

Background: The epithelial Na+ channel ENaC plays an important role in epithelial Na+ absorption.
Results: Lysine acetylation increased ENaC abundance and current by antagonizing its ubiquitination and degradation.
Conclusion: Acetylation regulates ENaC stability.
Significance: Lysine modification plays a central role in ENaC regulation, where it may contribute to the control of Na+ homeostasis and blood pressure.
Keywords: Epithelial Sodium Channel (ENaC), Histone Deacetylase (HDAC), Hypertension, Protein Degradation, Ubiquitylation (Ubiquitination), Amiloride, Lysine Acetylation, Epithelia
Abstract
The epithelial Na+ channel (ENaC) functions as a pathway for Na+ absorption in the kidney and lung, where it is crucial for Na+ homeostasis and blood pressure regulation. ENaC is regulated in part through signaling pathways that control the ubiquitination state of ENaC lysines. A defect in ubiquitination causes Liddle syndrome, an inherited form of hypertension. Here we determined that α-, β-, and γENaC are also substrates for lysine acetylation. Trichostatin A (TSA), a histone deacetylase inhibitor, enhanced ENaC acetylation and increased ENaC abundance in the total cell lysate and at the cell surface. Moreover, TSA increased ENaC current in Fischer rat thyroid and kidney collecting duct epithelia. We found that HDAC7 is expressed in the kidney collecting duct, supporting a potential role for this histone deacetylase in ENaC regulation. HDAC7 overexpression reduced ENaC abundance and ENaC current, whereas ENaC abundance and current were increased by silencing of HDAC7. ENaC and HDAC7 form a complex, as detected by coimmunoprecipitation. We observed a reciprocal relationship between acetylation and ubiquitination; TSA reduced ENaC ubiquitination, whereas HDAC7 increased ubiquitination. By reducing ENaC ubiquitination, TSA decreased the rate of ENaC degradation. Thus, acetylation increases epithelial Na+ absorption by antagonizing ENaC ubiquitination. This stabilizes ENaC, and hence, increases its abundance at the cell surface.
Introduction
The epithelial Na+ channel (ENaC),2 a heterotrimer of α, β, and γ subunits, functions as a pathway for Na+ transport across epithelial cells (1, 2). In the kidney collecting duct, ENaC plays an important role in extracellular Na+ and volume homeostasis, which is critical for blood pressure regulation (3). In the lung, ENaC controls the volume and composition of airway surface liquid (4).
Na+ absorption is regulated in large part through mechanisms that alter ENaC trafficking. Moreover, defects in ENaC trafficking cause disease. For example, an inherited form of hypertension, Liddle syndrome, is caused by mutations that disrupt PY motifs located in the C termini of β- or γENaC (3, 5). These motifs function as binding sites for the E3 ubiquitin ligase Nedd4-2 (6, 7). When bound to ENaC, Nedd4-2 catalyzes ubiquitination of lysines within the N termini of all three ENaC subunits (8). This reduces ENaC expression at the cell surface by increasing ENaC endocytosis and by targeting ENaC to lysosomes for degradation (9, 10). Conversely, ubiquitin hydrolases deubiquitinate ENaC, increasing surface expression by reducing endocytosis (USP2-45) and enhancing recycling (USP8) (11, 12). Ubiquitination also functions in the biosynthetic pathway, targeting incorrectly folded and unassembled ENaC subunits for endoplasmic reticulum-associated degradation, although the ubiquitin ligases that mediate this process have not been identified (13, 14).
ENaC ubiquitination occurs at multiple lysines located within the N termini of α-, β-, and γENaC (8, 15). It is clear that these lysines play an important role in ENaC regulation; mutation of all lysines largely recapitulates the effects of PY motif mutations that cause Liddle syndrome (15, 16). In addition to functioning as a substrate for ubiquitination, lysines can undergo other forms of post-translational modification. For example, lysines can be acetylated, whereby histone acetyltransferases catalyze transfer of an acetyl group from acetyl-CoA to the target lysine (17). Conversely, acetyl groups are removed by histone deacetylases (HDACs). As the names of these proteins imply, this process is best characterized in histones, where lysine acetylation/deacetylation regulates transcription by altering chromatin structure. Examples of acetylation of non-histone proteins are also emerging (18). Acetylation can alter protein function or stability by interfering with alternative lysine modifications (e.g. ubiquitination) or by altering protein interaction sites. For instance, acetylation stabilizes the tumor suppressor p53 by preventing Mdm2-mediated degradation (19). However, the situation can be more complex. HIF-1α, which mediates responses to hypoxia, is both stabilized (20) and destabilized (21) depending on which lysines are acetylated. Because lysines play a critical role in ENaC trafficking, we asked whether they might be targets for acetylation, and we investigated a potential role for lysine acetylation in ENaC regulation.
EXPERIMENTAL PROCEDURES
cDNA Constructs
Human α-, β-, and γENaC in pMT3 were cloned as described previously (22, 23). α-FLAG, β-FLAG, and γ-FLAG contain a FLAG epitope (DYKDDDDK) at the C terminus, and α-V5 contains a V5 epitope (GKPIPNPLLGLDST) at the C terminus. The cytoplasmic lysines were mutated to arginine in each subunit (K-R), as described previously (12). Mouse HDAC7 cDNA was provided by Ronald Evans (24), human HDAC9 cDNA was provided by Victoria Richon (25), and ubiquitin-HA was provided by Dirk Bohmann (26). HDAC7 siRNA was obtained from Dharmacon.
Cell Culture and Transfection
HEK 293T cells were cultured in Dulbecco's modified Eagle's medium. Fischer rat thyroid (FRT) cells were cultured on permeable filter supports (Millicell PCF, 0.4-μm pore size, 12-mm diameter) in F-12 Coon's medium with 5% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C. Cells were transfected with cDNAs and siRNAs using Lipofectamine 2000 (Invitrogen). Following transfection, 10 μm amiloride was added to the culture medium.
mpkCCDc14 cells (provided by Alain Vandewalle, Institut National de la Santé et de la Recherche Médicale) were cultured on permeable filter supports in equal volumes of DMEM and Ham's F12 with 60 nm sodium selenate, 5 mg/ml transferrin, 2 mm glutamine, 50 nm dexamethasone, 1 nm triiodothyronine, 10 ng/ml epidermal growth factor, 5 mg/ml insulin, 20 mm d-glucose, 2% FCS, and 20 mm HEPES, pH 7.4, as described previously (27). The cells were transfected with cDNA or siRNA using HVJ-E following the manufacturer's instructions (GenomONE) in medium lacking antibiotics. Six h after transfection, the cells were cultured in medium containing dexamethasone (50 nm) but not epidermal growth factor or insulin.
