

Background: Glycolysis in astrocytes may govern amyloid accumulation and cytotoxicity.
Results: Inhibiting astrocytic PFKFB3 results in an accumulation of amyloid protein and vulnerability to Aβ cytotoxicity. In APP transgenic mice, reactive astrogliosis parallels the increased Aβ burden and PFKFB3 activity in an age-dependent manner.
Conclusion: Astrocytic glycolysis is a key component for amyloid toxicity.
Significance: The bioenergetic mechanism in astrocytes may contribute to AD pathology.
Keywords: Alzheimer disease, amyloid precursor protein (APP), amyloid β (Aβ), astrocyte, glial cell, glycolysis, phosphofructokinase, reactive astrogliosis
Abstract
Alzheimer disease (AD) is characterized neuropathologically by synaptic disruption, neuronal loss, and deposition of amyloid β (Aβ) protein in brain structures that are critical for memory and cognition. There is increasing appreciation, however, that astrocytes, which are the major non-neuronal glial cells, may play an important role in AD pathogenesis. Unlike neurons, astrocytes are resistant to Aβ cytotoxicity, which may, in part, be related to their greater reliance on glycolytic metabolism. Here we show that, in cultures of human fetal astrocytes, pharmacological inhibition or molecular down-regulation of a main enzymatic regulator of glycolysis, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3), results in increased accumulation of Aβ within and around astrocytes and greater vulnerability of these cells to Aβ toxicity. We further investigated age-dependent changes in PFKFB3 and astrocytes in AD transgenic mice (TgCRND8) that overexpress human Aβ. Using a combination of Western blotting and immunohistochemistry, we identified an increase in glial fibrillary acidic protein expression in astrocytes that paralleled the escalation of the Aβ plaque burden in TgCRND8 mice in an age-dependent manner. Furthermore, PFKFB3 expression also demonstrated an increase in these mice, although at a later age (9 months) than GFAP and Aβ. Immunohistochemical staining showed significant reactive astrogliosis surrounding Aβ plaques with increased PFKFB3 activity in 12-month-old TgCRND8 mice, an age when AD pathology and behavioral deficits are fully manifested. These studies shed light on the unique bioenergetic mechanisms within astrocytes that may contribute to the development of AD pathology.
Introduction
Alzheimer disease (AD),2 the most common type of senile dementia, is a progressive neurodegenerative disorder characterized by a gradual deterioration of higher cognitive functions. At a structural level, the brains of AD patients typically exhibit amyloid β (Aβ) plaques, intracellular neurofibrillary tangles, vascular deposits of amyloid, synaptic disruption, and neuronal loss (1–3). A number of hypotheses have been advanced to explain the pathogenesis of this condition, including disruption of the cell cycle, deposition of misfolded proteins, preferential loss of brain cholinergic neurons, inflammation, oxidative stress, and vascular abnormalities (1). For the last two decades, the AD field has been dominated by the “amyloid hypothesis,” which posits that the accumulation and aggregation of amyloid result in alterations in synaptic function, activation of microglia, release of inflammatory mediators, and oxidative stress (4, 5). These mechanisms culminate in the neuronal dysfunction and degeneration responsible for cognitive deficits in AD. Recently an alternate view has arisen on the basis of a bioenergetics model, proposing that sporadic AD, comprising the majority of cases, is a metabolic disease and that the traditional neuron-centric view of this condition fails to take into account important contributions of non-neuronal cells, particularly astrocytes, to its pathogenesis (6–8).
The presence of reactive astrocytes around Aβ plaques suggests that this phenotype of cells may play an important role in AD pathogenesis (9, 10). Astrocytes can bind and internalize Aβ in vitro and in vivo and play an important role in plaque formation and maintenance through mechanisms that are not entirely clear (11–13). In contrast to neurons, which are highly vulnerable to Aβ exposure, astrocytes demonstrate relative resistance to Aβ toxicity (14, 15). A comparison of rat neurons and astrocytes reveals a differential bioenergetic response between the two cell types to experimental stimuli (such as nitric oxide or glutamate) that induce cellular stress (16, 17). Upon exposure to these stimuli, neurons undergo a rapid decline in ATP concentration, a collapse in mitochondrial potential (ΔΨm), and spontaneous apoptotic death. By contrast, astrocytes respond to cellular stressors by increasing their glycolytic metabolism to generate ATP and maintain ΔΨm. Further studies ascribe these different responses to the low levels in neurons of isoform 3 of 6-phosphofructo-2-kinase/fructose-2,6-biphosphosphatase (PFKFB3), a potent allosteric activator of 6-phosphofructo-1-kinase, which is the rate-limiting enzyme for glycolysis (16). Astrocytes, however, show an abundance of PFKFB3, and use of siRNA targeted against PFKFB3 abolishes the ability of these cells to up-regulate glycolysis in response to stimuli that induce cellular stress (18). On the basis of these observations, we hypothesized that, in astrocytes, impairment of bioenergetic mechanisms involving enzyme PFKFB3 renders these cells vulnerable to Aβ toxicity and promotes Aβ accumulation and plaque formation, a key feature of AD pathogenesis. In this study, we first identified basal levels of the glycolytic enzyme PFKFB3 in primary cell cultures of human fetal astrocytes (HFAs) and neurons. We next examined the role of PFKFB3 in the promotion of Aβ accumulation and plaque formation and the cellular viability of HFAs using pharmacological and siRNA approaches. Finally, we sought to examine the levels of PFKFB3 in TgCRND8 transgenic mice (expressing a mutant human amyloid precursor protein (APP)) versus age-matched wild-type mice and to correlate such alterations with age-dependent increases in Aβ plaques observed in TgCRND8 mice.
