate. Error bars denote S.E., and the asterisk indicates statistical significance (*, p < 0.05).
KB-R7943 also increased the rate of cell migration by 1.4–1.6-fold in LLC-PK1 cells (Fig. 7, A and B) accompanied by a dose-dependent increase in ERK1/2 phosphorylation (Fig. 7C). Likewise, primary human renal epithelial cells, HREpiC, showed a 2.8-fold increase in migration rate with 10 μm KB-R7943 treatment (Fig. 7D). Similar to MDCK and LLC-PK1 cells, NCX1 inhibition in HREpiC cells induced a 2.8-fold increase in ERK1/2 phosphorylation (Fig. 7E). Furthermore, MEK inhibitor PD98059 prevented cell migration induced by KB-R7943 (Fig. 7D), indicating that increased migration by NCX1 inhibition is ERK1/2-dependent. PI3K inhibitor LY294002 suppressed KB-R7943-induced cell migration rate (Fig. 7D) and ERK1/2 phosphorylation (Fig. 7E), suggesting that PI3K-dependent ERK1/2 activation is required for increased cell migration after NCX1 inhibition.
FIGURE 7.
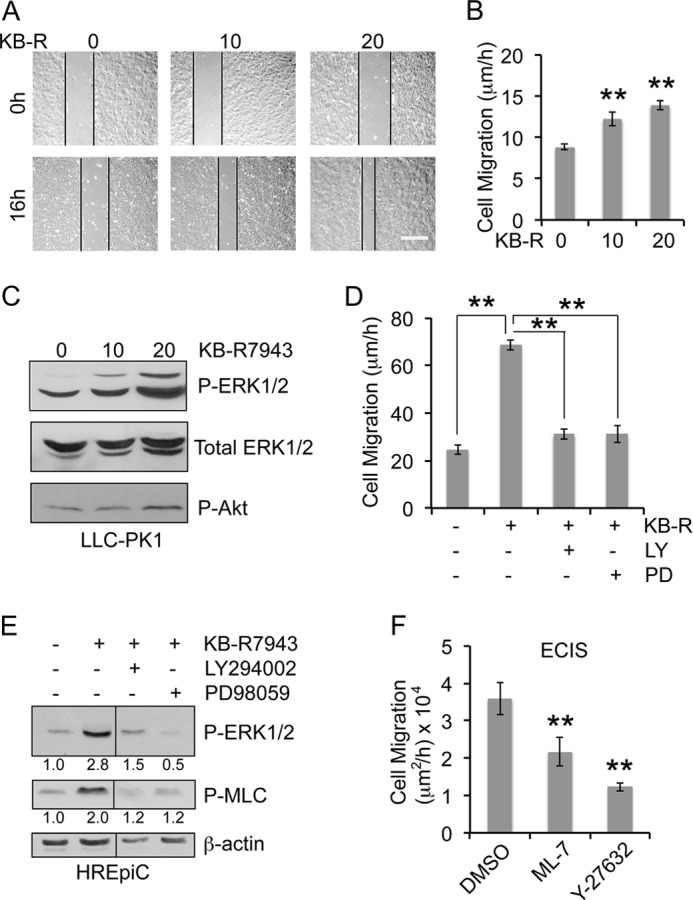
NCX1 inhibition by KB-R7943 increases ERK1/2 and MLC phosphorylation and rate of migration in renal epithelial cells in a PI3K-dependent manner. A, representative images of the wound in LLC-PK1 cells treated with different concentrations of KB-R7943 at 0 h and after 16 h. Scale bar = 100 μm. B, the graph represents the mean rate of migration calculated from three independent experiments in triplicate. Error bars denote S.E., and the asterisks indicate statistical significance (**, p < 0.005). C, corresponding immunoblots show the phosphorylation status of ERK1/2 and Akt with total ERK1/2 as the loading control in LLC-PK1 cells treated with the indicated concentrations of KB-R7943 for 16 h. D, the graph represents the average rate of migration of HREpiC cells treated with 10 μm KB-R7943 in the presence or absence of 10 μm LY294002 or 10 μm PD98059 for 10 h. Error bars denote S.E., and the asterisks indicate statistical significance (**, p < 0.005). E, corresponding immunoblots show the phosphorylation status of ERK1/2 and MLC with β-actin as loading control in HREpiC cells. Quantification from three independent experiments expressed as -fold change normalized to β-actin loading control are indicated below the blot. F, the graph shows the average rate of migration in β-KD cells treated with DMSO, 5 μm ML-7, or 5 μm Y-27632 for 16 h by ECIS wound healing assay from three independent experiments. Error bars denote S.E. (**, p < 0.005).