Biochemistry
Cells were treated with 700 nm trichostatin A (TSA) or vehicle (ethanol) for 24 h and then lysed in 150 mm NaCl, 50 mm Tris (pH 7.4), 0.5% Nonidet P-40, and protease inhibitor mixture (Sigma). To detect acetylated ENaC, 500 μg of protein was immunoprecipitated with anti-acetyl lysine antibody (Cell Signaling Technology) and immobilized protein A (Pierce). After extensive washing, immunoprecipitated proteins were eluted in SDS-PAGE sample loading buffer (100 mm dithiothreitol, 20% glycerol, 100 mm Tris-Cl, pH 6.8, and 4% SDS) at 95 °C for 5 min and then separated by SDS-PAGE. Cell lysates (10 μg) were also separated by SDS-PAGE. Proteins were detected by immunoblot with anti-FLAG M2 monoclonal antibody-peroxidase conjugate (Sigma) at 1: 5000 dilution and enhanced chemiluminescence (ECL Plus, GE Healthcare).
To detect the cell surface fraction of ENaC, transfected HEK 293T cells were washed with ice-cold PBS-CM (PBS with 1 mm CaCl2 and MgCl2) three times on ice, labeled with 0.5 mg/ml Sulfo-NHS-biotin (Pierce) in PBS-CM for 20 min on ice, and quenched with 100 mm glycine in PBS-CM for 10 min. After washing three times with PBS-CM, cells were lysed in 0.4% sodium deoxycholate, 1% Nonidet P-40, 63 mm EDTA, 50 mm Tris-HCl, pH 8, and protease inhibitor mixture. Biotin-labeled cell surface proteins were isolated by incubating cell lysate (200 μg) with immobilized NeutrAvidin beads (Pierce) for 12 h at 4 °C, and then detected by immunoblot (anti-FLAG M2 monoclonal antibody-peroxidase conjugate).
To detect interactions between αENaC and HDAC7, HEK 293T cells were transfected with αENaC-V5, HDAC7-HA, or both (total cDNA kept constant using cDNA encoding GFP). Following cell lysis in 150 mm NaCl, 50 mm Tris (pH 7.4), 0.5% Nonidet P-40, and protease inhibitor mixture (Sigma), we immunoprecipitated αENaC-V5 (1:300 anti-V5 antibody, Sigma) or HDAC7-HA (1:300 anti-HA antibody, Sigma). Following SDS-PAGE, we detected αENaC-V5 and HDAC7-HA by immunoblot.
To detect ubiquitinated ENaC, HEK 293T cells transfected with or without cDNAs encoding ubiquitin-HA, ENaC (α-FLAG, β-FLAG, and γ-FLAG), and HDAC7 were treated with N-acetyl-leucinyl-leucinyl-norleucinal (ALLN) (10 μm) for 12 h. They were lysed in 150 mm NaCl, 50 mm Tris (pH 7.4), 1% Triton X-100, and protease inhibitor mixture (Sigma). 800 μg of protein was immunoprecipitated with anti-HA antibody (1:300, Sigma) and immobilized protein A (Pierce), separated by SDS-PAGE, and then detected by immunoblot with anti-FLAG M2 monoclonal antibody-peroxidase conjugate (1:5000, Sigma) and quantitated by densitometry using ImageJ software.
To quantitate the rate of ENaC degradation, HEK 293T cells were treated with cycloheximide (10 μg/ml) for 0–3 h to inhibit protein synthesis, and αENaC was detected and quantitated by immunoblotting, as above. Proteins were detected by immunoblot in mouse kidney collecting duct epithelia (mpkCCD) cells using anti-HDAC7 antibody (Cell Signaling Technology) and anti-αENaC antibody (StressMarq Biosciences).
For renal tissue analysis, C57BL/6J mice and Sprague-Dawley rats were obtained from Harlan Laboratories and kept on a standard Harlan Teklad TD.96208 diet until the age of 8 weeks. Animal use and welfare procedures adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals following protocols reviewed and approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. Mice and rats were anesthetized with isoflurane, and the kidneys were flushed with PBS and then removed. Apical kidney sections corresponding to cortex were isolated (∼1 g) and then diced into small pieces with a razor blade. Glomeruli were isolated via differential sieving as described previously (28). Samples were pulse-sonicated in 10 mm NaCl, 10 mm Tris-HCl, (pH 7.5), 1% Nonidet P-40, and protease inhibitor mixture (Roche Applied Science) for 10 s and centrifuged at 10,000 × g for 10 min. Proteins were separated by SDS-PAGE and detected by immunoblot using HDAC7 antibody (Santa Cruz Biotechnology).
Immunohistochemistry
Sprague-Dawley rat kidneys were fixed in zinc formalin (24–48 h) and processed for paraffin embedding. Kidney sections were cut at 4 μm, dried, and deparaffinized for subsequent labeled streptavidin-biotin immunohistochemistry. The slides were treated with citrate buffer (pH 6) for a total of 35 min. The slides were blocked with a peroxidase block (Dako), avidin block (Vector Laboratories), biotin block (Vector Laboratories), and serum-free protein block (Dako). Tissue sections were incubated for 90 min with anti-HDAC7 antibody (Abcam) and then goat anti-rabbit biotinylated IgG (Biocare Medical) followed by streptavidin horseradish peroxidase (Biocare Medical) and visualized with diaminobenzidine (Dako). To identify collecting ducts, consecutive sections were stained with anti-aquaporin 2 antibody (Santa Cruz Biotechnology). All slides were counterstained with a Mayer hematoxylin (Dako), dehydrated, and mounted with permanent mounting medium (Sakura).