EXPERIMENT PROCEDURES
Cultures of fetal human cortical neurons (HFNs) and HFAs
Human cortical fetal neurons and astrocytes were isolated as described previously (19, 20). Briefly, neuronal cultures were prepared from gestational week 12–15 fetuses with approval of the Human Ethics Research Board at the University of Alberta. After we removed the meninges and blood vessels, the brain tissue was washed in minimum essential medium and mechanically dissociated by repeated trituration through a 20-gauge needle. Cells were centrifuged at 1500 × g for 10 min and resuspended in minimum essential medium with 10% heat-inactivated FBS, 0.2% N2 supplement, and 1% penicillin/streptomycin antibiotic solution (Pen-Strep, Gibco). Subsequently, cultures were treated with arabinofuranosylcytosine (25 μmol/liter) for 2 weeks. HFN cultures were grown in a 5% CO2 humidified incubator at 37 °C. To generate enriched primary astrocytes, the cell culture was incubated without arabinofuranosylcytosine. For immunohistochemical experiment, cells were plated for 2–7 days on glass coverslips precoated with poly-l-ornithine (Sigma) or 96-well plates. The age range for the most cells used in this study was 14–42 days in vitro (no more than 60 days in vitro for HFNs and no more than 90 days in vitro for HFAs). Soluble oligomeric Aβ1–42 and the reverse nonfunctional sequence peptide, Αβ42–1, were prepared according to protocols published previously (19), and the oligomerization state of the peptides was, therefore, not measured in this study.
Aβ Plaque and PFKFB3 Expression in Brain Regions of TgCRND8 APP-overexpressing Mice
Transgenic animals were obtained from the University of Alberta Centre for Prions and Protein Folding Diseases Animal Facility. Under halothane anesthesia, TgCRND8 APP mice (2, 4, 7, 9, and 12 months of age; n = 4 for each age group) and age-matched control mice (n = 4 for each age group) were decapitated, and the brains were removed quickly. The brains were hemisected, and one half of the brain was used for dissecting the cortex and hippocampus. Tissue was weighed and placed in cold radioimmune precipitation assay buffer (Thermo Scientific Inc., Rockford, IL) with protease inhibitors, and then proteins were isolated and measured using a Bio-Rad protein assay kit. Proteins were diluted with SDS sample buffer (New England Biolabs, Whitby, ON) and loaded at 20–30 μg/lane on 10–14% acrylamide gels, depending on the predicted protein size. Proteins were transferred to a nitrocellulose membrane and blocked with LI-COR blocking buffer. Nitrocellulose membranes were incubated with primary antibodies overnight at 4 °C on a shaker and subsequently incubated with IRDye 800CW goat anti-rabbit and IRDye 680CW goat anti-mouse secondary antibodies. The other halves of the brains from TgCRND8 and control mice were fixed for immunohistochemical analysis of brain sections from the cortex and hippocampus to determine Aβ plaque deposition, and the same sections were also double-stained using phospho-PFKFB3 antibody. For determination of Aβ plaque size and distribution, the Aβ (6E10)-immunostained sections were imaged using a Leica SP5 confocal microscope with a ×20/0.5 lens.
Fluorescent Immunohistochemistry
HFAs/HFNs on coverglass were fixed with 4% paraformaldehyde and stained with primary and secondary antibodies. The primary antibodies used were as follows: GFAP and rabbit anti-GFAP polyclonal antibody (Santa Cruz Biotechnology, Inc.); Aβ (6E10) mouse monoclonal antibody (Covance Inc., Montreal, Canada); phospho-PFKFB3 (Ser-467) rabbit monoclonal (Bioss Inc., Woburn, MA); and MAP2 mouse monoclonal antibody (Sigma). The secondary antibodies used were Alexa Fluor 350 goat anti-rabbit, Alexa Fluor 488 chicken anti-rabbit, and Alexa Fluor 546 goat anti-mouse antibody (Invitrogen). Photomicrographs of stained cells were taken using an Axioplan-2 fluorescence microscope with AxioVision software (Carl Zeiss Ltd., Toronto, ON, Canada) or a FV1000 confocal microscopy with Olympus FluoView software (F10-ASW). Astrocyte morphology and Aβ plaque size were further analyzed with ImageJ software, version 1.49 (http://imagej.nih.gov/ij/) (21). Three different batches of HFA/HFN cultures were used, and three to five different fields of view for each batch of cells were analyzed.
