

Background: Barley limit dextrinase (LD), the sole starch debranching enzyme active during seed germination, is regulated by an endogenous inhibitor (LDI).
Results: The crystal structure of the LD-LDI complex reveals a new and unexpected binding mode among cereal type inhibitors.
Conclusion: A hydrophobic cluster drives the picomolar affinity of LDI.
Significance: The molecular understanding of regulation of starch mobilization during germination is augmented.
Keywords: Crystal Structure, Enzyme Inhibitor, Plant Molecular Biology, Protein Complex, Protein-Protein Interaction, Barley Limit Dextrinase, Cereal Type Inhibitors, Debranching Enzyme, Seed Germination, Starch Metabolism Regulation
Abstract
Molecular details underlying regulation of starch mobilization in cereal seed endosperm remain unknown despite the paramount role of this process in plant growth. The structure of the complex between the starch debranching enzyme barley limit dextrinase (LD), hydrolyzing α-1,6-glucosidic linkages, and its endogenous inhibitor (LDI) was solved at 2.7 Å. The structure reveals an entirely new and unexpected binding mode of LDI as compared with previously solved complex structures of related cereal type family inhibitors (CTIs) bound to glycoside hydrolases but is structurally analogous to binding of dual specificity CTIs to proteases. Site-directed mutagenesis establishes that a hydrophobic cluster flanked by ionic interactions in the protein-protein interface is vital for the picomolar affinity of LDI to LD as assessed by analysis of binding by using surface plasmon resonance and also supported by LDI inhibition of the enzyme activity. A phylogenetic analysis identified four LDI-like proteins in cereals among the 45 sequences from monocot databases that could be classified as unique CTI sequences. The unprecedented binding mechanism shown here for LDI has likely evolved in cereals from a need for effective inhibition of debranching enzymes having characteristic open active site architecture. The findings give a mechanistic rationale for the potency of LD activity regulation and provide a molecular understanding of the debranching events associated with optimal starch mobilization and utilization during germination. This study unveils a hitherto not recognized structural basis for the features endowing diversity to CTIs.
Introduction
Starch, constituting the major carbohydrate and energy reserve in plant leaves, tubers, and seeds, is organized in insoluble supramolecular granules containing a mixture of two polysaccharides: amylopectin, an α-1,4-glucan with α-1,6 attached branches, and amylose, an essentially linear α-1,4-glucan (1). Starch has been identified as a prominent integrator in the regulatory network that adjusts plant growth to the available carbon supply (2). In addition to its central function in plant physiology, starch is the most important carbohydrate for food and feed (3), comprising up to two-thirds of the calorie intake in humans.
The mobilization of starch granules in barley grains during germination involves the concerted action of α- and β-amylase, α-glucosidase, and limit dextrinase (LD).5 LD provides the sole debranching activity relevant for degradation of stored starch during germination through hydrolysis of α-1,6-glucosidic linkages in branched malto-oligosaccharides (limit dextrins) liberated by the action of α- and β-amylases (4, 5). LD is designated as a pullulanase type I debranching enzyme because of its high activity on pullulan (6), a linear polysaccharide of α-1,6-linked maltotriose units, whereas it displays low activity toward amylopectin (7). Substantial LD activity has also been detected in the endosperm of developing rice (Oryza sativa) and maize (Zea mays) seeds, and LD is proposed to play a role in starch biosynthesis in rice, maize, and Arabidopsis (8–10). This is consistent with changes observed in starch structure in the developing barley grains elicited by antisense down-regulation of the endogenous LD inhibitor protein, LDI (11), and with changes in sorghum (Sorghum bicolor) starch digestibility that were positively correlated with LD activity (12).
LDI suppresses the LD activity in barley seeds during the early stages of germination (13–15). It belongs to the family of cereal type inhibitors (CTIs) sharing a common fold of four α-helices connected by irregular loops and stabilized by either four or five disulfide bonds (16). CTIs are widespread in cereals and participate in physiologically important processes in the seeds, e.g. regulation of endogenous hydrolases, as well as defense against pathogens and pests, mainly fungi and insects. Some CTIs have dual target enzyme specificity and inhibit both α-amylases and proteases, e.g. trypsin or chymotrypsin (17).
The regulation of LD activity by LDI is intimately associated to the intriguing double role that LD plays at the interface of starch synthesis and degradation. The molecular features that govern the formation of the regulatory LD-LDI complex are pivotal to promote our understanding of the regulation of starch metabolism in cereals. Here, a comprehensive analysis of the LD-LDI complex, covering x-ray crystal structure determination, binding kinetics, and van't Hoff thermodynamic characterization, is combined with mutational analysis of key residues at the interface in the protein-protein complex. The findings from this study also bring new insight into the functional versatility of the CTI protein scaffold by demonstrating a novel binding mode that overcomes the entropic penalty associated with the inhibition of a debranching enzyme that displays an open active site architecture. The exquisite mechanistic insight is discussed and reconciled with the upstream regulatory cascade that governs mobilization of starch in germinating barley seeds.
EXPERIMENTAL PROCEDURES
LD-LDI Protein Complex Formation and Crystallization
Recombinantly produced LD (6) and LDI (15) were mixed in a 1:4 molar ratio, and the LD-LDI complex was purified by size exclusion chromatography on a Hiload Superdex 200 26/60 column (GE Heathcare) in 50 mm Mes/NaOH (pH 6.6), 250 mm NaCl, 0.5 mm CaCl2 at a flow rate of 0.5 ml/min. The LD-LDI containing fractions were pooled and concentrated (Centricon, 30-kDa cutoff; Millipore) to A280 = 12.1. LD-LDI crystals were obtained using hanging drop vapor diffusion with equilibration of the droplet against a reservoir of 24% (w/v) PEG 8000 and 0.05 m KH2PO4. 0.5 μl of 0.1 m NAD was added to the crystallization droplet consisting of 2 μl of protein solution purified as described above added 2 μl of reservoir solution. By applying crystal seeding, new crystals appeared within 5 days at 22 °C. The crystals were mounted in elliptical Litholoops (Molecular Dimension) and cooled in N2(l) after addition of 25% (w/v) PEG3350, 0.05 m KH2PO4, and 10% glycerol to the droplet.
Data Collection, Molecular Replacement, and Structure Refinement
Final diffraction data were collected at the European Synchrotron Radiation Facility (Grenoble, France) microfocus Beamline ID23-2 (wavelength, 0.873 Å). Data were merged from four different sections of the crystal to minimize the effect of radiation damage on the data quality. The raw data were processed using MOSFLM (18) and merged and scaled using the program SCALA from the CCP4i program suite (19, 20). The resulting structure factors were used for molecular replacement using MOLREP from the CCP4i suite. The protein models were the protein moiety of HvLD-β-cyclodextrin (PDB code 2Y4S) and the α-helical parts of the bifunctional α-amylase/trypsin inhibitor from ragi (Eleusine coracana) (RBI; PDB code 1B1U). The model was refined using Refmac5 from the CCP4i program suite. Manual inspection, rebuilding, and addition of water molecules and ions were performed with Coot (21). Data analysis using xtriage (22) revealed that the crystal was twinned C2221 with a ≈ b and a refined twin fraction of 0.57. NCS (noncrystallographic symmetry) restraints were used in the initial stages of refinement, but not in the last refinement rounds. Twin refinement was applied throughout the refinement. In addition to the Coot validation functions, a final model geometry optimization was performed using the output from MolProbity (23). Coordinates and structure factors for the LD-LDI structure were deposited to the PDB with accession code 4CVW.
Bioinformatics
The phylogenetic tree was constructed with a set of 45 sequences found from BLAST searches with LDI (ABB88573), RBI (P01087), the 0.19 α-amylase inhibitor from wheat (Triticum aestivum) (0.19 AI; P01085), or the Hageman factor inhibitor from corn (Zea mays) (CHFI; P01088) against all sequences from monocots in the NCBI database. The sequences were selected following the criteria: Sequences with E values of <8 × 10−10 were pooled, and those with >95% identity were removed using the EMBOSS software suite (24). No sequences with E values of <8 × 10−10 were identified when dicot sequences from the NCBI database were searched. The resulting set of 45 sequences was aligned using MUSCLE from the MEGA version 5, and a neighbor-joining tree was constructed with 1000 bootstrap steps (25). The tree and alignment were visualized using Dendroscope and ESPript, respectively (26).