NCX1 Inhibition Mediates Migration by PI3K/ERK-dependent Increase in MLC Phosphorylation
Myosin light chain kinase and Rho-associated protein kinase are known substrates of ERK1/2 involved in increased cell migration (27). β-KD cells treated with ML-7 (myosin light chain kinase inhibitor) or Y-27632 (Rho-associated protein kinase inhibitor) showed reduced migration, supporting the involvement of myosin light chain kinase and Rho-associated protein kinase in mediating cell migration (Fig. 7F). Both myosin light chain kinase and Rho-associated protein kinase phosphorylate myosin light chain (MLC) and enhance migration (40, 41). KB-R7943 increased MLC phosphorylation by 2.0-fold in HREpiC cells (Fig. 7E). This increase was abrogated by inhibition of PI3K and ERK1/2, confirming that PI3K/ERK regulates KB-R9743-induced migration via MLC phosphorylation.
Overexpression of NCX1 in β-KD Cells Suppresses ERK1/2 Activation and Cell Migration
To confirm the role of NCX1 in the regulation of ERK1/2 and cell migration, we overexpressed NCX1 in β-KD cells (β-KD/NCX1). Immunoblot analysis showed that the levels of NCX1 were 2.0 ± 0.1-fold higher in β-KD/NCX1 cells (Fig. 8A). Higher NCX1 levels were accompanied by 75% reduction in ERK1/2 phosphorylation compared with β-KD cells. Unlike MDCK cells, β-KD cells do not show an increase in calcium response after KB-R7943 treatment. However, β-KD/NCX1 cells treated with KB-R7943 showed enhanced calcium response similar to MDCK, indicating that NCX1 activity was restored by overexpression of NCX1 (Fig. 8B). Finally, overexpression of NCX1 in β-KD cells suppressed migration rate in a wound-healing assay (Fig. 8C). Taken together, these data indicate that NCX1 expression in β-KD cells was sufficient to reduce calcium-induced ERK/12 activation and cell migration.
FIGURE 8.
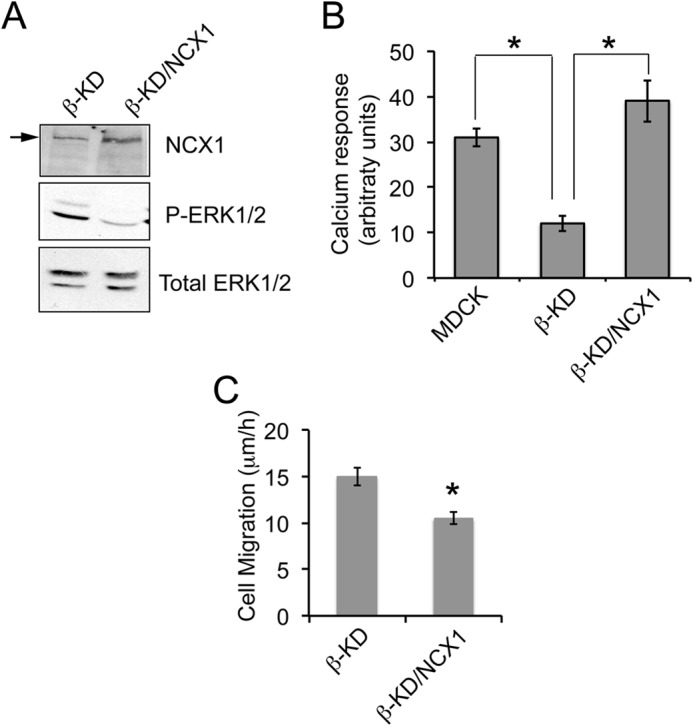
Overexpression of NCX1 in β-KD cells reduces ERK1/2 phosphorylation, exhibits response to NCX1 inhibitor, and decreases migration rate. A, representative immunoblots showing NCX1, phosphorylated ERK1/2, and total ERK1/2 levels in the indicated cell lines. B, the graph represents the mean amplitude of calcium response after treatment with 10 μm KB-R7943 in MDCK, β-KD, and β-KD/NCX1 from at least 15 individual cells measured by confocal microscopy. Similar data were obtained in three independent experiments. Error bars denote S.E., and the asterisks indicate statistical significance (*, p < 0.05). C, the graph depicts the average rate of migration as determined by wound healing assay from three independent experiments in triplicate. Error bars denote S.E., and the asterisk indicates statistical significance (*, p < 0.05).