Electrophysiology
Short-circuit current was measured in Ussing chambers using an EC-825 amplifier (Warner Instruments), digitized with a PowerLab interface (ADInstruments), and recorded and analyzed with Chart software (ADInstruments). The apical and basolateral surfaces were bathed in 135 mm NaCl, 1.2 mm CaCl2, 1.2 mm MgCl2, 2.4 mm K2HPO4, 0.6 mm KH2PO4, and 10 mm HEPES (pH 7.4) at 37 °C. Amiloride (10 μm) was added to the apical solution to quantitate ENaC current.
Statistics
All data are expressed as mean ± S.E. Differences were assessed by paired or unpaired t tests, with significance indicated by p < 0.05. Immunoblot and immunohistochemistry experiments were carried out three or more times.
RESULTS
ENaC Acetylation
To test whether ENaC subunits are substrates for lysine acetylation, we treated HEK 293 cells for 1 h with the class I and II HDAC inhibitor TSA. In the absence of TSA, we detected minimal acetylated α-, β-, and γENaC. TSA enhanced acetylation of all three subunits (Fig. 1). Mutation of all cytoplasmic lysines in γENaC abolished acetylation (Fig. 1). Thus, α-, β-, and γENaC are substrates for acetylation. Moreover, each subunit is deacetylated by one or more class I or II HDAC.
FIGURE 1.

ENaC acetylation. HEK 293 cells were transfected with cDNAs encoding α-FLAG, β-FLAG, or γ-FLAG (wild type or K-R) ENaC or empty plasmid (C) (2 μg), as indicated, and treated with TSA (700 nm) or vehicle (ethanol) for 24 h. Acetylated ENaC (ENaC-Ac) subunits were immunoprecipitated with anti-acetyl lysine antibody and immunoblotted with anti-FLAG antibody.
Because ENaC functions primarily as a heterotrimer of α-, β-, and γ subunits (22, 29), we asked whether acetylation would alter ENaC abundance when all three subunits were coexpressed. αENaC migrates as two bands when coexpressed with β- and γENaC: an ∼90-kDa full-length form and a proteolytically cleaved ∼70-kDa form (Fig. 2A) (30, 31). TSA increased the abundance of αENaC both in the total cell lysate and in the biotinylated cell surface fraction (Fig. 2A). Likewise, TSA increased the abundance of βENaC (which is not proteolytically cleaved) and γENaC (full-length ∼92-kDa form and cleaved ∼75-kDa form) in the cell lysate and at the cell surface (Fig. 2B).
FIGURE 2.
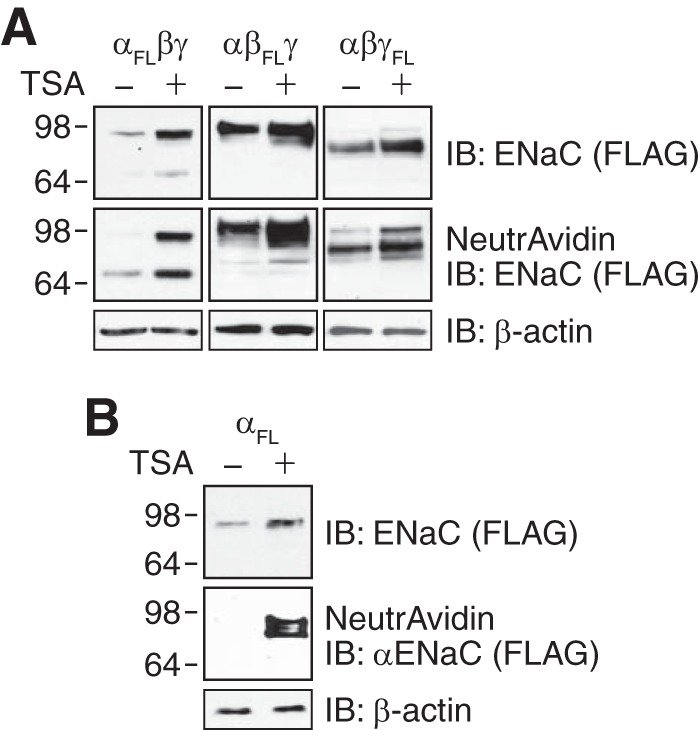
Acetylation increases ENaC abundance. A, immunoblots (IB) (anti-FLAG) of total (top panel) and biotinylated (middle panel) ENaC subunits in HEK 293 cells cotransfected with α-, β-, and γENaC (2 μg of each, one subunit contained FLAG epitope (FL)) and treated with TSA (700 nm) or vehicle (ethanol) for 24 h. Immunoblots for β-actin are shown in the bottom panel. B, immunoblots (anti-FLAG) of total (top panel) and biotinylated (middle panel) αENaC in HEK 293 cells transfected with αFLENaC (2 μg) and treated with TSA (700 nm) or vehicle (ethanol) for 24 h. Immunoblot for β-actin is shown in the bottom panel.
Although ENaC most commonly functions as a heterotrimer, αENaC can generate small amiloride-sensitive currents when expressed alone (23, 32). When we expressed αENaC without β- and γENaC, we detected αENaC protein in the total cell lysate but not at the cell surface (Fig. 2B). To test whether acetylation might alter αENaC abundance and localization, we treated cells with TSA. In addition to enhancing αENaC abundance in the cell lysate, TSA induced the appearance of αENaC at the cell surface (Fig. 2B). Thus, acetylation enhances the abundance of ENaC at the cell surface, both when it is assembled as a heterotrimer and when αENaC is expressed alone.
Acetylation Increases ENaC Current
Because acetylation increased ENaC abundance at the cell surface, we predicted that it would increase ENaC current. In FRT epithelia, coexpression of α-, β-, and γENaC generated transepithelial short-circuit Na+ current that was blocked by the ENaC inhibitor amiloride (Fig. 3A) (33). Treatment of the cells with TSA (24 h) increased amiloride-sensitive ENaC current (Fig. 3, A and B). This stimulation was abolished by mutation of all lysines in the ENaC cytoplasmic domains (Fig. 3B), consistent with a requirement for lysine acetylation.
FIGURE 3.