In-cell Western Blot and Western Blot Analyses
Intracellular signaling profiles were determined using LI-COR in-cell Western blot techniques. HFAs were seeded at 5000 cells/well in a 96-well plate. After culturing for 24 h, cells were pretreated for 24 with/without 3PO/PFK15 at the indicated concentrations, followed by treatment with Aβ1–42 in culture medium for another 24 h. Subsequently, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100, blocked with Odyssey blocking buffer (LI-COR), and stained with mouse Aβ (6E10) antibody. The secondary antibody was IRDye 800CW goat anti-mouse antibody, and Sapphire700 and DRAQ5 were used for cell number normalization (LI-COR). Plates were imaged using an Odyssey infrared imaging system (LI-COR), and the integrated intensity was normalized to the total cell number on the same well. For Western blot analysis, protein was loaded at 20–30 μg/lane. The primary antibodies used were PFKFB3 (rabbit polyclonal (Abcam, Toronto, ON), mouse monoclonal (Novus Canada, Oakville, ON), phospho-PFKFB3 (rabbit polyclonal, Bioss), GFAP (rabbit polyclonal, Invitrogen), β-actin (mouse monoclonal, Sigma), and β-tubulin (rabbit polyclonal, Cell Signaling Technology, Inc.). The secondary antibodies used were IRDye 680CW goat anti-mouse antibody and IRDye 800CW goat anti-rabbit antibody (LI-COR). Membranes were scanned and analyzed using the LI-COR Odyssey system.
Cell Death and Proliferation Assay: MTT Cell Death Assay
HFAs were seeded to 5000 cells/well in a 96-well plate in minimum essential medium, 10% FBS and incubated overnight. Cells in culture medium were preincubated for 24 h either with or without 3PO/PFK15, followed by treatment with Aβ1–42 or Aβ42–1 for 24 h. At the end of treatment, 20 μl of 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) was added to each well and incubated at 37 °C for 3 h. Medium was removed, 100 μl of MTT solvent (isopropanol with 4 mm HCl) was added to each well, and the plates were incubated for 30 min at room temperature on a rotating shaker. Plates were analyzed on a microplate reader at a 590-nm wavelength. The live/dead assay kit from Invitrogen was used according to the instructions of the manufacturer. HFAs were plated in 24-well plates and treated with 3PO and Aβ1–42 as indicated. Cells were analyzed with the GE Healthcare InCell analyzer, and 16 fields/well were imaged and analyzed further with the Investigate software package. The WST-1 proliferation assay kit from Roche was used according to the instructions of the manufacturer.
siRNA Transfection
Human PFKFB3 siRNAs used were Trilencer-27 for PFKFB3 (three unique 27-mer siRNA duplexes, OriGene Technologies, Inc., Rockville, MD) and negative control siRNA transfected into HFAs using Lipofectamine RNAiMAX or Lipofectamine 2000 reagent (Invitrogen).
Other Chemicals and Reagents
Cell culture reagents were obtained from Invitrogen unless stated otherwise. 3PO was from Merck Millipore (Billerica, MA). PFK15 was from Glixx Lab (Southborough, MA). DAPI was from Invitrogen. Fluorescent Aβ1–42 HiLyte Fluor 555 was from AnaSpec Inc. (Fremont, CA). Aβ1–42 and Aβ42–1 were from rPeptide (Bogart, GA).
Statistical Analysis
Values are means ± S.D. Significance was determined used unpaired Student's t test or one-way analysis of variance, followed by Tukey's test when appropriate, with Prism software (GraphPad Prism 5, GraphPad Software, San Diego, CA. p < 0.05 was considered significant.
RESULTS
Astrocytic Involvement in Aβ Clearance and Amyloid Plaque Formation
Application of fluorescent labeled Aβ1–42 to HFAs resulted in a concentration-dependent accumulation of the peptide over the surface of astrocytes to form amyloid plaques (Fig. 1, a and b). Clearance of the Aβ from the extracellular space and its internalization within astrocytic processes was also evident (Fig. 1a). Sustained exposure of HFAs to Aβ results in conversion to reactive astrocytes that were characterized by cellular hypertrophy and proliferation and increased synthesis of GFAP (Fig. 1, a and c). The presence of Aβ1–42 in culture dishes without HFAs did not result in amyloid plaque formation, nor did we observe any significant internal Aβ within HFAs that were not incubated with exogenous Aβ1–42 (data not shown).
FIGURE 1.

Uptake, accumulation, and amyloid plaque formation in HFAs exposed to Aβ. a, immunofluorescent staining of human fetal astrocytes cultured with fluorescent Aβ1–42 (1 μm, 24 h) and stained with Aβ (6E10) and GFAP antibody showing that Aβ1–42 is taken up into astrocytes and forms plaques that build up on the reactive astrocyte surface. b, in-cell-Western blot stained with Aβ (6E10) showing increased Aβ plaque formation followed by increasing extracellular Aβ1–42 concentration (Conc.). The Western blot shows that human fetal astrocytes exposed to Aβ1–42 become reactive, as characterized by increased GFAP protein expression (c, 1 μm, 24 h, n = 3).