Site-directed Mutagenesis, Production, and Purification of Wild Type LDI, LD, and LDI Variants
The single and double mutations in LDI were introduced following the manufacturer's protocol (QuikChange®; Stratagene) using the primers listed in Table 1. An N-terminally Glu-Phe elongated LDI variant (denoted as EF-LDI) resulted from a cloning procedure utilizing the EcoRI restriction site (15). The N-terminally truncated LDI variant (TLE deleted; denoted ΔE3LDI) was obtained as a side product from ΔV5LDI purification. The LDI variants were produced and purified essentially as described previously (15). The ΔV5LDI variant, however, was only purified by nickel-nitrilotriacetic acid column chromatography followed by buffer exchange to 10 mm Bicine/NaOH (pH 8.5) (Microcon, 3-kDa cutoff; Millipore). ΔV5LDI (770 μg/ml) gave a single band by SDS-PAGE analysis, and the N-terminal sequence was confirmed by automated N-terminal sequencing. The structural integrity of the LDI mutants was confirmed using circular dichroism (data not shown).
TABLE 1.
Mutagenesis primers for introduction of mutations in LDI and LD
Fw, forward primer; Rv, reverse primer.
| Primer name | Primer sequence (5′ to 3′) |
|---|---|
| LDI-ΔV5-Fw | GAGAAAAGAGAGGCTGAAGCTAAGGACGAGTGCCAACCAGGGGT |
| LDI-ΔV5-Rv | ACCCCTGGTTGGCACTCGTCCTTAGCTTCAGCCTCTCTTTTCTC |
| LDI-R34A-Fw | CGGGTCTGCGGCGCCGGTCCCAGCCGGCCC |
| LDI-R34A-Rv | GGGCCGGCTGGGACCGGCGCCGCAGACCCG |
| LDI-R38A-Fw | CGCGGTCCCAGCGCGCCCATGCTGGTGAAGG |
| LDI-R38A-Rv | CCTTCACCAGCATGGGCGCGCTGGGACCGCG |
| LDI-R38W-Fw | CGCGGTCCCAGCTGGCCCATGCTGGTGAAGGAG |
| LDI-R38W-Rv | CTCCTTCACCAGCATGGGCCAGCTGGGACCGCG |
| LDI-L41G-Fw | AGCCGGCCCATGGGGGTGAAGGAGCGGTGCTGC |
| LDI-L41G-Rv | GCAGCACCGCTCCTTCACCCCCATGGGCCGGCT |
| LDI-L41W-Fw | AGCCGGCCCATGTGGGTGAAGGAGCGGTGCTGC |
| LDI-L41W-Rv | GCAGCACCGCTCCTTCACCCACATGGGCCGGCT |
| LDI-V42D-Fw | CGGCCCATGCTGGATAAGGAGCGGTGCTGCCGG |
| LDI-V42D-Rv | CCGGCAGCACCGCTCCTTATCCAGCATGGGCCG |
| LDI-L41G-V42D-Fw | AGCCGGCCCATGGGGGATAAGGAGCGGTGCTGCCGG |
| LDI-L41G-V42D-Rv | CCGGCAGCACCGCTCCTTATCCCCCATGGGCCGGCT |
| LD-D730W-Fw | CCAAGTGAAAAGAACGAATGGAATTGGCCCCTGATGAAACC |
| LD-D730W-Rv | GGTTTCATCAGGGGCCAATTCCATTCGTTCTTTTCACTTGG |
| LD-D730R-Fw | CCAAGTGAAAAGAACGAACGTAATTGGCCCCTGATGAAACC |
| LD-D730R-Rv | GGTTTCATCAGGGGCCAATTACGTTCGTTCTTTTCACTTGG |
Recombinant LD WT and variants D730R and D730W were produced in Pichia pastoris and purified as described (6) (see Table 1 for list of primers). Enzyme kinetic constants of LD WT and variants were determined using a reducing sugar assay (6) with an LD concentration of 5.2 nm. The Michaelis constant, Km, and the catalytic constant/turnover number, kcat, were determined by fitting the Michaelis-Menten equation for uncompetitive substrate inhibition to the initial velocities, where Ki,s is the inhibition equilibrium constant. The fitting and plotting were done using the Enzyme Kinetics Module 1.0 of the program Sigmaplot 9.01 (Systat Software, Chicago, IL).
The effect of the double mutation L41G-V42D of LDI on the inhibitory activity was assayed using a modified version of the assay described above: LD was diluted in 20 mm sodium acetate (pH 5.5), 5 mm CaCl2, 0.005% Triton X-100 (100 μl), mixed with LDI in 1 mm Bicine/NaOH (pH 8.5), 0.005% Triton X-100 (100 μl) and incubated at room temperature (10 min). The reaction was initiated by addition of LD (assay concentration, 10 nm)/LDI (assay concentration, 0, 0.1, 1, or 5 μm) mixtures (110 μl) to 990 μl of substrate (0.4 mg/ml pullulan) pre-equilibrated at 37 °C (10 min). Aliquots (100 μl) were removed at 3-min intervals (0–15 min) and mixed with developing buffer (500 μl) (6) and Milli-Q water (400 μl).
Surface Plasmon Resonance Analysis
The LD-LDI interaction was analyzed by surface plasmon resonance (SPR; BIAcore® T100; GE Healthcare). Immobilization of WT LDI and variants on BIAcore CM5 sensor chips was performed by amine coupling according to the manufacturer's protocol using 1–10 μg/ml LDI in 5 mm sodium acetate (pH 4), to a final chip density of 200–400 response units. The standard analysis comprised 4 min of association, 15 min of dissociation, and two cycles (2 × 60 s) of regeneration with 10 mm glycine HCl (pH 1.5); flow rate 30 μl/min. The assays were run at 25 °C at seven LD concentrations (0.1–4 nm) in 10 mm Mes/NaOH (pH 6.0), 150 mm NaCl, 0.005% P-20 surfactant. In the analysis of the V42D and L41G_V42D LDI variants, the concentration of LD was increased to 1–1000 nm and 0.5–8 μm, respectively. Furthermore, the concentration of the LD variants D730R and D730W were 0.4–48 and 0.1–8 nm, respectively. Mass transfer limitations were shown not to be relevant by comparing with rate constants obtained with 60 μl/min flow rate. Reference cell sensorgrams were corrected for refractive index changes, for possible nonspecific LD binding to the cell surface, and for drift specific for the sample cell. The effect of ionic strength on the binding kinetics of WT LD and LDI was determined at the standard analysis conditions, but with 0.075–1 m NaCl, whereas the pH dependence was evaluated using 10 mm of sodium acetate (pH 5.0–5.5), Mes/NaOH (pH 6.0–6.5), Hepes/NaOH (pH 7.0–7.5), glycine/NaOH (pH 8.0–9.0), and Bicine/NaOH (pH 9.5–10.0). Temperature dependence was measured at nine temperatures (10–45 °C) for five LD concentrations (0.4–8 nm).
Two independent data sets were collected for all conditions, and all conditions were analyzed in duplicate, except for 0.4 nm LD samples that were analyzed in quadruplicates and served as a control to assess the response level changes during the course of the experiment. The corrected sensorgrams were analyzed using BIAcore T100 Evaluation Software (ver. 1.1). A 1:1 binding model (27) accounting for possible mass-transport limitations was applied by nonlinear regression fitting to the sensorgrams to determine the association (kon, m−1 s−1), the dissociation rate constant (koff, s−1), and KD given by koff/kon.
Thermodynamic parameters at 25 °C and standard conditions were calculated from nonlinear van't Hoff analysis (BIAcore T100 Evaluation Software version 1.1) using kinetic data obtained in the 10–35 °C range.