DISCUSSION
Our data show that reduced NCX1 function in β-KD cells was due to drastic reduction in NCX1 membrane localization. In addition, the data suggest that Na,K-β associates with NCX1, and its abundance is affected in the knockdown of Na,K-β in β-KD cells. Furthermore, reduced NCX1 activity in these cells was associated with increased cell migration via calcium/calmodulin/PI3K-mediated ERK1/2 activation. Treatment of MDCK, LLC-PK1, and HREpiC cells with NCX1 inhibitor increased intracellular calcium, ERK1/2 phosphorylation, and enhanced migration, suggesting that the increased migration in β-KD cells is via suppression of NCX1 activity. This KB-R7943-induced migration was associated with PI3K- and ERK1/2-dependent phosphorylation of MLC. Furthermore, β-KD cells with NCX1 overexpression restored NCX1 activity, resulting in reduced ERK1/2 phosphorylation and migration. A model summarizing the mechanism of migration induced by reduced NCX1 activity is outlined (Fig. 9).
FIGURE 9.
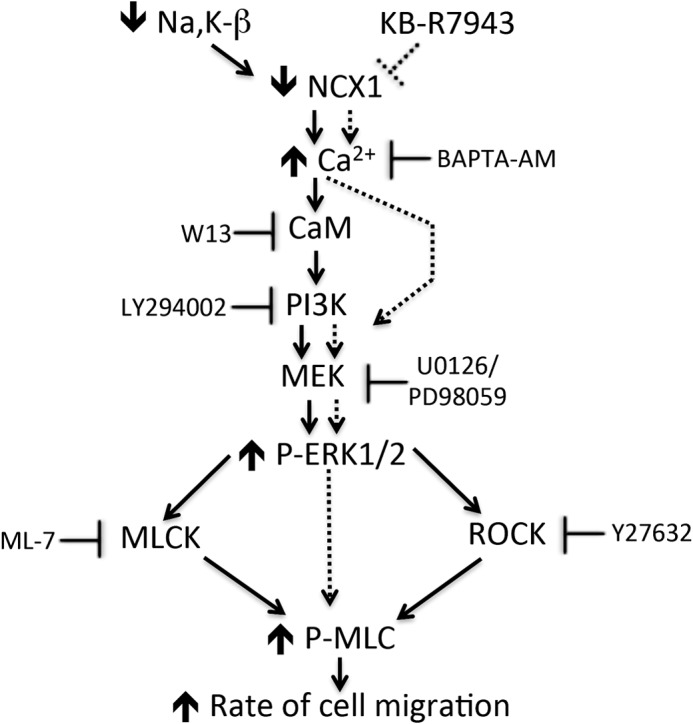
Schematic model of regulation of migration by reduced protein expression or functional inhibition of NCX1 in renal epithelial cells. The solid arrows indicate the consequences of the reduction in the NCX1 protein level, whereas the dotted lines indicate the consequences of functional inhibition of NCX1. ROCK, Rho-associated protein kinase.
Na,K-β, the regulatory subunit of Na,K-ATPase, is important for co-translational transport and membrane insertion of Na,K-α (42). In addition, Na,K-β has been shown to interact with other membrane transporters such as Na+-K+-Cl− co-transporter (43) and large conductance Ca2+-activated K+ channels (44) and mediate membrane targeting of these transporters. In our study Na,K-β associated with NCX1, and Na,K-β knockdown reduced NCX1 protein, suggesting that it plays a similar role in aiding the trafficking and stabilization of NCX1 at the membrane. Because there is a reduction in the total NCX1 protein, whereas the rate of newly synthesized NCX1 is unaltered in β-KD cells, it is likely that NCX1 is targeted for degradation in β-KD cells.
NCX1 is a bidirectional ion counter transporter that can function either in the Ca2+ efflux or Ca2+ influx mode. NCX1 activity and the directionality of ion transport is driven by the membrane potential and the transmembrane electrochemical gradients of Na+ and Ca2+ (45–47). The major function of NCX1 in the kidney is Ca2+ extrusion, where it plays an important role in net transepithelial Ca2+ absorption and is responsible for the majority of Ca2+ extrusion (48). The importance of NCX1 in Ca2+ efflux is evident by the rise in intracellular calcium in β-KD cells, which express reduced levels of NCX1. Although bi-directional NCX1 inhibitor KB-R7943 is more potent in inhibiting the reverse than the forward mode (49), its sensitivity is dictated by the splice variant of NCX1 expressed by the cells. The splice variant expressed in the kidney, NCX1.3 (50), is inhibited in its forward mode by KB-R7943 with IC50 values of 2–3 μm (51). Treatment with KB-R7943 in Ca2+-free medium also caused an increase in intracellular calcium (Fig. 6B). Taken together, the Ca2+ efflux activity of NCX1 is involved in the regulation of cell migration in our study.