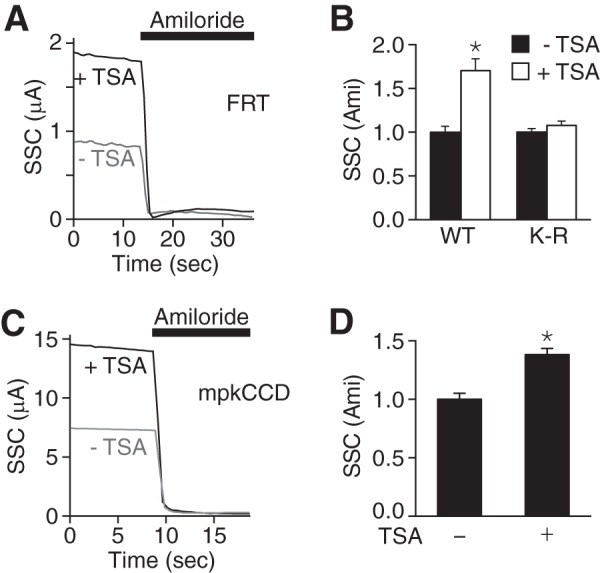
Acetylation increases ENaC current. A, superimposed representative short-circuit current traces in FRT epithelia transfected with α-, β-, and γENaC (0.5 μg of each) and treated with TSA (50 nm, black) or vehicle (ethanol, gray) for 24 h. Amiloride (10 μm) was added to the apical bathing solution, as indicated by the black bar. B, quantification of amiloride-sensitive short-circuit currents (relative to −TSA groups) in FRT epithelia transfected with wild type ENaC or cytoplasmic K-R mutant ENaC (K-R) (mean ± S.E., n = 12–14; *, p < 0.0001). Non-normalized currents in the absence of TSA were: wild type, 2.82 ± 0.2 μA/cm2; K-R, 0.73 ± 0.03 μA/cm2. C, representative short-circuit current traces in mpkCCD epithelia treated with TSA (50 nm, black) or vehicle (ethanol, gray) for 24 h. D, quantification of amiloride-sensitive short-circuit currents in mpkCCD epithelia (relative to −TSA group) (mean ± S.E., n = 14; *, p < 0.009).
We also tested the effect of TSA on endogenous ENaC current in mpkCCD (27). We found that TSA increased amiloride-sensitive transepithelial short-circuit current (Fig. 3, C and D), similar to results in FRT epithelia. Together, the data indicate that lysine acetylation increases ENaC current.
HDAC7 Regulates ENaC
TSA increases acetylation by inhibiting class I and II HDACs. We tested HDAC7 as a candidate to regulate ENaC. HDAC7 protein was present in total kidney lysates from rat and mouse, but comparatively little HDAC7 was detected in glomeruli (Fig. 4A). By immunohistochemistry in rat kidney, we found that HDAC7 staining was most prominent in the collecting duct; this segment was identified by the presence of aquaporin 2 (Fig. 4B). Importantly, the collecting duct is a site for ENaC-mediated Na+ absorption.
FIGURE 4.
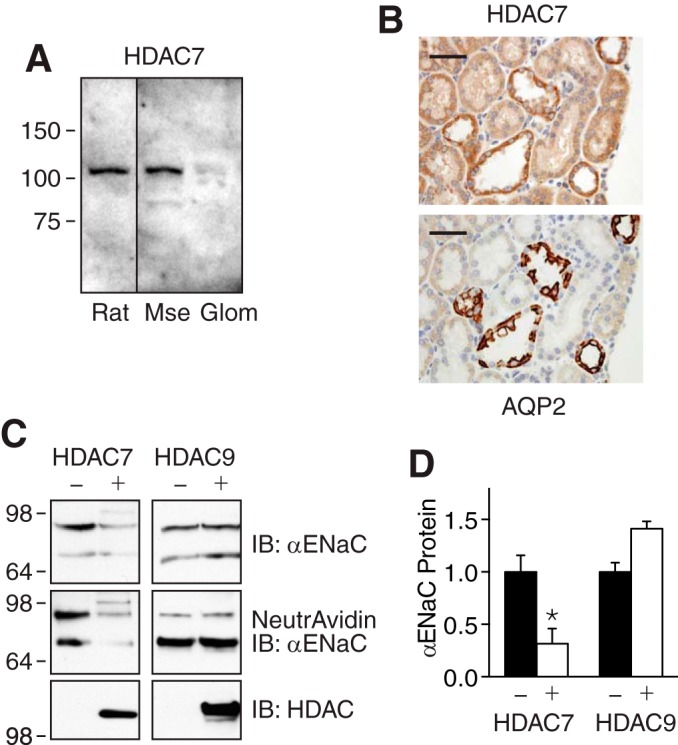
HDAC7 reduces ENaC abundance. A, HDAC7 immunoblots in kidney lysates from rat and mouse (Mse) and isolated glomeruli (Glom). B, immunohistochemistry of rat kidney sections using HDAC7 antibody (top panel) or aquaporin 2 (AQP2) antibody (bottom panel). Scale bars are 50 μm. C, immunoblots (IB) (anti-FLAG) of total (top panel) and biotinylated (middle panel) αENaC in HEK 293 cells cotransfected with αFL-, β-, and γENaC (1 μg of each) with or without HDAC7 or HDAC9 (3 μg). Immunoblots for HDAC7 or HDAC9 are shown in the bottom panels. D, quantification of αENaC in total cell lysate in cells cotransfected with or without HDAC7 or HDAC9 (relative to −HDAC group) (mean ± S.E., n = 5; *, p < 0.005).
To test the effect of HDAC7 on ENaC protein abundance, we coexpressed HDAC7 and ENaC in HEK 293 cells. HDAC7 reduced αENaC (coexpressed with β- and γENaC) abundance in the total cell lysate (Fig. 4, C and D) and at the cell surface (Fig. 4C). In contrast, HDAC9 did not reduce αENaC abundance (Fig. 4, C and D).
We tested whether HDAC7 would alter ENaC current. In FRT epithelia, HDAC7 decreased amiloride-sensitive short-circuit current (Fig. 5, A and B). In mpkCCD epithelia, overexpression of HDAC7 (Fig. 5C) reduced endogenous ENaC current (Fig. 5D). Conversely, HDAC7 siRNA reduced expression of endogenous HDAC7 by 78% (Fig. 5E), which increased αENaC abundance (Fig. 5E) and ENaC current (Fig. 5F). A second HDAC7 siRNA produced a similar increase in ENaC current (Fig. 5F).
FIGURE 5.