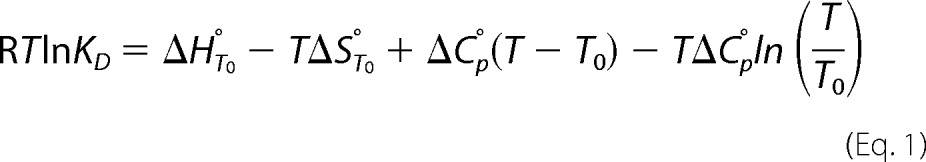 |
Differential Scanning Calorimetry
Thermal stability of LD, LDI, and the LD-LDI complex was measured using a VP-DSC MicroCalorimeter (MicroCal; cell volume = 0.5206 ml). LD, LDI, or LD-LDI samples (0.4 or 1 mg/ml; 2 ml) were dialyzed at 4 °C (Spectra/Por dialysis tubings, 3500-Da cutoff; Spectrum Laboratories) against 1 liter of 20 mm sodium phosphate (pH 6.0, 6.5, or 8.0), or 20 mm sodium phosphate (pH 6.5), 150 mm NaCl. Protein and buffer samples (for baseline scans and reference cell) were degassed prior to scanning at 20–120 °C (1 °C/min). The reversibility of LDI unfolding was evaluated by rapid cooling and rescanning. Thermograms were background corrected and analyzed using Origin ver. 7 software (OriginLab).
RESULTS
Three-dimensional Structure of the LD-LDI Complex Reveals a New CTI Binding Mode
The crystal structure of the LD-LDI complex was determined to 2.7 Å resolution (Fig. 1A and Table 2). The asymmetric unit contained two LD-LDI complexes; complex I comprising chains A (LD; residues 2–884) and C (LDI; residues 8–108) and complex II comprising chains B (LD; residues 2–885) and D (LDI; residues 6–107). The LDI and LD structures from complexes I and II were very similar and superimposed with an RMSD of 0.2 and 0.3 Å, respectively, based on Cα atoms.
FIGURE 1.

Crystal structure of the complex between LD and LDI. A, overall structure of the LD-LDI complex. LDI is shown in orange. The four LD domains, the CBM21-like N-domain (residues 2–124), the carbohydrate-binding module 48 (CBM48; residues 125–230), the catalytic domain (residues 231–774), and the C-domain (residues 775–884), are depicted in red, green, gray, and blue, respectively. The LD catalytic residues, Asp473, Glu510, and Asp642 are shown as black sticks. Calcium ions are presented as purple spheres. The start and end points of the two unresolved short loops of the N-domain are indicated by asterisks. B, close-up on the LDI structure (orange) and the interaction surface with LD (electrostatic potential: blue and red represent positive and negative potential, respectively). LDI structural elements and cysteines are labeled. C–E, amino acid residues in LDI subjected to mutational analysis (orange sticks and ribbon) and their interaction partners in LD (white sticks and ribbon). C, LDI Arg34 from loop 1 interacts with LD Glu729 and Asp730. D, LDI Arg38 interacts directly with the LD catalytic nucleophile Asp473 and general acid/base Glu510. E, two hydrophobic residues, Leu41 and Val42 in LDI are in contact with LD Trp512, Phe514, and Phe553. F, the electrostatic potential of the solvent-accessible surface of LDI in the area where LD Asp730 interacts with Arg34 and Arg84 from LDI.
TABLE 2.
Data collection and refinement statistics for the LD-LDI complex
The values in parentheses are referring to the outer resolution shell.
| Data collection | |
| Beamline | ESRF ID23-2 |
| Wavelength (Å) | 0.873 |
| Resolution range (Å) | 158–2.67 (2.85–2.67) |
| Space group | C2221 |
| Unit cell parameters (Å) | |
| a | 167.0 |
| b | 168.6 |
| c | 157.7 |
| No. of reflections | 294,667 (40,824) |
| No. unique reflections | 60,740 (8,784) |
| Wilson B-factor (Å2) | 24.83 |
| Completeness (%) | 99.4 (99.7) |
| Mean (I)/σ(mean(I)) | 8.0 (2.2) |
| Redundancy | 4.9 (4.4) |
| Rmergea | 0.176 (0.677) |
| Rpimb | 0.089 (0.348) |
| Complex molecules/asymmetric unit | 2 |
| Refinement | |
| Used reflections | 57,661 |
| Rcryst/Rfree (%) | 25.7/29.3 |
| Atoms | |
| Amino acid residues | 15126 |
| Calcium ions | 4 |
| Water molecules | 99 |
| RMSD from ideality | |
| Bond length (Å) | 0.006 |
| Torsion angle (°) | 0.847 |
| Ramachandran plot (%) | |
| Allowed | 99.69 |
| Disallowed | 0.31 |
| MolProbity scorec | 1.46 |
a Rmerge = ΣhklΣi|Ii(hkl) − <I(hkl)>|/ΣhklΣiIi(hkl), where Ii(hkl) is the intensity of the ith observation of reflection hkl, and <I(hkl)> is the average over all observations of reflection hkl.
b Rpim is the multiplicity weighted Rmerge (54).
c MolProbity score is a log-weighted combination of clash score, percentage residues in nonfavored Ramachandran regions and percentage bad side chain rotamers, giving one number that reflects the crystallographic resolution at which those values would be expected (23).
The three-dimensional structure of LDI was composed of four α-helices (α1–4) twisted into a right-handed superhelix, where the α-helices are connected by long loops and stabilized by four disulfides: Cys9–Cys57, Cys23–Cys46, Cys32–Cys87, and Cys47–Cys105 (Fig. 1B). This fold is common to CTIs (16), and the overall structure of LDI is similar to the three other structure-determined CTIs: the bifunctional α-amylase/trypsin inhibitor from ragi (E. coracana) (RBI; PDB codes 1B1U and 1TMQ), the Hageman factor inhibitor from corn (Z. mays) (CHFI; PDB code 1BEA), and the 0.19 α-amylase inhibitor from wheat (T. aestivum) (0.19 AI; PDB code 1HSS) with RMSDs of the Cα atoms between LDI and these inhibitors in the 0.8–1.2 Å range (Table 3). Differences between the four CTI structures were essentially confined to loops and the N- and C-terminal regions (Fig. 2).
TABLE 3.
Structural similarities between core Cα atoms of LDI (chain C; 101 Cα atoms) and related proteins
| The bifunctional trypsin/α-amylase inhibitor from ragi (RBI) | Inhibitor of α-amylase from yellow meal worm in complex with RBI (TMA:RBI) | Corn Hageman factor inhibitor (CHFI) | α-Amylase inhibitor from wheat (0.19 AI) | |
|---|---|---|---|---|
| PDB code | 1B1U | 1TMQ: chain B | 1BEA: chain A | 1HSS: chain A |
| Reference | Ref. 43 | Ref. 30 | Ref. 55 | Ref. 56 |
| RMSD (Å) | 0.86 | 0.83 | 0.78 | 1.18 |
| No. of Cα-atoms in superposition | 117 | 117 | 116 | 111 |
| Sequence identity (%) | 48.4 | 48.4 | 46.9 | 25.8 |
| Sequence similarity (%) | 57.8 | 57.8 | 53.1 | 37.1 |
FIGURE 2.

Structural alignment of the structure-determined cereal type inhibitors and the LDI (orange). A and B, bifunctional α-amylase/trypsin inhibitor from ragi (A, RBI; blue; PDB code 1B1U) and RBI from the complex with α-amylase from yellow mealworm (B, purple; PDB code 1TMQ). The structural differences at the N terminus are marked with a dashed oval. C and D, CHFI (C, green; PDB code 1BEA) and 0.19 α-amylase inhibitor from wheat (D, red; PDB code 1HSS). The loop involved in protease inhibition by RBI and the CHFI is marked with an oval. See Table 3 for details of the structural similarities between LDI and the structure-determined CTIs.
The overall LD structure in the LD-LDI complex was identical to that of free LD (PDB code 4AIO) superimposing with an RMSD of Cα atoms of 0.4 Å. However, three short flexible loops in the N-terminal domain of free LD (28) were too flexible to be modeled in LD-LDI as seen also for LD-cyclodextrin complexes (29). The catalytic site residues Asp473, Glu510, and Asp642 from free LD and from LD in complex with LDI were essentially superimposable, but two residues at the active site, Phe553 and Arg697, adopted different rotamers in LD-LDI compared with free LD (Fig. 3).