Many ion transporters regulate cell migration by maintaining housekeeping functions inside the cell such as membrane potential, cell volume, intracellular calcium concentration, and pH (52). NCX1 has been reported to promote the migration of a diverse group of non-epithelial cells such as endothelial cells, oligodendrocytes, myofibroblasts, microglia, and pancreatic cancer cells (53–60). Inhibition or knockdown of NCX1 reduced intracellular calcium and prevented migration. This apparent contrast to our findings could be explained by the directionality of Ca2+ flux by NCX1 in these cell types. This would suggest that NCX1 regulation of cell migration might be dependent on the directionality of ion transport. However, the underlying commonality in these reports and ours is that a rise in intracellular calcium was required for increase in cell migration. Migration is a calcium-dependent process as most of the regulators of motility that coordinate focal adhesion dynamics and interaction with cytoskeletal proteins are calcium-sensitive (61, 62).
Ouabain, a specific inhibitor of Na,K-ATPase activity, leads to an increase in intracellular sodium. This increase reduces the driving force for the sodium influx through NCX1, subsequently resulting in an increase in intracellular calcium (24). NCX1 knock-out mice conclusively demonstrated that the ouabain-induced increase in intracellular calcium and enhanced cardiac contractility requires NCX1 activity (63). Therefore, ouabain sensitivity is dependent on NCX1 as well as Na,K-ATPase. Our previous studies showed that the Na,K-β KO cardiomyocytes and hearts with greatly reduced NCX1 were insensitive to ouabain-induced calcium transients and cardiac contractility (22). Consistent with these observations, β-KD with reduced NCX1 protein did not respond to ouabain, further confirming that NCX1 functions in the Ca2+ efflux mode.
Migrating cells have a calcium gradient inside the cell with high calcium levels toward the retracting end and low calcium at the leading end (64), thus making it amenable to respond to calcium sparks that trigger forward movement. Other ion transporters involved in migration have a polarized distribution. The localization of NCX1 in a migrating cell has not been determined. Studies are currently under way in our laboratory to determine the role of NCX1 in directional movement.
PI3K is a dual specificity kinase that possesses serine/threonine protein kinase activity in addition to its well known lipid kinase activity (31, 32). The lipid kinase activity is responsible for Akt phosphorylation, and the protein kinase activity mediates ERK1/2 activation by direct phosphorylation of MEK (33). In this study we observed calcium-dependent ERK1/2 activation in β-KD cells and after NCX1 inhibition; however, Akt was not dependent on calcium, indicating that the protein kinase activity of PI3K is involved in calcium-dependent ERK1/2 activation. A previous study has shown that direct binding of calmodulin to the SH2 domain of p85 subunit of PI3K results in increase of its lipid kinase activity (35); however, the effect on protein kinase activity was not tested. Increased lipid kinase activity in gain-of-function oncogenic mutants of PI3K p110 was accompanied by enhanced protein kinase activity (65). Therefore, it is tempting to speculate that calmodulin, by increasing the PI3K lipid kinase activity, also induces protein kinase activity of PI3K in renal epithelial cells. However, the exact mechanism by which calmodulin activates PI3K protein kinase activity remains to be determined.
Our previous studies have demonstrated that replenishing Na,K-β in highly motile Moloney sarcoma virus transformed-MDCK cells reduced cell motility (13, 15) and that knockdown of Na,K-β prevented epithelial lumen formation via PI3K-dependent ERK1/2 activation (18). This is the first report showing that knockdown of Na,K-β in MDCK cells increased rate of cell migration and that increased intracellular calcium after NCX1 reduction was required for ERK1/2 activation and enhanced migration in β-KD cells. Thus, it is likely that reduced NCX1 is a downstream mediator of the β-KD phenotype.
The role of Na,K-β as a motility and tumor suppressor in conjunction with reduced expression of Na,K-β in several carcinomas has been established (13, 14, 66–68). Similar to Na,K-β levels, NCX1 transcript level is also reduced in various subtypes of renal carcinoma (data not shown). Therefore, given the role of NCX1 in cell migration in epithelial cells, it is likely that reduced expression of NCX1 is also a prognostic indicator of carcinoma progression. Experiments are in progress to address the alternate functions of NCX1 in renal cancers.
Acknowledgments
We thank Dr. Kenneth Philipson, UCLA, for the generous gift of NCX1 expression plasmid. We also thank Dr. Donna Woulfe and Aasma Khan, University of Delaware, for help with analysis of flow cytometry data and Dr. Mary E. Boggs, University of Delaware, for useful suggestions on calcium measurement techniques. pWZL-neo Δ-p85 plasmid was a gift from Willian Hahn (Addgene plasmid #10888).