Aβ Plaque Formation Depends on Astrocyte Glycolytic Enzyme PFKFB3 Function
Astrocytes express much higher levels of PFKFB3 than neurons and demonstrate low levels of APC/C-Cdh1 activity. APC/C-Cdh1 is an E3 ubiquitin ligase that degrades PFKFB3 following ubiquitination. In contrast, neurons express low levels of PFKFB3 but higher levels of APC/C-Cdh1 activity. These distinctions result in astrocytes functioning at a high glycolysis rate and neurons being extremely sensitive to energy depletion and degeneration (22). In our cell culture model, HFAs indeed demonstrated a significantly higher expression of PFKFB3 than HFNs (Fig. 2a). In addition to increased levels of GFAP, as shown in Fig. 1c, HFAs exposed to Aβ1–42 also displayed increased expression of the key glycolytic enzyme PFKFB3, which is indicative of heightened glycolytic activity. Another amyloidogenic peptide, human amylin, also increased astrocytic PFKFB3 expression (Fig. 2b). Interestingly, after inhibition of glycolysis with PFK15, a potent PFKFB3 inhibitor (23), HFAs also demonstrated increased expression of GFAP, consistent with reactive astrocytes (Fig. 2, c and d). Because Aβ plaque formation on astrocytes depends on extracellular Aβ concentration in vitro (Fig. 1b), we sought to examine whether inhibition of glycolysis influenced Aβ deposition. Following inhibition of glycolysis with either 3PO or PFK15, both PFKFB3 inhibitors, the intensity of GFAP staining in HFAs also increased significantly with formation of larger Aβ plaques (Fig. 2, e–g). The Aβ density curves were also shifted to the left (Fig. 3a). Furthermore, Aβ plaque formation increased in a concentration-dependent manner following applications of progressively higher concentrations of the glycolytic inhibitors 3PO or PFK15 while maintaining the same concentration of Aβ1–42 in the extracellular milieu (Fig 3, b–d). Astrocytes in HFA cultures that became reactive also showed morphological cellular changes and increased GFAP staining after exposure to either Aβ or an impairment of glucose metabolism with application of glycolytic inhibitors (Fig 3d).
FIGURE 2.

Glycolytic activity affects Aβ uptake and plaque formation in human astrocytes. a, Western blot analysis showing significantly different basal levels of the glycolytic enzyme PFKFB3 in HFNs and HFAs (n = 6). b, HFAs increase PFKFB3 protein expression after exposure to Aβ1–42 (1 μm) or human amylin (hAmylin, another amyloidogenic peptide, 1 μm) for 24 h. c, HFAs become “reactive”; that is, characterized by increased GFAP protein expression after metabolic stress, such as inhibition of glycolysis with the PFKFB3 inhibitor PFK15 (20 μm, 24 h, n = 3). d, representative photographs showing that HFA cultures become reactive and demonstrate morphological changes and an increase in GFAP staining after exposure to Aβ or an impairment in glucose metabolism with application of the glycolytic inhibitors 3PO (20 μm) or PFK15 (10 μm), which further increased Aβ buildup and plaque formation (e, f, and g). The results shown are from five independent experiments. h, the glycolytic metabolic stress not only increases Aβ buildup but also results in the formation of larger Aβ plaques. *, p < 0.05; **, p < 0.01. Scale bars = 20 μm.
FIGURE 3.

Aβ plaque formation in human fetal astrocytes is correlated with PFKFB3 activity. a, in-cell Western blot stained with Aβ (6E10) showing that deteriorated astrocyte metabolism enhances Aβ plaque formation, followed by glycolysis inhibition with the PFKFB3 inhibitors 3PO (20 μm) or PFK15 (10 μm). Conc, concentration. b and c, Aβ plaque formation increases with increasing concentrations of glycolysis inhibitor 3PO (b) or PFK15 (c) (Aβ1–42 concentration, 1 μm). d, using confocal microscopy quantification, Aβ fluorescence increased significantly after glycolysis inhibition with 3PO (20 μm, n = 9). *, p < 0.05.
Glycolytic Inhibition Renders Astrocytes Vulnerable to Aβ Toxicity
In comparison with neurons, astrocytes under normal glucose metabolic conditions are relatively resistant to Aβ cytotoxicity (14). However, an inhibition of glycolytic metabolism in astrocytes renders them vulnerable to cell death upon exposure to Aβ. In HFAs pretreated with PFKFB3 inhibitors (3PO or PFK15), an increase in cell death was observed using three distinct assays of cell viability (Fig. 4, a–d). As shown in Fig. 4, a–d, the effects of applying either 3PO or PFK15 were dose-dependent. We further confirmed that these changes in astrocyte survival were PFKFB3-specific by using siRNA to specifically down-regulate PFKFB3 protein expression (Fig. 4, e and f). In HFA cultures where PFKFB3 activity was down-regulated in this manner, an increase in GFAP (indicative of their change to reactive astrocytes) and cell death was observed upon exposure to Aβ at concentrations that normally did not influence cell survival (Fig. 4e).
FIGURE 4.

PFKFB3 activity affects Aβ cytotoxicity in human fetal astrocytes. a, in-cell analysis imaging the live/dead assay with calcein (green, live cells) and ethidium homodimer I (red, dead cells) with human fetal astrocytes cultured with 3PO (20 μm) and Aβ1–42 (1 μm) for 48 h. b, using the live/dead assay and quantifying with InCell analyzer, resting human astrocytes are not sensitive to Aβ cytotoxicity. However, astrocytes had increased sensitivity to Aβ cytotoxicity after PFKFB3 activity was inhibited with 3PO. c, using an MTT proliferation assay, increased Aβ induced astrocyte cell death after metabolic stress when PFKFB3 activity was inhibited with PFK15 or 3PO. d, another proliferation assay (WST-1) showed similar increased Aβ toxicity following glycolysis inhibition with 3PO (20 μm, 24 h) but not the reverse sequence peptide Aβ42–1. e and f, the increased Aβ cytotoxicity after PFKFB3 inhibition is PFKFB3-specific because it is down-regulated with PFKFB3 siRNA transfection in these cells. *, p < 0.05.