FIGURE 3.

Structural alignment of the active site residues from barley LD in complex with LDI and from free LD (PDB code 4AIO). Residues of complexed LD (white sticks), which are in contact with LDI (distance <4.0 Å), are superimposed with the corresponding residues of free LD (green sticks). The residues, which adopt different rotamers, are marked with dotted ovals.
The active site region of LD features a wide cleft, which was buried by LDI upon complex formation, resulting in the loss of 1325 Å2 of solvent-accessible surface. LDI residues from α1 and α2, loop regions 1 and 3, as well as the segment connecting C9 with α1 were in contact with LD (Fig. 1B and Table 4). A number of side chain rearrangements were observed in LD outside the active site region in the LD-LDI interface (intermolecular distance <4 Å). Thus, Asn551, Arg582, Glu726, and Asp730 occurred as different rotamers than in free LD (Fig. 3).
TABLE 4.
Contacts and hydrogen bonds between LDI and LD calculated by the PDBePISA Interface Server (57)
The distances are for the A and C chain complex; values in parenthesis are for the B and D chain complex. ND, not detected.
| Total interface contacts (distance < 4.0 Å) between LDI and LD |
Salt bridges/hydrogen bonds (distance ≤ 3.5 Å) between LDI and LD |
|||
|---|---|---|---|---|
| LDI | LD | LDI | LD | Distance |
| Å | ||||
| Gly12 | Arg582 | Gly12 O | Arg582 Nη2 | 3.5 (3.5) |
| Pro16 | Phe620 | His17 Nϵ2 | Asp579 Oδ2 | 2.8 (3.0) |
| His17 | Gly575, Gln574, Asp579 | Asn18 Nδ2 | Tyr573 O | 3.3 (3.4) |
| Asn18 | Phe572, Tyr573 | Thr22 Oγ1 | Asn551 Nδ2 | 3.3 (3.5) |
| Ala21 | Asn555, Gln558 | Thr25 Oγ1 | Phe553 O | 2.7 (2.9) |
| Thr22 | Asn551, Phe553 | Arg34 Nη1 | Asp730 Oδ1 | 2.7 (2.9) |
| Thr25 | Phe553 | Asp730 Oδ2 | 3.1 (3.0) | |
| Ile28 | Asp730 | Arg34 Nη2 | Asp730 Oδ1 | 4.0 (3.5) |
| Arg34 | Glu726, Glu729, Asp730 | Gly35 O | Lys727 NZ | 3.5 (3.6) |
| Gly35 | Arg697, Glu726, Lys727 | Gly35 N | Glu726 O | 3.5 (ND) |
| Pro36 | Phe553, Arg697, Lys727 | Ser37 N | Asp698 Oδ2 | 3.7 (3.5) |
| Ser37 | Asp698, Lys727 | Ser37 Oγ | Asp698 Oδ2 | 2.9 (2.7) |
| Arg38 | Ala438, Asp473, Leu474, Glu510 | Arg38 Nη1 | Glu510 Oϵ1 | 3.3 (3.5) |
| Met40 | Phe514 | Glu510 Oϵ2 | 3.5 (3.4) | |
| Leu41 | Trp512, Phe514 | Arg38 Nη2 | Glu510 Oϵ1 | ND (3.4) |
| Val42 | Phe553 | Asp473 Oδ1 | 3.0 (3.2) | |
| Glu44 | Asp513, Phe514, Ala515, Arg519 | Asp473 Oδ2 | 3.5 (3.9) | |
| Arg45 | Asp545, Asn551 | Glu44 Oϵ1 | Ala515 N | 3.4 (2.8) |
| Ala52 | Glu621 | Arg45 Nη2 | Asp545 Oδ2 | 3.7 (3.5) |
| Val77 | Tyr573 | Arg84 Nη1 | Asp730 O | 3.2 (3.3) |
| Asp78 | Tyr573 | Arg85 Nϵ | Glu729 Oϵ2 | 3.0 (3.6) |
| Arg84 | Asp730 | Arg85 Nη1 | Glu729 Oϵ1 | ND (3.1) |
| Arg85 | Glu729 | Glu729 Oϵ2 | ND (2.9) | |
| Arg85 Nη2 | Glu729 Oϵ1 | 3.1 (3.6) | ||
The N-terminal segment of RBI has been shown to be essential for the interaction with yellow mealworm α-amylase (30) in contrast to LD-LDI, where the LDI N terminus is not involved in the interaction (Fig. 1B). This observation was confirmed by LDI variants with extended or shortened N terminus, where the binding to LD was barely influenced (Fig. 4 and Table 5). In fact, LDI and LDI-like proteins lack the N-terminal Ser-Val dipeptide that is essential for binding of RBI to yellow meal worm α-amylase, as illustrated by a multiple alignment of protein sequences from monocots (Poaceae) found by using the sequences of LDI, RBI, CHFI, and 0.19 AI in a protein BLAST search in the NCBI database (Fig. 5 and Table 6). The three putative LDI-like inhibitors from wheat, rice, and Brachypodium distachyon identified from this BLAST search all lack the dipeptide and have one less cysteine residue than the previously characterized CTI α-amylase/protease inhibitors, which contain five disulfides (Fig. 6a).
FIGURE 4.

Representative plots of single SPR data sets used for obtaining the data shown in Table 5. A, 1:1 binding model (black lines) fitted to the SPR data (orange dashed line) from LD binding to different LDI variants including wild type. B, top panel, sensorgram from the SPR analysis of LD binding to the LDI-V42D variant with the points used for the steady state fit indicated by ×. Bottom panel, steady state plot of data from a triple determination. C, top panel, sensorgram from SPR analysis of LD binding to the LDI-L41GV42D variant. The points used for the steady state fit are indicated by ×. Bottom panel, steady state plot of data from a double determination. D, 1:1 binding model (black) fitted to SPR data (orange dashed line) from analysis of the binding of the two LD variants to wild type LDI.
TABLE 5.
Key residues of the LD-LDI interaction analyzed by SPR
The values show Kinetics of LD binding to LDI WT and to LDI variants and binding of LD variants to WT LDI measured by SPR at 25 °C, pH 6.0, 150 mm NaCl (see Fig. 4 for representative plots of the SPR data sets).
| kon | koff | KD | Relative KD | ΔGa | ΔΔGb | ΔGsolvc | |
|---|---|---|---|---|---|---|---|
| m− s−1 | s−1 | m | kJ mol−1 | kJ mol−1 | kJ mol−1 | ||
| LDI variant | |||||||
| WT | (1.5 ± 0.02)·106 | (6.4 ± 0.20)·10−5 | (4.2 ± 0.20)·10−11 | 1 | −59.2 | 0 | −19.7 |
| R34A | (9.4 ± 0.3)·105 | (8.7 ± 0.11)·10−4 | (9.2 ± 1.4)·10−10 | 22 | −51.5 | 7.7 | −19.3 |
| R38A | (1.2 ± 0.01)·106 | (1.7 ± 0.09)·10−3 | (1.4 ± 0.06)·10−9 | 33 | −50.5 | 8.8 | −24.8 |
| R38W | (1.0 ± 0.01)·106 | (5.2 ± 0.11)·10−4 | (5.2 ± 0.1)·10−10 | 12 | −52.9 | 6.2 | −31.5 |
| L41G | (1.3 ± 0.08)·106 | (5.5 ± 0.38)·10−3 | (4.2 ± 0.04)·10−9 | 100 | −47.8 | 11.4 | −15.5 |
| L41W | (8.9 ± 1.3)·105 | (6.5 ± 0.5)·10−5 | (7.4 ± 0.53)·10−11 | 1.8 | −57.8 | 1.4 | −21.0 |
| V42Dd | (1.7 ± 0.05)·10−7 | 4048 | −38.6 | 20.6 | −16.4 | ||
| L41G-V42Dd,e | (2.0 ± 0.09)·10−5 | 476190 | −26.8 | 32.4 | −12.2 | ||
| ΔV5LDIf | (1.0 ± 0.0)·106 | (5.7 ± 0.2)·10−5 | (5.5 ± 0.2)·10−11 | 1.3 | −58.5 | 0.7 | |
| ΔE3LDIf | (9.1 ± 0.1)·105 | (6.6 ± 0.1)·10−5 | (7.2 ± 0.3)·10−11 | 1.7 | −57.8 | 1.4 | |
| EF-LDIf,g | 1.4·106 | 1.1·10−4 | 8.4·10−11 | 2 | −57.5 | 1.7 | |
| LD variant | |||||||
| D730W | (1.1 ± 0.04)·106 | (3.5 ± 0.4)·10−4 | (3.3 ± 0.2)·10−10 | 7.9 | −54.1 | 5.1 | |
| D730R | (1.5 ± 0.07)·105 | (1.1 ± 0.04)·10−3 | (7.2 ± 0.04)·10−9 | 171 | −46.4 | 12.8 | |
a ΔG based on the measured KD.
b ΔΔG = ΔGLDI variant − ΔGLDI-WT or ΔΔG = ΔGLD variant − ΔGLD-WT.
c Solvation energy gain at complex formation calculated using the PDBePISA server (57). In silico mutations for these calculations were introduced using Coot (21).
d The results are based on steady state kinetics.
e See Table 7 for residual LD activity in the presence of LDI variant L41G-V42D.
f N-terminally truncated or extended LDI variants.
g Result from single experiment.