Note Added in Proof
Figs. 5D and 7E did not conform with the JBC policy that figures assembled from separate images should indicate the borders between the images in the version of this article that was published on March 13, 2015 as a Paper in Press. Both figures were prepared from separate sections of a single immunoblot. The borders between the sections are now indicated with dividing lines. The revisions do not change the interpretation of the results or the conclusions.
This work was supported, in whole or in part, by National Institutes of Health Grants 8 P20GM103446-13 (to the Delaware Bioimaging Center (IDeA Networks of Biomedical Research Excellence (INBRE)-NIGMS), P20GM103464 (to the Cell Science Core of the Center for Pediatric Research), and U54GM104941 (to DE-CTR ACCEL). This work was also supported by American Heart Association Grant 10SDG260011 and by the Nemours Foundation.
- SERCA
- sarco/endoplasmic reticulum calcium transport ATPase
- NCX1
- sodium-calcium exchanger 1
- Na,K-α
- Na,K-ATPase α-subunit
- Na,K-β
- Na,K-ATPase β-subunit
- MDCK
- Madin-Darby canine kidney
- PMCA
- plasma membrane calcium ATPase
- β-KD
- β knockdown
- β-KD/R
- β-KD cells rescued by overexpression of shRNA resistant Na,K-β
- HREpiC
- human primary renal epithelial cells
- ECIS
- electrical cell-substrate impedance sensing
- MLC
- myosin light chain.
REFERENCES
- 1. Monteith G. R., Davis F. M., Roberts-Thomson S. J. (2012) Calcium channels and pumps in cancer: changes and consequences. J. Biol. Chem. 287, 31666–31673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Monteith G. R., McAndrew D., Faddy H. M., Roberts-Thomson S. J. (2007) Calcium and cancer: targeting Ca2+ transport. Nat. Rev. Cancer 7, 519–530 [DOI] [PubMed] [Google Scholar]
- 3. Parsons J. T., Horwitz A. R., Schwartz M. A. (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 11, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsavaler L., Shapero M. H., Morkowski S., Laus R. (2001) Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 61, 3760–3769 [PubMed] [Google Scholar]
- 5. Brouland J. P., Gélébart P., Kovàcs T., Enouf J., Grossmann J., Papp B. (2005) The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am J. Pathol 167, 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldhaber J. I., Philipson K. D. (2013) Cardiac sodium-calcium exchange and efficient excitation-contraction coupling: implications for heart disease. Adv. Exp. Med. Biol. 961, 355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matsuda T., Takuma K., Baba A. (1997) Na+-Ca2+ exchanger: physiology and pharmacology. Jpn. J. Pharmacol. 74, 1–20 [DOI] [PubMed] [Google Scholar]
- 8. Lytton J. (2007) Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem. J. 406, 365–382 [DOI] [PubMed] [Google Scholar]
- 9. Hilge M., Aelen J., Vuister G. W. (2006) Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol. Cell 22, 15–25 [DOI] [PubMed] [Google Scholar]
- 10. Reeves J. P. (1998) Na+/Ca2+ exchange and cellular Ca2+ homeostasis. J. Bioenerg. Biomembr. 30, 151–160 [DOI] [PubMed] [Google Scholar]
- 11. Chambers K. F., Pearson J. F., Pellacani D., Aziz N., Gužvić M., Klein C. A., Lang S. H. (2011) Stromal upregulation of lateral epithelial adhesions: gene expression analysis of signalling pathways in prostate epithelium. J. Biomed. Sci. 18, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J., Jin H. B., Sun Y. M., Su Y., Wang L. F. (2010) KB-R7943 inhibits high glucose-induced endothelial ICAM-1 expression and monocyte-endothelial adhesion. Biochem. Biophys. Res. Commun. 392, 516–519 [DOI] [PubMed] [Google Scholar]