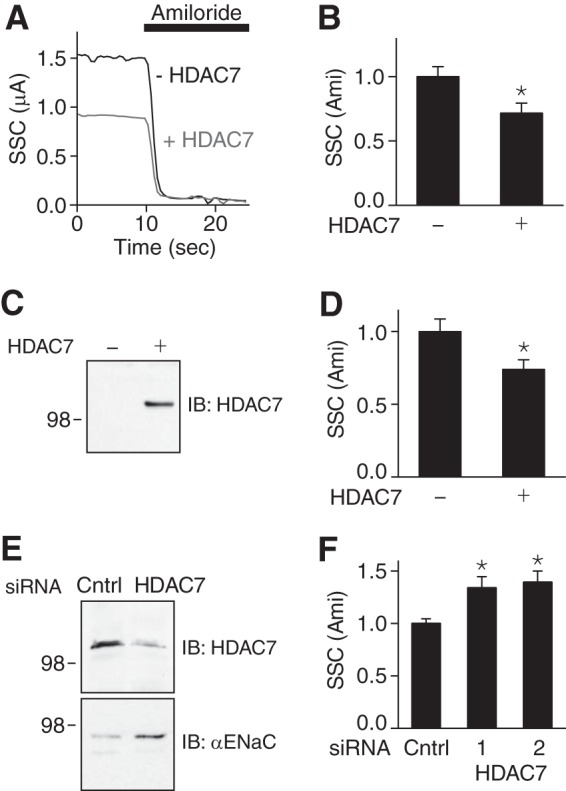
HDAC7 reduces ENaC current. A, superimposed representative short-circuit current traces in FRT epithelia cotransfected with α-, β-, and γENaC (0.25 μg of each) and HDAC7 or GFP (−HDAC7)(0.75 μg). Amiloride (10 μm) was added to the bathing solution, as indicated by the black bar. B, quantification of amiloride-sensitive short-circuit currents in FRT epithelia (relative to −HDAC7 group) (mean ± S.E., n = 11; *, p < 0.01). C, immunoblot (IB) (anti-FLAG) of HDAC7-FLAG in mpkCCD epithelia transfected with HDAC7-FLAG (+) or GFP (−)(2.8 μg). D, quantification of amiloride-sensitive short-circuit currents in mpkCCD epithelia transfected with HDAC7 or GFP (relative to −HDAC7 group) (mean ± S.E., n = 15–16; *, p < 0.015). E, immunoblot of endogenous HDAC7 and αENaC in mpkCCD epithelia transfected with HDAC7 siRNA or non-targeting control (Cntrl) siRNA. F, quantification of amiloride-sensitive short-circuit currents in mpkCCD epithelia transfected with two HDAC7 siRNAs or control siRNA (relative to control siRNA group) (mean ± S.E., n = 14–22; *, p < 0.009).
We asked whether ENaC and HDAC7 form a complex. When we immunoprecipitated αENaC, we detected coprecipitated HDAC7 in HEK 293 cells that were cotransfected with ENaC and HDAC7, but not in cells transfected with either protein alone (Fig. 6). Using the reciprocal strategy, we found that αENaC coprecipitated with HDAC7 (Fig. 6). These findings indicate that ENaC and HDAC7 interact with one another to form a complex. Together, the data support a role for HDAC7 in ENaC regulation.
FIGURE 6.
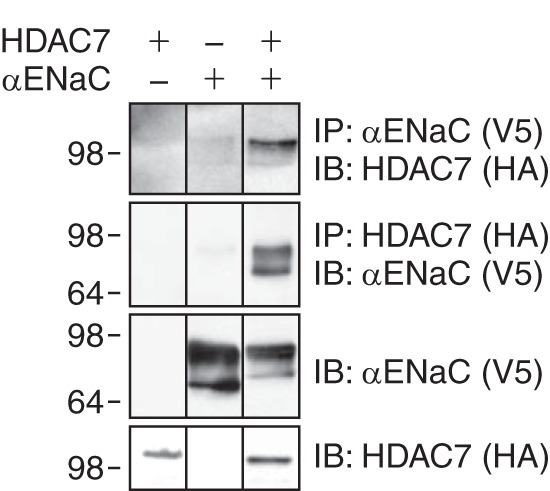
HDAC7 and αENaC form a complex. Coimmunoprecipitation of αENaC-V5 and HDAC7-HA in HEK 293 cells transfected with cDNAs encoding αENaC-V5, HDAC7-HA, or both was performed. Cell lysates were immunoprecipitated (IP) and immunoblotted (IB) as indicated.
Acetylation Antagonizes ENaC Ubiquitination
Because acetylation and ubiquitination both occur on lysine residues, we hypothesized that acetylation might alter ENaC ubiquitination. In HEK 293 cells expressing α-, β-, and γENaC, we detected ubiquitinated αENaC in the total cell lysate (Fig. 7A, top panel). When we increased ENaC acetylation with TSA, αENaC ubiquitination was reduced (Fig. 7A, top panel, and 7B). We also detected and quantitated the cell surface fraction of ubiquitinated αENaC (labeled with biotin); TSA reduced ubiquitination of cell surface αENaC (Fig. 7A, middle panel, and 7B). As a reciprocal approach, we overexpressed HDAC7 to reduce acetylation. HDAC7 increased αENaC ubiquitination both in the total cell lysate and in the cell surface fraction (Fig. 7, C and D). Thus, we determined that there is an opposing relationship between acetylation and ubiquitination.
FIGURE 7.