FIGURE 5.


Multiple alignment of 45 protein sequences identified from BLAST searches with the sequences of LDI, RBI, 0.19 AI, and CHFI against the monocot sequence database. Sequence features are labeled in the subalignment in Fig. 6. See Table 6 for information about each sequence included.
TABLE 6.
Source organisms and accession numbers of sequences included in the multiple sequence alignments (Figs. 5 and 6A) and the phylogenetic tree (Fig. 6B)
| No. | Group | Accession ID | Organism name |
|---|---|---|---|
| 1 | LDI-like inhibitors | ABB88573 | Hordeum vulgare |
| 2 | CAA68248 | Triticum aestivum | |
| 3 | XP_003561291 | Brachypodium distachyon | |
| 10 | ABK34477 | Oryza sativa Indica gr. | |
| 29 | NP_001147201 | Zea mays | |
| 27 | XP_002459556 | Sorghum bicolor | |
| 9 | NP_001059199 | Oryza sativa Japonica gr. | |
| 15 | XP_002459322 | Sorghum bicolor | |
| 12 | P81367 | Sorghum bicolor | |
| 11 | XP_002461685 | Sorghum bicolor | |
| 7 | XP_002461687 | Sorghum bicolor | |
| 42 | P01088 | Zea mays | |
| 4 | P01087 | Eleusine coracana | |
| 18 | XP_002461684 | Sorghum bicolor | |
| 14 | XP_002459323.1 | Sorghum bicolor | |
| 28 | S51811 | Triticum aestivum | |
| 44 | CMx subunits of tetrameric α-amylase/protease inhibitors | 1312252B | Secale cereale |
| 5 | CAA11030 | Hordeum vulgare | |
| 8 | CAA67193 | Hordeum vulgare | |
| 6 | CAA11028 | Hordeum vulgare subsp. spontane | |
| 25 | AAZ67071 | Secale cereale | |
| 19 | CAA42453 | Triticum aestivum | |
| 20 | P32936 | Hordeum vulgare | |
| 24 | P17314 | Triticum aestivum | |
| 13 | CAA49536.1 | Hordeum vulgare | |
| 23 | P34951.2 | Hordeum vulgare | |
| 16 | P83207 | Triticum aestivum | |
| 22 | P28041 | Hordeum vulgare | |
| 21 | P16850 | Triticum aestivum | |
| 17 | P16851 | Triticum aestivum | |
| 32 | NP_001059191 | Oryza sativa Japonica gr. | |
| 31 | NP_001059192 | Oryza sativa Japonica gr. | |
| 30 | XP_003561293 | Brachypodium distachyon | |
| 26 | 1208404A | Hordeum vulgare | |
| 33 | Q01881 | Oryza sativa Japonica gr. | |
| 43 | 0.28 AI monomeric inhibitor | P01083 | Triticum aestivum |
| 41 | Dimeric α-amylase inhibitors | ACP40903 | Eremopyrum bonaepartis |
| 39 | ACP40883 | Triticum timopheevii subsp. armeniacum | |
| 36 | ACP40915 | Secale cereale | |
| 38 | ABI54565 | Aegilops sharonensis | |
| 40 | ACP40690 | Triticum dicoccoides | |
| 37 | ACP40801 | Triticum dicoccoides | |
| 45 | P01085 | Triticum aestivum | |
| 35 | ACP40906 | Eremopyrum bonaepartis | |
| 34 | ACP40674 | Triticum dicoccoides | |
FIGURE 6.

LDI and related CTIs. A, multiple sequence alignment of eight CTIs including the three structure-determined proteins: LDI, wheat pUP88, protein from Brachypodium, RBI, rice LDI, CHFI, and dimeric and monomeric wheat α-amylase inhibitors 0.19 AI and 0.28 AI. The secondary structure of LDI is indicated above the alignment, disulfides are numbered 1–4 (below alignment), and the fifth cysteine pair (lacking in LDI-like proteins) is indicated by a green × under the conserved cysteine and a red box around the nonconserved cysteine, as well as an ×. The LDI residues mutated in this study are marked by asterisks. RBI and CHFI residues involved in α-amylase binding or trypsin inhibition are indicated by black boxes. B, phylogenetic tree based on multiple sequence alignment of 45 protein sequences from monocots related to LDI, RBI, 0.19 AI, and CHFI. The clustering of the CTIs is based on the annotation of characterized CTIs: LDI-like proteins (this study), CMx subunits of heterotetrameric α-amylase/protease inhibitors, and mono- and dimeric α-amylase inhibitors. The characterized proteins are indicated by asterisks. See Fig. 5 for the full multiple sequence alignment and Table 6 for source organisms and accession numbers of sequences included in the phylogenetic tree.
Kinetics, van't Hoff Energetics, and Complex Stability
The affinity of LD for LDI was determined to KD = 30 pm at 25 °C and pH 6.5, which was the pH optimal for binding, as measured by SPR. The KD increased 12–15-fold at pH 5.0 or 10.0, largely because of increases in the dissociation rate constant, koff (Fig. 7). Furthermore, KD decreased at lower temperatures (10–20 °C) but increased about 6-fold at 45 °C as compared with KD at 25 °C, mainly because of a higher koff (Fig. 7). By contrast, KD was independent of ionic strength in the range 75–1000 mm NaCl. A van't Hoff nonlinear thermodynamic analysis using SPR data from the 10–35 °C temperature range (Fig. 8a) showed that LD-LDI complex formation was accompanied by a large decrease in heat capacity (ΔCp° = −3.2 kJ K−1 mol−1) and driven by a free energy change (ΔG° = −57 kJ mol−1) originating from equally favorable entropy (−TΔS° = −30 kJ mol−1) and enthalpy (ΔH° = −27 kJ mol−1) changes.
FIGURE 7.

SPR analysis of the pH (A) and temperature (B) dependence of the formation of the LD-LDI complex.
FIGURE 8.