- 13. Barwe S. P., Anilkumar G., Moon S. Y., Zheng Y., Whitelegge J. P., Rajasekaran S. A., Rajasekaran A. K. (2005) Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 16, 1082–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Inge L. J., Rajasekaran S. A., Yoshimoto K., Mischel P. S., McBride W., Landaw E., Rajasekaran A. K. (2008) Evidence for a potential tumor suppressor role for the Na,K-ATPase β1-subunit. Histol. Histopathol. 23, 459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rajasekaran S. A., Palmer L. G., Quan K., Harper J. F., Ball W. J., Jr., Bander N. H., Peralta Soler A., Rajasekaran A. K. (2001) Na,K- ATPase β-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol. Biol. Cell 12, 279–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barwe S. P., Kim S., Rajasekaran S. A., Bowie J. U., Rajasekaran A. K. (2007) Janus model of the Na,K-ATPase β-subunit transmembrane domain: distinct faces mediate α/β assembly and β-β homo-oligomerization. J. Mol. Biol. 365, 706–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kitamura N., Ikekita M., Sato T., Akimoto Y., Hatanaka Y., Kawakami H., Inomata M., Furukawa K. (2005) Mouse Na+/K+-ATPase β1-subunit has a K+-dependent cell adhesion activity for β-GlcNAc- terminating glycans. Proc. Natl. Acad. Sci. U.S.A. 102, 2796–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barwe S. P., Skay A., McSpadden R., Huynh T. P., Langhans S. A., Inge L. J., Rajasekaran A. K. (2012) Na,K-ATPase β-subunit cis homo-oligomerization is necessary for epithelial lumen formation in mammalian cells. J. Cell Sci. 125, 5711–5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ottolia M., John S., Ren X., Philipson K. D. (2007) Fluorescent Na+-Ca2+ exchangers: electrophysiological and optical characterization. J. Biol. Chem. 282, 3695–3701 [DOI] [PubMed] [Google Scholar]
- 20. Khan A., Li D., Ibrahim S., Smyth E., Woulfe D. S. (2014) The physical association of the P2Y12 receptor with PAR4 regulates arrestin-mediated Akt activation. Mol. Pharmacol. 86, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li W., Duncan R. L., Karin N. J., Farach-Carson M. C. (1997) 1,25(OH)2D3 enhances PTH-induced Ca2+ transients in preosteoblasts by activating L-type Ca2+ channels. Am. J. Physiol. 273, E599–E605 [DOI] [PubMed] [Google Scholar]
- 22. Barwe S. P., Jordan M. C., Skay A., Inge L., Rajasekaran S. A., Wolle D., Johnson C. L., Neco P., Fang K., Rozengurt N., Goldhaber J. I., Roos K. P., Rajasekaran A. K. (2009) Dysfunction of ouabain-induced cardiac contractility in mice with heart-specific ablation of Na,K-ATPase β1-subunit. J. Mol. Cell. Cardiol. 47, 552–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rajasekaran S. A., Gopal J., Willis D., Espineda C., Twiss J. L., Rajasekaran A. K. (2004) Na,K-ATPase β1-subunit increases the translation efficiency of the α1-subunit in MSV-MDCK cells. Mol. Biol. Cell 15, 3224–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blaustein M. P. (1993) Physiological effects of endogenous ouabain: control of intracellular Ca2+ stores and cell responsiveness. Am. J. Physiol. 264, C1367–C1387 [DOI] [PubMed] [Google Scholar]
- 25. Keese C. R., Wegener J., Walker S. R., Giaever I. (2004) Electrical wound-healing assay for cells in vitro. Proc. Natl. Acad. Sci. U.S.A. 101, 1554–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Treloar K. K., Simpson M. J. (2013) Sensitivity of edge detection methods for quantifying cell migration assays. PloS ONE 8, e67389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang C., Jacobson K., Schaller M. D. (2004) MAP kinases and cell migration. J. Cell Sci. 117, 4619–4628 [DOI] [PubMed] [Google Scholar]
- 28. Qian Y., Corum L., Meng Q., Blenis J., Zheng J. Z., Shi X., Flynn D. C., Jiang B. H. (2004) PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration. Am. J. Physiol. Cell Physiol. 286, C153–C163 [DOI] [PubMed] [Google Scholar]
- 29. Zhou G. L., Zhuo Y., King C. C., Fryer B. H., Bokoch G. M., Field J. (2003) Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Mol. Cell. Biol. 23, 8058–8069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao J. J., Gjoerup O. V., Subramanian R. R., Cheng Y., Chen W., Roberts T. M., Hahn W. C. (2003) Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer cell 3, 483–495 [DOI] [PubMed] [Google Scholar]
- 31. Dhand R., Hiles I., Panayotou G., Roche S., Fry M. J., Gout I., Totty N. F., Truong O., Vicendo P., Yonezawa K., (1994) PI 3-kinase is a dual specificity enzyme: autoregulation by an intrinsic protein-serine kinase activity. EMBO J. 13, 522–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carpenter C. L., Auger K. R., Duckworth B. C., Hou W. M., Schaffhausen B., Cantley L. C. (1993) A tightly associated serine/threonine protein kinase regulates phosphoinositide 3-kinase activity. Mol. Cell. Biol. 13, 1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bondeva T., Pirola L., Bulgarelli-Leva G., Rubio I., Wetzker R., Wymann M. P. (1998) Bifurcation of lipid and protein kinase signals of PI3Kγ to the protein kinases PKB and MAPK. Science 282, 293–296 [DOI] [PubMed] [Google Scholar]