In co-cultures of HFAs and HFNs, where astrocytes provide a homeostatic environment to keep neurons healthy (20), an inhibition of PFKFB3 function (application of 3PO or PFK15), HFAs demonstrated reactive astrogliosis (Fig. 5, a–c). These reactive HFAs developed increased vulnerability when exposed to Aβ (they became more reactive with stronger GFAP staining, Fig. 5, a–c) and a decreased capability to maintain glial-neuronal homeostasis that resulted in neuronal death (Fig. 5, a and d).
FIGURE 5.

PFKFB3 activity is important for Aβ cytotoxicity in a mixed culture of human fetal neurons and astrocytes. a, in cocultured HFNs and HFAs, HFAs maintain a homeostatic environment and keep HFNs healthy. b and c, after b and c) and have decreased capability to maintain the microenvironment, resulting in a gradual loss of HFNs. d, when these stressed HFAs are exposed to Aβ, they become more sensitive to Aβ cytotoxicity, resulting in a further loss of their capability to support neurons and, hence, increased neuronal cell death (3PO, 20 μm; PFK15, 10 μm; Aβ1–42, 1 μm; 24 h). *, p < 0.05; **, p < 0.01. Scale bar = 20 μm).
PFKFB3 Activity Is Involved in Aβ Plaque Formation in the Transgenic AD Mouse Model
TgCRND8 mice carry combined APP Swedish (K670M/N671L) and Indiana (V717F) mutations, resulting in an aggressive neuropathology evident by 3 months of age, and, by 6 months of age, animals demonstrate large numbers of diffuse and plaque amyloid deposits (24). Therefore, in TgCRND8 mice, we examined age-dependent increases in reactive astrocytes (as measured by GFAP expression) and the glycolytic profile (as measured by PFKFB3 expression). In the same animals, we also assessed the amyloid burden with immunohistochemistry and Western blot analysis. In TgCRND8 mice, an increase in Aβ load was seen as early as 2 month of age (Fig. 6a), followed by increased GFAP protein expression from about 4 months of age, indicative of reactive astrogliosis. An increase in PFKFB3 protein expression followed at a later age, after 9 months. Immunohistochemical staining showed significant reactive astrogliosis surrounding Aβ plaque with increased PFKFB3 activity in these aged (12-month-old) AD mice (Fig. 6, b and c).
FIGURE 6.

Temporal changes in Aβ, GFAP, and PFKFB3 expression in AD transgenic mice (TgCRND8). a, Western blot analyses for GFAP, PFKFB3, and Aβ protein expression from the cortex of transgenic AD mouse brain (TgCRND8) compared with the age-matched wild type (n = 4 for each group). Quantitative changes in GFAP, PFKFB3, and Aβ protein expression in wild-type and TgCRND8 mice over time (in months) are depicted in the left panels. b, immunofluorescence staining of 12-month-old AD mouse brain with Aβ (6E10) and GFAP antibodies showing Aβ plaque surrounded closely by enlarged and stronger GFAP-stained astrocytes (right panels), whereas the age-matched wild-type mouse brain shows no significant plaque formation and reactive astrogliosis (left panels). c, in AD mouse brain (right panels), increased phospho-PFKFB3 (green) surrounds and is colocalized with Aβ plaques (red), shown as yellow, whereas the age-matched wild-type mouse brain shows weak PFKFB3 activity (n = 4 for each group). *, p < 0.05; **, p < 0.01.
To mimic the above in vivo sequence of events in our HFA culture model system, we first incubated HFAs with soluble oligomeric Aβ1–42 for 12 h, which resulted in early amyloid plaque formation. Subsequent applications of the glycolytic inhibitor PFK15 resulted in a concentration-dependent acceleration and increase in amyloid plaque formation (Fig. 7).
FIGURE 7.

Delayed glycolysis inhibition further enhances plaque formation in HFAs. a, HFAs cultured with 200 nm Fluor-Aβ1–42 for 12 h forms Aβ plaques. b and c, with continued application of Fluor-Aβ1–42, delayed inhibition of glycolysis with the PFKFB3 inhibitor PFK15 (1 or 5 μm) for another 24 h results in accelerated and increased amyloid plaque formation. The increase in the plaque-covered area (d) and plaque size and distribution (e and f) appear to be correlated with the concentration of PFK15 that is applied. Results from three separate experiments with different batches of cultured HFAs were analyzed. *, p < 0.05 compared with the Aβ control group. Scale bar = 100 μm.