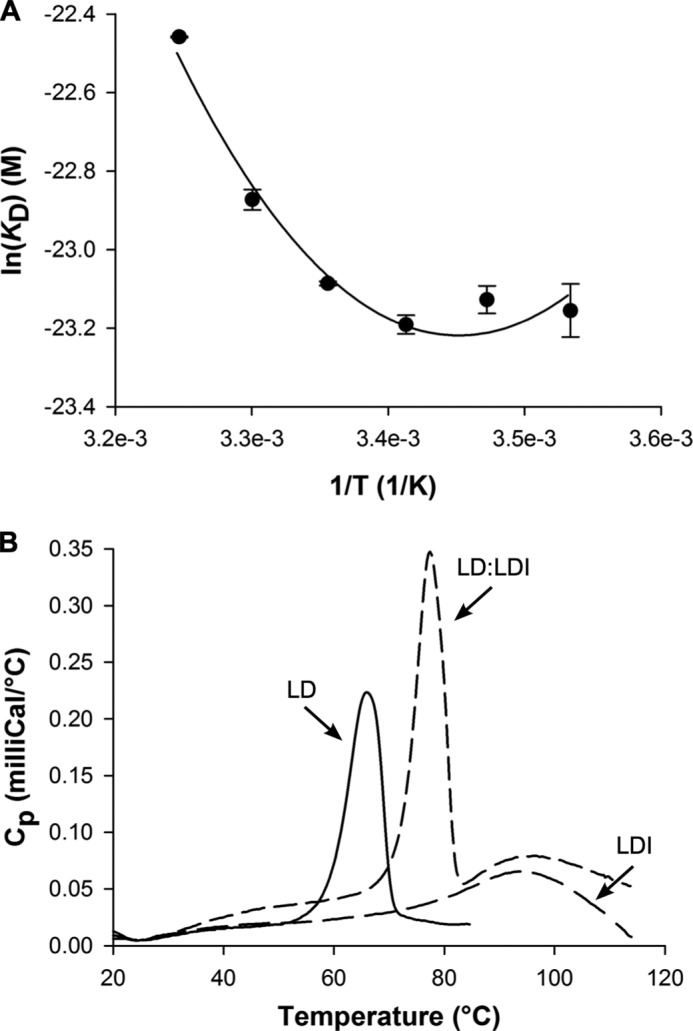
van't Hoff energetics and conformational stability of LD-LDI. A, van't Hoff Plot of the fitting of the nonlinear function to the SPR data in the temperature range 10–35 °C. B, differential scanning calorimetry thermograms of LD (5 μm), LDI (25 μm), and LD-LDI (5 μm:25 μm) unfolding at pH 6.5.
The conformational stability of free LD, LDI, and the LD-LDI complex was assessed by differential scanning calorimetry. The thermogram of LD featured a single peak with an assigned unfolding temperature (Tm) of 65.9 °C, whereas the calorimetric trace of LDI showed a very broad peak with Tm of 97.4 °C (Fig. 8b). As judged from the lack of thermal transition area recovery after rescanning, the unfolding of LD and LDI was irreversible, which precluded a full thermodynamic analysis. The thermogram of LD-LDI revealed two peaks with Tm values of 77.4 and ∼100 °C, respectively. The first transition was ascribed to the dissociation of the complex and unfolding of LD, whereas the second probably originates from unfolding of LDI at higher temperature (Fig. 8b). Thus, the LD-LDI complex provides substantial stabilization to LD as manifested in a Tm increase of more than 11 °C as compared with free LD.
Key Residues of LD-LDI Complex Formation
An examination of the LD-LDI interface for residues involved in the complex formation guided the selection of Arg34, Arg38, Leu41, and Val42 in LDI and Asp730 in LD for mutational analysis. LDI Arg34 forms an ionic network with Glu729 and Asp730 (Fig. 1c) at the entrance of the LD active site. LDI Arg38 has contacts with both the catalytic nucleophile (Asp473) and the general acid/base catalyst (Glu510) of LD (Fig. 1d and Table 4). In addition to these ionic/polar interactions, LDI residues Leu41 and Val42 establish a central hydrophobic cluster jointly with LD residues Trp512, Phe514, and Phe553 (Fig. 1e). Trp512 is situated at LD substrate binding subsite +2 (29) and belongs to a conserved motif in glycoside hydrolase family 13 (GH13) that also includes the general acid-base catalyst (31). LD D730 binds in a positively charged pocket on the surface of LDI via a hydrogen bond (2.7 Å) and a salt bridge (4.0 Å) to Arg34 and a hydrogen bond to Arg84 (3.2 Å) (Fig. 1c and Fig. 1f). Furthermore, LD Asp730 adopts different rotamers in the free LD and LD bound to LDI as mentioned above (Fig. 3).
In total, seven variants of LDI were made based on the protein-protein interactions listed above: the single R34A (probe for charge and size effects), R38A, R38W (probe for charge effects), L41G (probe for size effects), L41W (probe for size effects), V42D (probe for charge effects), and the L41G-V42D variants. In addition, the mutations D730R and D730W were introduced in LD to probe an electrostatic interaction with LDI at a distant position from the active site (23 Å from the catalytic nucleophile). Binding of the LDI variants to WT LD and the binding of WT LDI to the LD variants were evaluated by SPR. Changes in the association rate constant (kon) were modest for all the LDI variants as compared with the WT. By contrast, koff increases of varying magnitude were measured for all LDI variants with the exception of LDI-L41W (Table 5). The KD values for LDI-V42D and LDI-L41G-V42D were determined from steady state data because koff was too fast to allow determination of the binding kinetics. Thus, the KD of L41G and V42D increased by 100–400-fold, whereas R34A, R38A, and R38W caused modest 12–33-fold increases. The combined mutation of L41G and V42D led to a dramatic KD increase of 5 × 105-fold. Although no residual activity was detected for LD in the presence of WT LDI at a 1:1 molar ratio (15), 500-fold molar excess of the double mutant LDI-L41G-V42D was not sufficient to abolish LD activity (27% residual activity was measured) (Table 7). This poor inhibitory activity fits well with the KD of 20 μm determined by SPR and emphasized the pivotal role of LDI Leu41 and Val42 in their joint contribution to LD-LDI complex formation. Finally, the KD values of LD-D730W and LD-D730R binding to WT LDI were 8- and 171-fold higher, respectively, than for WT LD (Table 5). Although LD-D730R showed a 10-fold decrease, the kon of LD D730W was only reduced 1.5-fold. The two mutations of LD had no significant effect on the kinetics of LD acting on pullulan: the Km for both variants was 0.24 ± 0.05 mg/ml (0.16 ± 0.02 mg/ml for WT LD), whereas kcat was 94 ± 16 s−1 for LD-D730R and 85 ± 14 s−1 for LD-D730W (78 ± 10 s−1 for WT LD).
TABLE 7.
Residual LD activity (10 nm in assay) in the presence of different ratios of the double mutant LDI-L41G-V42D
| LDI:LD molar ratio | Residual activity |
|---|---|
| % | |
| 0 | 100 ± 1.8 |
| 10 | 93.5 ± 2.1 |
| 100 | 78.3 ± 0.8 |
| 500 | 27.1 ± 0.3 |
DISCUSSION
This is the first study exploring the binding of a proteinaceous inhibitor of a starch debranching enzyme, which is found both in seeds and leaves of many plants including maize (8, 32), mung bean (33), oat (34, 35), pea (36), rice (32), sorghum (12), spinach (37), sugar beet (38), and wheat (39). LD inhibitory activity measured previously in wheat seed extracts (40) may stem from an LDI-like protein (pUP88; CAA68248) annotated in the present study. Relatively high levels of LD inhibitory activity were also reported for rye and triticale and low levels in oat (40). Therefore close homologues of LDI perform similar important functional roles in other cereals, underlining the broad relevance of the present study.
A Novel Inhibitory Binding Mode of CTIs to Glycoside Hydrolases
Only two crystal structures of LDI homologues of the CTI family in complex with α-amylases have been reported, i.e. the complex formed by α-amylase from yellow meal worm (TMA) and RBI, the LDI homologue from ragi (30), and the complex between pig pancreatic α-amylase and the wheat inhibitor 0.28 AI; the structural coordinates of the latter complex are not deposited in the PDB (41). Very surprisingly, superimposition of the complexes of LDI (LD-LDI) and its homologue from ragi (TMA-RBI; PDB code 1TMQ) showed their binding modes to be entirely different (Fig. 9), although both of these target enzymes belong to GH13 and hence share a similar fold of the catalytic domain, as well as active site amino acid motifs (31).
FIGURE 9.