- 34. Rondinone C. M., Carvalho E., Rahn T., Manganiello V. C., Degerman E., Smith U. P. (2000) Phosphorylation of PDE3B by phosphatidylinositol 3-kinase associated with the insulin receptor. J. Biol. Chem. 275, 10093–10098 [DOI] [PubMed] [Google Scholar]
- 35. Joyal J. L., Burks D. J., Pons S., Matter W. F., Vlahos C. J., White M. F., Sacks D. B. (1997) Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem. 272, 28183–28186 [DOI] [PubMed] [Google Scholar]
- 36. Rotfeld H., Hillman P., Ickowicz D., Breitbart H. (2014) PKA and CaMKII mediate PI3K activation in bovine sperm by inhibition of the PKC/PP1 cascade. Reproduction 147, 347–356 [DOI] [PubMed] [Google Scholar]
- 37. Easley C. A., 4th, Brown C. M., Horwitz A. F., Tombes R. M. (2008) CaMK-II promotes focal adhesion turnover and cell motility by inducing tyrosine dephosphorylation of FAK and paxillin. Cell Motil. Cytoskeleton 65, 662–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amran M. S., Homma N., Hashimoto K. (2003) Pharmacology of KB-R7943: a Na+-Ca2+ exchange inhibitor. Cardiovasc. Drug Rev. 21, 255–276 [DOI] [PubMed] [Google Scholar]
- 39. Billman G. E. (2001) KB-R7943: Kanebo. Curr. Opin. Investig. Drugs 2, 1740–1745 [PubMed] [Google Scholar]
- 40. Totsukawa G., Yamakita Y., Yamashiro S., Hartshorne D. J., Sasaki Y., Matsumura F. (2000) Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 150, 797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ikebe M., Hartshorne D. J. (1985) Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J. Biol. Chem. 260, 10027–10031 [PubMed] [Google Scholar]
- 42. Geering K. (2001) The functional role of β subunits in oligomeric P-type ATPases. J. Bioenerg. Biomembr. 33, 425–438 [DOI] [PubMed] [Google Scholar]
- 43. Carmosino M., Torretta S., Procino G., Timperio A., Zolla L., Svelto M. (2014) Na+/K+-ATPase β1-subunit is recruited in Na-K-2Cl co-transporter isoform 2 multiprotein complexes in rat kidneys: possible role in blood pressure regulation. J. Hypertens. 32, 1842–1853 [DOI] [PubMed] [Google Scholar]
- 44. Jha S., Dryer S. E. (2009) The β1 subunit of Na+/K+-ATPase interacts with BKCa channels and affects their steady-state expression on the cell surface. FEBS Lett. 583, 3109–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blaustein M. P., Lederer W. J. (1999) Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79, 763–854 [DOI] [PubMed] [Google Scholar]
- 46. Philipson K. D., Nicoll D. A. (2000) Sodium-calcium exchange: a molecular perspective. Annu. Rev. Physiol. 62, 111–133 [DOI] [PubMed] [Google Scholar]
- 47. Liao J., Li H., Zeng W., Sauer D. B., Belmares R., Jiang Y. (2012) Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 335, 686–690 [DOI] [PubMed] [Google Scholar]
- 48. Kip S. N., Strehler E. E. (2003) Characterization of PMCA isoforms and their contribution to transcellular Ca2+ flux in MDCK cells. Am. J. Physiol. Renal Physiol. 284, F122–F132 [DOI] [PubMed] [Google Scholar]
- 49. Iwamoto T., Watano T., Shigekawa M. (1996) A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J. Biol. Chem. 271, 22391–22397 [DOI] [PubMed] [Google Scholar]
- 50. Dunn J., Elias C. L., Le H. D., Omelchenko A., Hryshko L. V., Lytton J. (2002) The molecular determinants of ionic regulatory differences between brain and kidney Na+/Ca2+ exchanger (NCX1) isoforms. J. Biol. Chem. 277, 33957–33962 [DOI] [PubMed] [Google Scholar]
- 51. Hamming K. S., Soliman D., Webster N. J., Searle G. J., Matemisz L. C., Liknes D. A., Dai X. Q., Pulinilkunnil T., Riedel M. J., Dyck J. R., Macdonald P. E., Light P. E. (2010) Inhibition of β-cell sodium-calcium exchange enhances glucose-dependent elevations in cytoplasmic calcium and insulin secretion. Diabetes 59, 1686–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schwab A., Fabian A., Hanley P. J., Stock C. (2012) Role of ion channels and transporters in cell migration. Physiol. Rev. 92, 1865–1913 [DOI] [PubMed] [Google Scholar]