DISCUSSION
Bioenergetic mechanisms and neuronal-astrocytic interplay are increasingly viewed as important elements in the pathogenesis of aging and AD (7, 10). Brain glucose uptake and metabolism both decline with the aging process and are accompanied by a decrease in glycolysis (25, 26). This hypometabolism of glucose as an energy substrate is also viewed as an early event in the genesis of AD (27) and may, in fact, represent a general phenomenon for many neurodegenerative diseases (28, 29). For decades, AD has been considered to arise mainly because of specific dysfunction and pathology affecting neurons. This neuron-centric view has been challenged recently by a “neuroenergetic hypothesis” (30), which serves to incorporate changes in the metabolic machinery of astrocytes as an important step in the neurodegenerative processes that unfold during AD (31–33). In this study, we demonstrate, for the first time, that, in HFAs, impairment of glycolysis promotes an increase in amyloid aggregation and internalization within these glial cells. Furthermore, inhibition of glycolysis renders the normally resistant human astrocytes vulnerable to Aβ toxicity. Finally, in a transgenic mouse model of AD, we identify that increased reactive astrogliosis (as measured by GFAP expression) and amyloid deposition precede the increase in the key glycolytic enzyme PFKFB3.
Astrocytes undergo functional decline with age (senescence) that can be recapitulated, in part, when such cells are exposed in vitro to stressful stimuli such as oxidative stress or exposure to Aβ peptides (34). When exposed to Aβ, in addition to internalization and accumulation of this peptide, HFAs demonstrate a change in metabolic phenotype characterized by up-regulation of PFKFB3, which is the rate-limiting enzyme for glycolysis via the 6-phosphofructo-1-kinase pathway. This initial increase in glycolysis within HFAs may be viewed as a compensatory homeostatic mechanism to produce increased energy substrate in the form of lactate, which provides an additional source of energy for neurons under conditions when oxidative phosphorylation activity is impaired, i.e. in the presence of Aβ or other stressors. Impairment of the glycolytic enzyme PFKFB3, either pharmacologically (with 3PO or PFK15) or by silencing PFKFB3 at the mRNA level with siRNA transfection of HFAs, resulted in significant cell death of astrocytes when exposed to Aβ, as measured using three independent cell survival assays.
Reactive astrogliosis is a well characterized spectrum of morphological and biochemical changes that occur in astrocytes in response to a variety of insults (35) and has also been identified in the context of stress-induced astrocyte senescence (36). In the brains of AD patients, an increase in glycolytic enzymes is correlated with elevated GFAP expression in astrocytes, suggesting that up-regulation of glycolysis may, in part, be a result of reactive astrocytosis that develops with advancing AD (37). In a recent study, several GFAP isoforms have been shown to be up-regulated in a concerted manner in the human AD brain (38). Our results also show an overall increase in total GFAP levels after exposure to Aβ1–42 (Fig. 1c) following compromise of glycolysis in cultured HFAs (Fig. 2c) and, additionally, in an age-dependent manner in the AD mouse brain (Fig. 6a). We suspect that the multiple bands we observed on the Western blots correspond to different GFAP isoforms described in a recent report (38), although we did not characterize this further. Interestingly, such changes in glycolysis also correlate spatially with Aβ deposition in PET imaging studies on the human brain (39). Our in vivo data from transgenic AD mice (TgCRND8) that overexpress amyloid and develop cognitive impairment in an age-dependent manner suggest that PFKFB3, a key regulatory enzyme for the initiation of glycolysis, may play an important role in AD pathogenesis. We observed an increase in amyloid burden in TgCRND8 mice that preceded similar increases in GFAP expression in AD mice over age-matched wild-type controls. At ∼7 months, we noticed an increase in PFKFB3 levels in the brains of TgCRND8 mice compared with control mice. Immunohistochemical staining confirmed a close anatomical relationship between reactive astrocytes and PFKFB3 expression within the same cells that surround and are intermingled with amyloid plaques (Fig. 6, b and c). The sequence of temporal changes in amyloid levels and GFAP and PFKFB3 activity observed in transgenic AD mice fit with our in vitro data in HFA cultures and suggest that sustained amyloid exposure of astrocytes leads to increased reactive astrogliosis followed by a slower induction of glycolysis, as measured by PFKFB3 levels. Our histological analysis in TgCRND8 AD mice does not allow us to exclude contributions of neuronal PFKFB3 in disease progression. However, basal levels of PFKFB3 in the neurons under normal conditions are quite low compared with astrocytes and neuronal glycolysis, therefore accounting for only a small portion of the energy demand (16, 17). On the basis of our in vitro and in vivo data, the impact of Aβ on astrocytic bioenergetic mechanisms and the consequences of this interplay on neuronal survival are summarized in Fig. 8.
FIGURE 8.
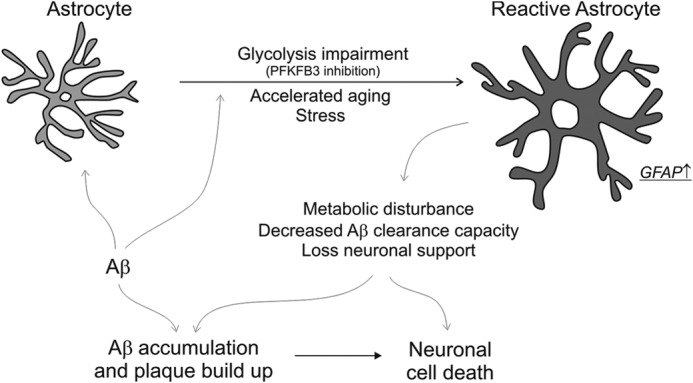
Astrocytic bioenergetic mechanisms contribute to amyloid accumulation and neuronal death. The schematic depicts the transformation of astrocytes to a reactive state when exposed to Aβ protein, a process that is augmented by loss of glycolytic function (PFKFB3 inhibition), accelerated aging, or cellular stress. The homeostatic disturbance that ensues in reactive astrocytes (identified by an increase in GFAP) results in a loss of neuronal metabolic support and an inability to clear Aβ protein. These changes lead to accumulation of Aβ and adversely impact neuronal function, resulting in neurodegeneration.