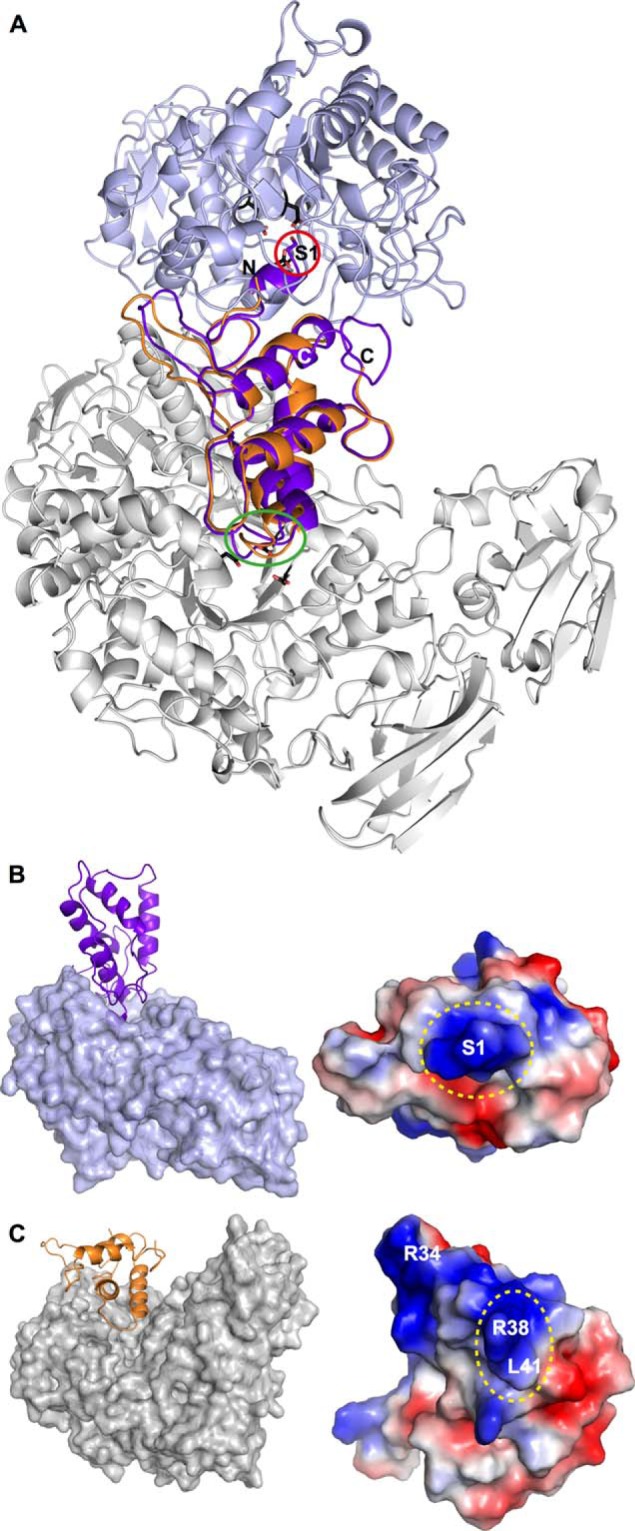
Comparison of LDI and RBI binding to LD and TMA. A, the two CTIs, LDI (orange) and RBI (purple) superimpose with an RMSD of 0.8 Å, but the binding modes of LDI to LD (gray) and RBI to TMA (light blue) are completely different. The N-terminal serine of RBI (purple stick and red circle) interacts with the catalytic site residues of TMA (black sticks), whereas the N terminus of LDI has no contact with LD. The catalytic site residues of LD are shown as black sticks. The trypsin-binding loop (harboring Arg34 and Leu35) of RBI is marked with a green oval. B, binding mode of RBI (purple) to TMA (light blue surface) and the electrostatic potential of the RBI interaction surface. Blue and red represent positive and negative potential, respectively. The N terminus of RBI is marked with a dotted oval. C, binding mode of LDI (orange) to LD (gray surface) and the electrostatic potential of the LDI interaction surface. The Leu41–Val42 hotspot of the interaction is marked with a dotted oval (Val42 is not visible).
First, the N-terminal segment of RBI (Ser1–Ala11) exerts a key role in inhibition by blocking the active site of its α-amylase target (TMA). Thus, the N-terminal sequence 1SVGTS5 assumes a 310-helical conformation upon binding to TMA but is unstructured in the free RBI (42, 43). This rearrangement appears essential for the inhibition mechanism that involves contacts between the N-terminal amino group of RBI with the three acidic residues at the active site in TMA (30). Essentially the same binding mode is reported for an LDI homologue from wheat with its porcine α-amylase target (0.28 AI) (41). By contrast, the N-terminal segment of LDI (1TLESVKDE8) has no contact with LD (Fig. 2b), consistent with the lack of significant electron density for the first five residues, indicating that this region is disordered. Remarkably, the close homologues of LDI are either having an additional N-terminal di/tripeptide (wheat pUP88 and LDI itself) preceding the functionally important serine residues observed in α-amylase inhibitors or the serine residue is not present at the N terminus (B. distachyon and rice) (Fig. 6a). This is in excellent agreement with a different binding mode of the LDI-like proteins targeting debranching enzymes as opposed to those inhibitors targeting α-amylases, which have a narrower active site cleft.
Five of the RBI residues (Pro52, Cys55, Val67, Thr69, and Pro70) from the TMA binding regions (Pro52–Cys55, Arg61, Val67–Ser71, Thr107–Gly110, and Leu115–Leu117) (30) are conserved in LDI (Fig. 6a), but these residues make no contact with LD in the LD-LDI complex (Table 4). The variable residues flanking these five residues in LDI probably contribute to the lack of α-amylase inhibition (44) as known for the RBI binding mode. The LD-LDI binding mode vice versa cannot be applied to α-amylases because of steric clashes between their narrow cleft-shaped active site and LDI. Based on the phylogeny (Fig. 6b) and the missing N-terminal dipeptide motif, we propose that the proteins that confer inhibition of LD-like debranching enzymes form a clade among the CTI proteins, which we designate as LDI-like (Fig. 6b).
LDI Loop 1 Involvement in Protein-Protein Interactions
Certain CTI proteins have dual targets (17). The LDI homologue RBI is a dual α-amylase/trypsin inhibitor, and its inhibition of trypsin is effectuated by a trypsin-binding loop located at the opposite side of the α-amylase-binding site. Arg34 and Leu35 from this loop interact with the active site in trypsin (Fig. 9a) (30). Notably, the sequence of the RBI trypsin-binding loop is not conserved in the corresponding LDI loop 1 (Ser37 and Arg38 correspond to Arg34 and Leu35, respectively, in RBI) (Fig. 6a), which is central in the LD-LDI complex contacts (Fig. 6a). LDI indeed fails to inhibit trypsin (44). In addition, no equivalent to the hydrophobic patch demonstrated here to be critical for the LD-LDI complex formation exists in the RBI:trypsin interface. This asserts a diversity of interactions mediated by CTI proteins and a fine-tuning of their surfaces to match their physiologically relevant targets.
In addition to being part of the LD-LDI interface, the LDI loop 1 has been shown to be a recognition site for the regulatory eukaryotic 14-3-3 proteins, which are proteins that bind to specific phosphorylated domains of target proteins including signaling proteins and metabolic enzymes (45). Strikingly, this site will only be available for regulation by the 14-3-3 proteins in free LDI. The interaction could have implications for LDI involvement in regulatory networks, but the biological relevance of the reported LDI-14-3-3 protein interaction is not known.
Inhibitory Mechanism of LDI
The large drop in heat capacity (ΔCp° = −3.2 kJ K−1 mol−1) and the favorable entropy change associated with LD-LDI formation (approximately 50% of the total free energy of binding) could be caused by the burial of substantial hydrophobic solvent-accessible surface area in the complex (46). This is consistent with the LD-LDI structure, where LDI Arg38 binding to the catalytic site of LD is flanked by a hydrophobic patch containing Leu41 and Val42 shown to be crucial for affinity to LD (Fig. 9c). For both LDI and RBI, a patch with positively charged surface potential becomes buried in the complex interface, but the insertion of RBI into the narrower active site cleft of the α-amylase (Fig. 9b) occurs without concurrent intermolecular hydrophobic patch matching. Thermodynamic data are not available for the TMA-RBI interaction; however, the ordering of the N-terminal segment of RBI upon binding to TMA possibly includes a significant entropic penalty and a concomitant enthalpic compensation. Binding of porcine pancreas α-amylase to the LDI homologue from wheat (0.19 AI) was driven by a favorable change in both entropy and enthalpy, albeit with a larger enthalpic contribution (47), suggesting that polar interactions are more important for that complex than for LD-LDI. The liberation of ordered solvent molecules accompanying hydrophobic cluster formation at the LD-LDI interface provides an important entropic favorable contribution to the complex formation, which seems to be a characteristic energetic determinant that distinguishes the open active site architecture of debranching enzymes as opposed to the more narrow active site of α-amylases.