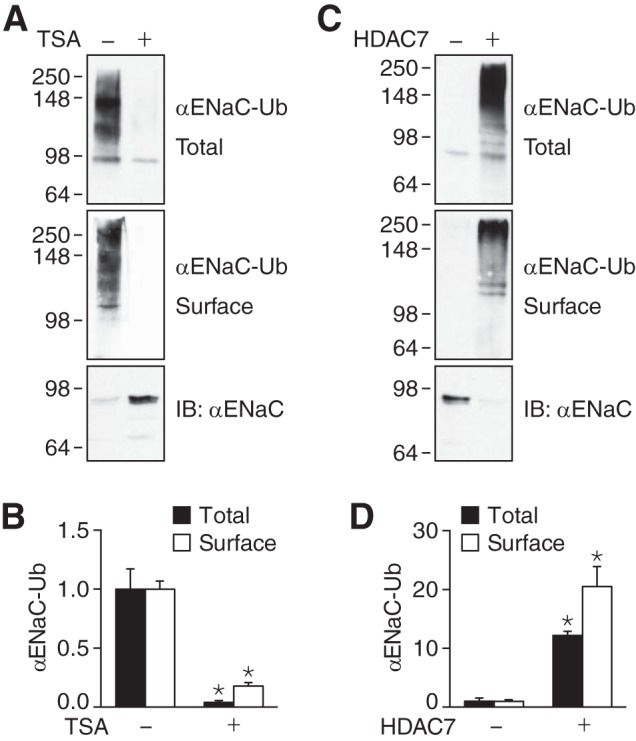
Acetylation antagonizes ENaC ubiquitination. A and C, immunoblots (IB) of ubiquitinated αENaC (αENaC-Ub) in total cell lysate (top panels) (immunoprecipitation of αFL; immunoblotting of ubiquitin-HA and biotinylated cell surface fraction (middle panels) (immunoprecipitation of αFL, followed by pulldown of biotinylated proteins with immobilized NeutrAvidin, and then immunoblotting of ubiquitin-HA), and total αENaC in the cell lysates (bottom panels). HEK 293 cells were transfected with αFL-, β-, and γENaC (1 μg of each) and ubiquitin-HA (3 μg) and treated with TSA (700 nm for 24 h, +) or vehicle (ethanol, −) (A), or cotransfected with HDAC7 (+) or GFP (−) (C). B and D, the quantity of ubiquitinated αENaC (relative to −TSA (B) or −HDAC7 (D)) is plotted (mean ± S.E., n = 4–6; *, p < 0.007). In panel C, we used shorter exposures than in panel A to allow us to detect increased ubiquitination induced by HDAC7.
Acetylation Reduces ENaC Degradation
Previous work indicates that ubiquitination induces ENaC degradation (8, 15, 34). We hypothesized that by reducing ubiquitination, acetylation might decrease ENaC degradation. To test this hypothesis, we treated cells with or without TSA, and then quantitated the rate of αENaC (coexpressed with β- and γENaC) degradation using a cycloheximide chase assay. In the absence of TSA, αENaC protein disappeared with a half-life of ∼45 min (Fig. 8, A and B). TSA reduced the ENaC degradation rate, increasing the half-life to ∼150 min. This indicates that acetylation stabilizes ENaC, which explains the increase in ENaC abundance at the cell surface.
FIGURE 8.
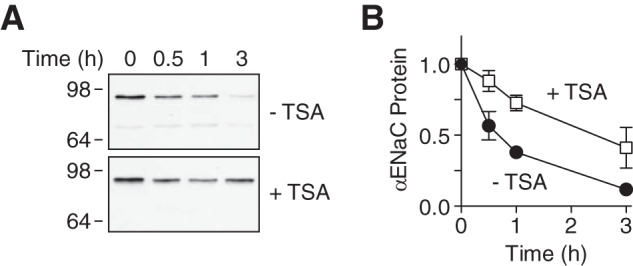
Acetylation reduces ENaC degradation. A, immunoblots of αENaC-FLAG in HEK 293 cells cotransfected with β- and γENaC and treated with TSA (700 nm) or vehicle for 24 h. To inhibit protein synthesis, the cells were treated with cycloheximide (10 μg/ml) for 0–3 h prior to lysis. B, quantification of αENaC protein relative to 0 min time point (mean ± S.E.; n = 5–7).
DISCUSSION
In this work, we define a role for lysine acetylation in the regulation of ENaC. We found that acetylation reduced ENaC degradation, enhancing its abundance at the cell surface. As a result, acetylation increased ENaC current in epithelia.
Lysines are substrates for both acetylation and ubiquitination, which had opposing effects on ENaC degradation. Thus, they function as a critical convergence point for ENaC regulation. In contrast to the stabilizing effect we observed for acetylation, ubiquitination reduces ENaC abundance by increasing its degradation (8, 15). Ubiquitination is a dynamic process; the ubiquitination state is determined by the relative activity of E3 ubiquitin ligases, such as Nedd4-2 (8, 35), and ubiquitin hydrolases, including USP2-45 (36) and USP8 (12). Here we showed that the ubiquitination state of ENaC is also modulated by lysine acetylation. Acetylation antagonized ENaC ubiquitination; maneuvers that increased acetylation reduced ubiquitination, whereas deacetylation had the opposite effect. The simplest model is one in which ubiquitin ligases and histone acetyltransferases compete for a common set of ENaC lysines. When acetylated, these lysines would no longer be susceptible to ubiquitination. However, there are other potential explanations for the data. Acetylation might reduce ubiquitination indirectly by targeting a different subset of lysines. In this case, acetylation could induce conformational changes that reduce accessibility of ubiquitination sites. Alternatively, acetylation could enhance ENaC deubiquitination by altering accessibility or binding of ubiquitin hydrolases.
By opposing ubiquitination, acetylation could regulate ENaC at multiple cellular locations. In the biosynthetic pathway, ubiquitination targets misfolded or incorrectly assembled channels for endoplasmic reticulum-associated degradation (13). In this location, acetylation could increase ENaC delivery to the cell surface. At the cell surface, ubiquitination functions as a signal to induce ENaC endocytosis (9). Once it reaches the endosome, ubiquitination mediates sorting between lysosomal degradation and recycling pathways (9, 37). Thus, acetylation could regulate ENaC in the endocytic pathway, reducing endocytosis and/or increasing recycling. Although additional work will be required to distinguish between these possibilities, our data begin to hint at a role for acetylation in the biosynthetic pathway. First, acetylation stabilized ENaC in the total cell lysate, which predominantly reflects ENaC located in the biosynthetic pathway. Second, when acetylated, we found that αENaC trafficked to the cell surface in the absence of the other two subunits. Without acetylation, efficient forward trafficking requires coassembly of αENaC with β- and γENaC; when αENaC is expressed alone, it is ubiquitinated and targeted to the proteasome for degradation (13–15). The functional relevance of this observation is unclear. It has been suggested that αENaC homomultimers might be responsible for the high conductance non-selective cation currents observed in alveolar epithelial cells, which contrast with the highly Na+-selective low conductance current produced by αβγENaC (38, 39). Perhaps acetylation underlies the trafficking of these αENaC channels to the cell surface.