Neoplastic transformation in cells causes a marked increase in glycolytic activity even under normoxic conditions, a phenomenon originally termed the Warburg effect (40). Recently, small molecule inhibitors of PFKFB3, such as 3PO and PFK15, have been developed to promote cytostasis and inhibit tumorigenic growth in vivo (23, 41). On the basis of our studies, an undesired consequence of this strategy for cancer treatment may consist of impairment of astroglial glycolytic metabolism and, as a result, a downstream decrease in neuronal viability that depends intimately on healthy, synergistic neuron-astrocyte interactions.
Acknowledgments
We thank Jing Yang for assistance with transgenic TgCRND8 mice and Dave MacTavish and Beipei Shi for technical assistance.
This research was supported by grants from Alberta Bio-Solutions (Alberta Prion Research Institute), the Alzheimer's Society of Alberta and Nunavut, and the University Hospital Foundation (FoMD/UHF medical grants competition).
- AD
- Alzheimer disease
- Aβ
- amyloid β
- PFKFB3
- 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase
- HFA
- fetal human cortical astrocyte
- HFN
- fetal human cortical neuron
- GFAP
- glial fibrillary acidic protein
- 3PO
- 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one
- PFK15
- 1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Querfurth H. W., LaFerla F. M. (2010) Alzheimer's disease. N. Engl. J. Med. 362, 329–344 [DOI] [PubMed] [Google Scholar]
- 2. Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T. (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skovronsky D. M., Lee V. M., Trojanowski J. Q. (2006) Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu. Rev. Pathol. 1, 151–170 [DOI] [PubMed] [Google Scholar]
- 4. Selkoe D. J. (2008) Biochemistry and molecular biology of amyloid β-protein and the mechanism of Alzheimer's disease. Handb. Clin. Neurol. 89, 245–260 [DOI] [PubMed] [Google Scholar]
- 5. Hardy J. (2009) The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J. Neurochem. 110, 1129–1134 [DOI] [PubMed] [Google Scholar]
- 6. Swerdlow R. H. (2007) Is aging part of Alzheimer's disease, or is Alzheimer's disease part of aging? Neurobiol. Aging 28, 1465–1480 [DOI] [PubMed] [Google Scholar]
- 7. Demetrius L. A., Driver J. (2013) Alzheimer's as a metabolic disease. Biogerontology 14, 641–649 [DOI] [PubMed] [Google Scholar]
- 8. Demetrius L. A., Simon D. K. (2012) An inverse Warburg effect and the origin of Alzheimer's disease. Biogerontology 13, 583–594 [DOI] [PubMed] [Google Scholar]
- 9. Maragakis N. J., Rothstein J. D. (2006) Mechanisms of disease: astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2, 679–689 [DOI] [PubMed] [Google Scholar]
- 10. Sofroniew M. V., Vinters H. V. (2010) Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagele R. G., D'Andrea M. R., Lee H., Venkataraman V., Wang H. Y. (2003) Astrocytes accumulate A β 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971, 197–209 [DOI] [PubMed] [Google Scholar]
- 12. Nielsen H. M., Veerhuis R., Holmqvist B., Janciauskiene S. (2009) Binding and uptake of A β1–42 by primary human astrocytes in vitro. Glia 57, 978–988 [DOI] [PubMed] [Google Scholar]
- 13. Friedrich R. P., Tepper K., Rönicke R., Soom M., Westermann M., Reymann K., Kaether C., Fändrich M. (2010) Mechanism of amyloid plaque formation suggests an intracellular basis of Aβ pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 1942–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pike C. J., Burdick D., Walencewicz A. J., Glabe C. G., Cotman C. W. (1993) Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 13, 1676–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garwood C. J., Pooler A. M., Atherton J., Hanger D. P., Noble W. (2011) Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2, e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bolaños J. P., Almeida A., Moncada S. (2010) Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci. 35, 145–149 [DOI] [PubMed] [Google Scholar]
- 17. Allaman I., Bélanger M., Magistretti P. J. (2011) Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 34, 76–87 [DOI] [PubMed] [Google Scholar]
- 18. Herrero-Mendez A., Almeida A., Fernández E., Maestre C., Moncada S., Bolaños J. P. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747–752 [DOI] [PubMed] [Google Scholar]
- 19. Jhamandas J. H., Li Z., Westaway D., Yang J., Jassar S., MacTavish D. (2011) Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. Am. J. Pathol. 178, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fu W., Ruangkittisakul A., MacTavish D., Baker G. B., Ballanyi K., Jhamandas J. H. (2013) Activity and metabolism-related Ca2+ and mitochondrial dynamics in co-cultured human fetal cortical neurons and astrocytes. Neuroscience 250, 520–535 [DOI] [PubMed] [Google Scholar]