- 53. Tong X. P., Li X. Y., Zhou B., Shen W., Zhang Z. J., Xu T. L., Duan S. (2009) Ca2+ signaling evoked by activation of Na+ channels and Na+/Ca2+ exchangers is required for GABA-induced NG2 cell migration. J. Cell Biol. 186, 113–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sakamoto K., Owada Y., Shikama Y., Wada I., Waguri S., Iwamoto T., Kimura J. (2009) Involvement of Na+/Ca2+ exchanger in migration and contraction of rat cultured tendon fibroblasts. J. Physiol. 587, 5345–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Raizman J. E., Komljenovic J., Chang R., Deng C., Bedosky K. M., Rattan S. G., Cunnington R. H., Freed D. H., Dixon I. M. (2007) The participation of the Na+-Ca2+ exchanger in primary cardiac myofibroblast migration, contraction, and proliferation. J. Cell. Physiol. 213, 540–551 [DOI] [PubMed] [Google Scholar]
- 56. Kemény L. V., Schnúr A., Czepán M., Rakonczay Z., Jr., Gál E., Lonovics J., Lázár G., Simonka Z., Venglovecz V., Maléth J., Judák L., Németh I. B., Szabó K., Almássy J., Virág L., Geisz A., Tiszlavicz L., Yule D. I., Wittmann T., Varró A., Hegyi P. (2013) Na+/Ca2+ exchangers regulate the migration and proliferation of human gastric myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G552–G563 [DOI] [PubMed] [Google Scholar]
- 57. Ifuku M., Färber K., Okuno Y., Yamakawa Y., Miyamoto T., Nolte C., Merrino V. F., Kita S., Iwamoto T., Komuro I., Wang B., Cheung G., Ishikawa E., Ooboshi H., Bader M., Wada K., Kettenmann H., Noda M. (2007) Bradykinin-induced microglial migration mediated by B1-bradykinin receptors depends on Ca2+ influx via reverse-mode activity of the Na+/Ca2+ exchanger. J. Neurosci. 27, 13065–13073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dong H., Shim K. N., Li J. M., Estrema C., Ornelas T. A., Nguyen F., Liu S., Ramamoorthy S. L., Ho S., Carethers J. M., Chow J. Y. (2010) Molecular mechanisms underlying Ca2+-mediated motility of human pancreatic duct cells. Am. J. Physiol. Cell Physiol. 299, C1493–C1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chifflet S., Justet C., Hernández J. A., Nin V., Escande C., Benech J. C. (2012) Early and late calcium waves during wound healing in corneal endothelial cells. Wound Repair Regen. 20, 28–37 [DOI] [PubMed] [Google Scholar]
- 60. Andrikopoulos P., Baba A., Matsuda T., Djamgoz M. B., Yaqoob M. M., Eccles S. A. (2011) Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J. Biol. Chem. 286, 37919–37931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Prevarskaya N., Skryma R., Shuba Y. (2013) Targeting Ca2+ transport in cancer: close reality or long perspective? Expert Opin. Ther. Targets 17, 225–241 [DOI] [PubMed] [Google Scholar]
- 62. Wiegert J. S., Bading H. (2011) Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 49, 296–305 [DOI] [PubMed] [Google Scholar]
- 63. Reuter H., Henderson S. A., Han T., Ross R. S., Goldhaber J. I., Philipson K. D. (2002) The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ. Res. 90, 305–308 [DOI] [PubMed] [Google Scholar]
- 64. Schwab A., Finsterwalder F., Kersting U., Danker T., Oberleithner H. (1997) Intracellular Ca2+ distribution in migrating transformed epithelial cells. Pflugers Arch. 434, 70–76 [DOI] [PubMed] [Google Scholar]
- 65. Buchanan C. M., Dickson J. M., Lee W. J., Guthridge M. A., Kendall J. D., Shepherd P. R. (2013) Oncogenic mutations of p110α isoform of PI 3-kinase upregulate its protein kinase activity. PloS ONE 8, e71337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vagin O., Tokhtaeva E., Sachs G. (2006) The role of the β1 subunit of the Na,K-ATPase and its glycosylation in cell-cell adhesion. J. Biol. Chem. 281, 39573–39587 [DOI] [PubMed] [Google Scholar]
- 67. Rajasekaran S. A., Ball W. J., Jr., Bander N. H., Liu H., Pardee J. D., Rajasekaran A. K. (1999) Reduced expression of β-subunit of Na,K- ATPase in human clear-cell renal cell carcinoma. J. Urol. 162, 574–580 [PubMed] [Google Scholar]
- 68. Rajasekaran S. A., Barwe S. P., Rajasekaran A. K. (2005) Multiple functions of Na,K-ATPase in epithelial cells. Semin. Nephrol. 25, 328–334 [DOI] [PubMed] [Google Scholar]