The acetylation state of proteins is controlled in part by the activity of HDACs, which deacetylate their targets. Here we defined a role for HDAC7 in the regulation of ENaC surface expression. We found that HDAC7 is expressed in the kidney collecting duct, which is a critical site for ENaC-mediated Na+ transport. Moreover, knockdown of HDAC7 increased ENaC current, whereas HDAC7 overexpression had the opposite effect. Previous work indicates that HDAC7 has diverse functional effects. HDAC7 contributes to regulation of bone remodeling, T lymphocyte selection/activity, myocyte migration/differentiation, and modulation of enzymes involved in intermediate metabolism (40–43). HDAC7 is thought to contribute to the pathogenesis of diseases including cystic fibrosis (44) and α-1 antitrypsin deficiency (45). Most of these effects are mediated by its activity as a transcriptional corepressor. In contrast, HDAC7 regulates ENaC through a post-translational mechanism, enhancing ENaC ubiquitination. Although our data support a role for HDAC7, it seems possible that additional HDACs will also contribute to ENaC regulation.
ENaC plays an essential role in controlling extracellular Na+ and fluid balance. A defect in ENaC ubiquitination causes an inherited form of hypertension (Liddle syndrome), emphasizing the critical role that ubiquitination plays in the regulation of epithelial Na+ transport. In this work, we have identified an additional level of complexity in the signaling pathways that control ENaC ubiquitination. Elucidation of these pathways may provide a deeper understanding of diseases including hypertension, and targeting them may lead to new and more effective treatments.
Acknowledgments
We thank Abigail Hamilton and Diane Olson (University of Iowa), Vladislav Levchenko (Medical College of Wisconsin), and Shanna Dennis (Sponsored Projects for Undergraduate Research (SPUR) Student, St. Norbert College) for technical assistance. We acknowledge the University of Iowa DNA Core Facility for reagents and DNA sequencing.
This work was supported by National Institutes of Health Grants HL058812 and HL072256 (to P. M. S.) and HL108880 (A. S.), Department of Veterans Affairs Biomedical Laboratory Research and Development Service Grant BX001862 (to P. M. S.), and an American Heart Association Postdoctoral Research Fellowship (to P. B.).
- ENaC
- epithelial Na+ channel
- HDAC
- histone deacetylase
- TSA
- trichostatin A
- FRT
- Fischer rat thyroid
- mpkCCD
- mouse kidney collecting duct epithelia.
REFERENCES
- 1. Schild L. (2004) The epithelial sodium channel: from molecule to disease. Rev. Physiol. Biochem. Pharmacol. 151, 93–107 [DOI] [PubMed] [Google Scholar]
- 2. Snyder P. M. (2005) Minireview: regulation of epithelial Na+ channel trafficking. Endocrinology 146, 5079–5085 [DOI] [PubMed] [Google Scholar]
- 3. Lifton R. P. (1996) Molecular genetics of human blood pressure variation. Science 272, 676–680 [DOI] [PubMed] [Google Scholar]
- 4. Boucher R. C., Cotton C. U., Gatzy J. T., Knowles M. R., Yankaskas J. R. (1988) Evidence for reduced Cl− and increased Na+ permeability in cystic fibrosis human primary cell cultures. J. Physiol. 405, 77–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Snyder P. M., Price M. P., McDonald F. J., Adams C. M., Volk K. A., Zeiher B. G., Stokes J. B., Welsh M. J. (1995) Mechanism by which Liddle's syndrome mutations increase activity of a human epithelial Na+ channel. Cell 83, 969–978 [DOI] [PubMed] [Google Scholar]
- 6. Goulet C. C., Volk K. A., Adams C. M., Prince L. S., Stokes J. B., Snyder P. M. (1998) Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle's syndrome. J. Biol. Chem. 273, 30012–30017 [DOI] [PubMed] [Google Scholar]
- 7. Staub O., Dho S., Henry P., Correa J., Ishikawa T., McGlade J., Rotin D. (1996) WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J. 15, 2371–2380 [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou R., Patel S. V., Snyder P. M. (2007) Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 282, 20207–20212 [DOI] [PubMed] [Google Scholar]
- 9. Kabra R., Knight K. K., Zhou R., Snyder P. M. (2008) Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J. Biol. Chem. 283, 6033–6039 [DOI] [PubMed] [Google Scholar]
- 10. Lu C., Pribanic S., Debonneville A., Jiang C., Rotin D. (2007) The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 8, 1246–1264 [DOI] [PubMed] [Google Scholar]
- 11. Oberfeld B., Ruffieux-Daidié D., Vitagliano J. J., Pos K. M., Verrey F., Staub O. (2011) Ubiquitin-specific protease 2-45 (Usp2-45) binds to epithelial Na+ channel (ENaC)-ubiquitylating enzyme Nedd4-2. Am. J. Physiol. Renal Physiol. 301, F189–F196 [DOI] [PubMed] [Google Scholar]
- 12. Zhou R., Tomkovicz V. R., Butler P. L., Ochoa L. A., Peterson Z. J., Snyder P. M. (2013) Ubiquitin-specific peptidase 8 (USP8) regulates endosomal trafficking of the epithelial Na+ channel. J. Biol. Chem. 288, 5389–5397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buck T. M., Kolb A. R., Boyd C. R., Kleyman T. R., Brodsky J. L. (2010) The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol. Biol. Cell 21, 1047–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malik B., Schlanger L., Al-Khalili O., Bao H. F., Yue G., Price S. R., Mitch W. E., Eaton D. C. (2001) ENac degradation in A6 cells by the ubiquitin-proteosome proteolytic pathway. J. Biol. Chem. 276, 12903–12910 [DOI] [PubMed] [Google Scholar]
- 15. Staub O., Gautschi I., Ishikawa T., Breitschopf K., Ciechanover A., Schild L., Rotin D. (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 16, 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ruffieux-Daidié D., Staub O. (2011) Intracellular ubiquitylation of the epithelial Na+ channel controls extracellular proteolytic channel activation via conformational change. J. Biol. Chem. 286, 2416–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sadoul K., Boyault C., Pabion M., Khochbin S. (2008) Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie 90, 306–312 [DOI] [PubMed] [Google Scholar]