- 21. Grathwohl S. A., Kälin R. E., Bolmont T., Prokop S., Winkelmann G., Kaeser S. A., Odenthal J., Radde R., Eldh T., Gandy S., Aguzzi A., Staufenbiel M., Mathews P. M., Wolburg H., Heppner F. L., Jucker M. (2009) Formation and maintenance of Alzheimer's disease β-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Almeida A., Moncada S., Bolaños J. P. (2004) Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat. Cell Biol. 6, 45–51 [DOI] [PubMed] [Google Scholar]
- 23. Clem B. F., O'Neal J., Tapolsky G., Clem A. L., Imbert-Fernandez Y., Kerr D. A., 2nd, Klarer A. C., Redman R., Miller D. M., Trent J. O., Telang S., Chesney J. (2013) Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 12, 1461–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chishti M. A., Yang D. S., Janus C., Phinney A. L., Horne P., Pearson J., Strome R., Zuker N., Loukides J., French J., Turner S., Lozza G., Grilli M., Kunicki S., Morissette C., Paquette J., Gervais F., Bergeron C., Fraser P. E., Carlson G. A., George-Hyslop P. S., Westaway D. (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570 [DOI] [PubMed] [Google Scholar]
- 25. Ding F., Yao J., Rettberg J. R., Chen S., Brinton R. D. (2013) Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer's mouse brain: implication for bioenergetic intervention. PLoS ONE 8, e79977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yao J., Rettberg J. R., Klosinski L. P., Cadenas E., Brinton R. D. (2011) Shift in brain metabolism in late onset Alzheimer's disease: implications for biomarkers and therapeutic interventions. Mol. Aspects Med. 32, 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomi M., Zhao Y., Thamotharan S., Shin B. C., Devaskar S. U. (2013) Early life nutrient restriction impairs blood-brain metabolic profile and neurobehavior predisposing to Alzheimer's disease with aging. Brain Res. 1495, 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ferreira I. L., Resende R., Ferreiro E., Rego A. C., Pereira C. F. (2010) Multiple defects in energy metabolism in Alzheimer's disease. Curr. Drug Targets 11, 1193–1206 [DOI] [PubMed] [Google Scholar]
- 29. Liang W. S., Reiman E. M., Valla J., Dunckley T., Beach T. G., Grover A., Niedzielko T. L., Schneider L. E., Mastroeni D., Caselli R., Kukull W., Morris J. C., Hulette C. M., Schmechel D., Rogers J., Stephan D. A. (2008) Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 4441–4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Demetrius L. A., Simon D. K. (2013) The inverse association of cancer and Alzheimer's: a bioenergetic mechanism. J. R. Soc. Interface 10, 20130006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heneka M. T., Rodríguez J. J., Verkhratsky A. (2010) Neuroglia in neurodegeneration. Brain Res. Rev. 63, 189–211 [DOI] [PubMed] [Google Scholar]
- 32. Rossi D., Volterra A. (2009) Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res. Bull. 80, 224–232 [DOI] [PubMed] [Google Scholar]
- 33. Verkhratsky A., Sofroniew M. V., Messing A., deLanerolle N. C., Rempe D., Rodríguez J. J., Nedergaard M. (2012) Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro. 4, e00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lynch A. M., Murphy K. J., Deighan B. F., O'Reilly J. A., Gun'ko Y. K., Cowley T. R., Gonzalez-Reyes R. E., Lynch M. A. (2010) The impact of glial activation in the aging brain. Aging Dis. 1, 262–278 [PMC free article] [PubMed] [Google Scholar]
- 35. Hamby M. E., Sofroniew M. V. (2010) Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7, 494–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhat R., Crowe E. P., Bitto A., Moh M., Katsetos C. D., Garcia F. U., Johnson F. B., Trojanowski J. Q., Sell C., Torres C. (2012) Astrocyte senescence as a component of Alzheimer's disease. PLoS ONE 7, e45069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bigl M., Brückner M. K., Arendt T., Bigl V., Eschrich K. (1999) Activities of key glycolytic enzymes in the brains of patients with Alzheimer's disease. J. Neural. Transm. 106, 499–511 [DOI] [PubMed] [Google Scholar]
- 38. Kamphuis W., Middeldorp J., Kooijman L., Sluijs J. A., Kooi E. J., Moeton M., Freriks M., Mizee M. R., Hol E. M. (2014) Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer's disease. Neurobiol. Aging 35, 492–510 [DOI] [PubMed] [Google Scholar]
- 39. Vlassenko A. G., Vaishnavi S. N., Couture L., Sacco D., Shannon B. J., Mach R. H., Morris J. C., Raichle M. E., Mintun M. A. (2010) Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc. Natl. Acad. Sci. U.S.A. 107, 17763–17767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koppenol W. H., Bounds P. L., Dang C. V. (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337 [DOI] [PubMed] [Google Scholar]
- 41. Clem B., Telang S., Clem A., Yalcin A., Meier J., Simmons A., Rasku M. A., Arumugam S., Dean W. L., Eaton J., Lane A., Trent J. O., Chesney J. (2008) Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 7, 110–120 [DOI] [PubMed] [Google Scholar]