- 19. Li M., Luo J., Brooks C. L., Gu W. (2002) Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 277, 50607–50611 [DOI] [PubMed] [Google Scholar]
- 20. Geng H., Liu Q., Xue C., David L. L., Beer T. M., Thomas G. V., Dai M. S., Qian D. Z. (2012) HIF1α protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 287, 35496–35505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jeong J. W., Bae M. K., Ahn M. Y., Kim S. H., Sohn T. K., Bae M. H., Yoo M. A., Song E. J., Lee K. J., Kim K. W. (2002) Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 111, 709–720 [DOI] [PubMed] [Google Scholar]
- 22. McDonald F. J., Price M. P., Snyder P. M., Welsh M. J. (1995) Cloning and expression of the β- and γ-subunits of the human epithelial sodium channel. Am. J. Physiol. 268, C1157–C1163 [DOI] [PubMed] [Google Scholar]
- 23. McDonald F. J., Snyder P. M., McCray P. B., Jr., Welsh M. J. (1994) Cloning, expression, and tissue distribution of a human amiloride-sensitive Na+ channel. Am. J. Physiol. 266, L728–L734 [DOI] [PubMed] [Google Scholar]
- 24. Kao H. Y., Downes M., Ordentlich P., Evans R. M. (2000) Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 14, 55–66 [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou X., Marks P. A., Rifkind R. A., Richon V. M. (2001) Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. U.S.A. 98, 10572–10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Treier M., Staszewski L. M., Bohmann D. (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell 78, 787–798 [DOI] [PubMed] [Google Scholar]
- 27. Bens M., Vallet V., Cluzeaud F., Pascual-Letallec L., Kahn A., Rafestin-Oblin M. E., Rossier B. C., Vandewalle A. (1999) Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J. Am. Soc. Nephrol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 28. Ilatovskaya D. V., Palygin O., Chubinskiy-Nadezhdin V., Negulyaev Y. A., Ma R., Birnbaumer L., Staruschenko A. (2014) Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int. 86, 506–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Canessa C. M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J. D., Rossier B. C. (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467 [DOI] [PubMed] [Google Scholar]
- 30. Hughey R. P., Mueller G. M., Bruns J. B., Kinlough C. L., Poland P. A., Harkleroad K. L., Carattino M. D., Kleyman T. R. (2003) Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J. Biol. Chem. 278, 37073–37082 [DOI] [PubMed] [Google Scholar]
- 31. Knight K. K., Olson D. R., Zhou R., Snyder P. M. (2006) Liddle's syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc. Natl. Acad. Sci. U.S.A. 103, 2805–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Canessa C. M., Horisberger J. D., Rossier B. C. (1993) Epithelial sodium channel related to proteins involved in neurodegeneration. Nature 361, 467–470 [DOI] [PubMed] [Google Scholar]
- 33. Snyder P. M. (2000) Liddle's syndrome mutations disrupt cAMP-mediated translocation of the epithelial Na+ channel to the cell surface. J. Clin. Invest. 105, 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Malik B., Yue Q., Yue G., Chen X. J., Price S. R., Mitch W. E., Eaton D. C. (2005) Role of Nedd4-2 and polyubiquitination in epithelial sodium channel degradation in untransfected renal A6 cells expressing endogenous ENaC subunits. Am. J. Physiol. Renal Physiol. 289, F107–F116 [DOI] [PubMed] [Google Scholar]
- 35. Kamynina E., Debonneville C., Bens M., Vandewalle A., Staub O. (2001) A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J. 15, 204–214 [DOI] [PubMed] [Google Scholar]
- 36. Fakitsas P., Adam G., Daidié D., van Bemmelen M. X., Fouladkou F., Patrignani A., Wagner U., Warth R., Camargo S. M., Staub O., Verrey F. (2007) Early aldosterone-induced gene product regulates the epithelial sodium channel by deubiquitylation. J. Am. Soc. Nephrol. 18, 1084–1092 [DOI] [PubMed] [Google Scholar]
- 37. Zhou R., Kabra R., Olson D. R., Piper R. C., Snyder P. M. (2010) Hrs controls sorting of the epithelial Na+ channel between endosomal degradation and recycling pathways. J. Biol. Chem. 285, 30523–30530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jain L., Chen X. J., Malik B., Al-Khalili O., Eaton D. C. (1999) Antisense oligonucleotides against the α-subunit of ENaC decrease lung epithelial cation-channel activity. Am. J. Physiol. 276, L1046–L1051 [DOI] [PubMed] [Google Scholar]
- 39. Jain L., Chen X. J., Ramosevac S., Brown L. A., Eaton D. C. (2001) Expression of highly selective sodium channels in alveolar type II cells is determined by culture conditions. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L646–L658 [DOI] [PubMed] [Google Scholar]
- 40. Gao C., Liu Y., Lam M., Kao H. Y. (2010) Histone deacetylase 7 (HDAC7) regulates myocyte migration and differentiation. Biochim. Biophys. Acta 1803, 1186–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jin Z., Wei W., Dechow P. C., Wan Y. (2013) HDAC7 inhibits osteoclastogenesis by reversing RANKL-triggered β-catenin switch. Mol. Endocrinol. 27, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kasler H. G., Young B. D., Mottet D., Lim H. W., Collins A. M., Olson E. N., Verdin E. (2011) Histone deacetylase 7 regulates cell survival and TCR signaling in CD4/CD8 double-positive thymocytes. J. Immunol. 186, 4782–4793 [DOI] [PubMed] [Google Scholar]
- 44. Hutt D. M., Herman D., Rodrigues A. P., Noel S., Pilewski J. M., Matteson J., Hoch B., Kellner W., Kelly J. W., Schmidt A., Thomas P. J., Matsumura Y., Skach W. R., Gentzsch M., Riordan J. R., Sorscher E. J., Okiyoneda T., Yates J. R., 3rd, Lukacs G. L., Frizzell R. A., Manning G., Gottesfeld J. M., Balch W. E. (2010) Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 6, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bouchecareilh M., Hutt D. M., Szajner P., Flotte T. R., Balch W. E. (2012) Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)-mediated correction of α1-antitrypsin deficiency. J. Biol. Chem. 287, 38265–38278 [DOI] [PMC free article] [PubMed] [Google Scholar]