Electrostatic interactions seem less critical for the LD-LDI complex formation than was expected based on the positive surface potential patch of LDI (Fig. 9c), and the observed contact between LDI Arg38 and the acidic LD catalytic site residues. This interpretation is further supported by the insensitivity of LD-LDI affinity to ionic strength and by the LDI mutational data (Table 5). Nonetheless, charged interactions are likely to contribute to electrostatic steering at the molecular encounter distance (48). Indeed the largest decrease in kon among the present mutants occurred because of electrostatic complementarity being lost for LD-D730R. LD Asp730 was <4 Å from a positively charged patch at the periphery of LDI (Arg34) in LD-LDI (Fig. 1c and f). Concordantly, KD is pH-dependent (Fig. 7a), possibly because of charge disruption of hydrophilic patches.
Biological Implications of LD Inhibition
During the first 24 h of germination, LD and LDI are spatially separated, residing mainly in the aleurone layer and in the endosperm of the seed, respectively (49). LD has no classical signal peptide to direct secretion and is released slowly from aleurone cells (4, 49, 50). Possibly, the degradative damage to the aleurone cell walls occurring 24–48 h after germination onset allows diffusion of LD into the starchy endosperm (50). When LD encounters LDI at this stage, further hydrolysis of branched dextrins products from starch degradation catalyzed by the abundant α- and β-amylases will not take place. Consequently, undesirable germination occurring because of fluctuations in environmental factors (e.g. premature sprouting) at an otherwise unfavorable state can be brought to a halt without complete exhaustion of the carbohydrate reserve, and furthermore the LD activity will be protected for more favorable times by conformational stabilization of LD as it forms a complex with the LDI as judged by a marked increase in unfolding temperature of LD (ΔTm = 11 °C) in the complex as compared with the free form of the enzyme. The full activity of LD can only be unleashed when inactivation of LDI takes place. The amount of intact LDI is reduced to below the detection limit concomitant with a reported increase in the level of free LD during germination (51). The loss of LDI, maybe caused by proteolysis (51) is probably facilitated by thioredoxin h reduction of disulfides in LDI, which per se results in LDI inactivation (15). The abundances of CTIs in general decrease during germination (52).
The binding mode of LDI to LD illustrates an extraordinary versatility of CTIs featuring in a variety of regulatory and defense related protein-protein interactions in plants. The impressive conformational stability of LDI together with possibility for integration with redox regulatory networks provides a possible rationale for conservation of the structural scaffold of the CTIs through a divergent evolution of the individual protein subfamilies. The outcome of this is a multitude of roles played by CTIs in regulation of plant development as seen in the present study and for previously described defense mechanisms (53).
Acknowledgments
Anne Blicher (Technical University of Denmark) is acknowledged for amino acid and N-terminal sequence analyses. We thank Monica Palcic and Nathalie Juge and members of our research groups for fruitful discussions, as well as Jose A. Cuesta Seijo (Carlsberg Laboratory) for collection of the final data set at the European Synchrotron Radiation Facility. The European Synchrotron Radiation Facility and its staff, as well as MAX II Laboratory, Lund University, and MAX laboratory staff, are acknowledged for beam time and assistance. The DANSCATT program from the Danish National Research Council is thanked for travel support to the respective beam lines. Novo Nordisk is thanked for circular dichroism instrument time. We acknowledge Bent Sigurskjold (Copenhagen University), who lent us the differential scanning calorimeter.
This work was supported by the Carlsberg Foundation, the Danish Council for Independent Research, Natural Sciences, Technical University of Denmark Ph.D. stipends (to M. B. V.-C. and M. S. M.), and an Oticon Foundation M.Sc. scholarship (to J. M. J.).
The atomic coordinates and structure factors (code 4CVW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LD
- barley limit dextrinase
- LDI
- barley limit dextrinase inhibitor
- CTI
- cereal type family inhibitor
- RBI
- bifunctional α-amylase/trypsin inhibitor from ragi
- 0.19 AI
- 0.19 α-amylase inhibitor from wheat
- CHFI
- Hageman factor inhibitor from corn
- SPR
- surface plasmon resonance
- TMA
- α-amylase from yellow meal worm
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- Bicine
- N,N-bis(2-hydroxyethyl)glycine.
REFERENCES
- 1. Tester R. F., Karkalas J., Qi X. (2004) Starch: composition, fine structure and architecture. J. Cereal Sci. 39, 151–165 [Google Scholar]
- 2. Sulpice R., Pyl E. T., Ishihara H., Trenkamp S., Steinfath M., Witucka-Wall H., Gibon Y., Usadel B., Poree F., Piques M. C., Von Korff M., Steinhauser M. C., Keurentjes J. J., Guenther M., Hoehne M., Selbig J., Fernie A. R., Altmann T., Stitt M. (2009) Starch as a major integrator in the regulation of plant growth. Proc. Natl. Acad. Sci. U.S.A. 106, 10348–10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zeeman S. C., Kossmann J., Smith A. M. (2010) Starch: Its metabolism, evolution, and biotechnological modification in plants. Annu. Rev. Plant Biol. 61, 209–234 [DOI] [PubMed] [Google Scholar]
- 4. Burton R. A., Zhang X. Q., Hrmova M., Fincher G. B. (1999) A single limit dextrinase gene is expressed both in the developing endosperm and in germinated grains of barley. Plant Physiol. 119, 859–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kristensen M., Lok F., Planchot V., Svendsen I., Leah R., Svensson B. (1999) Isolation and characterization of the gene encoding the starch debranching enzyme limit dextrinase from germinating barley. Biochim. Biophys. Acta 1431, 538–546 [DOI] [PubMed] [Google Scholar]
- 6. Vester-Christensen M. B., Hachem M. A., Naested H., Svensson B. (2010) Secretory expression of functional barley limit dextrinase by Pichia pastoris using high cell-density fermentation. Protein Expr. Purif. 69, 112–119 [DOI] [PubMed] [Google Scholar]
- 7. Møller M. S., Windahl M. S., Sim L., Bøjstrup M., Abou Hachem M., Hindsgaul O., Palcic M., Svensson B., Henriksen A. (2015) Oligosaccharide and substrate binding in the starch debranching enzyme barley limit dextrinase. J. Mol. Biol. 427, 1263–1277 [DOI] [PubMed] [Google Scholar]
- 8. Dinges J. R., Colleoni C., James M. G., Myers A. M. (2003) Mutational analysis of the pullulanase-type debranching enzyme of maize indicates multiple functions in starch metabolism. Plant Cell 15, 666–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wattebled F., Planchot V., Dong Y., Szydlowski N., Pontoire B., Devin A., Ball S., D'Hulst C. (2008) Further evidence for the mandatory nature of polysaccharide debranching for the aggregation of semicrystalline starch and for overlapping functions of debranching enzymes in Arabidopsis leaves. Plant Physiol. 148, 1309–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujita N., Toyosawa Y., Utsumi Y., Higuchi T., Hanashiro I., Ikegami A., Akuzawa S., Yoshida M., Mori A., Inomata K., Itoh R., Miyao A., Hirochika H., Satoh H., Nakamura Y. (2009) Characterization of pullulanase (PUL)-deficient mutants of rice (Oryza sativa L.) and the function of PUL on starch biosynthesis in the developing rice endosperm. J. Exp. Bot. 60, 1009–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stahl Y., Coates S., Bryce J. H., Morris P. C. (2004) Antisense downregulation of the barley limit dextrinase inhibitor modulates starch granule size distribution, starch composition and amylopectin structure. Plant J. 39, 599–611 [DOI] [PubMed] [Google Scholar]
- 12. Gilding E. K., Frère C. H., Cruickshank A., Rada A. K., Prentis P. J., Mudge A. M., Mace E. S., Jordan D. R., Godwin I. D. (2013) Allelic variation at a single gene increases food value in a drought-tolerant staple cereal. Nat. Commun. 4, 1483. [DOI] [PubMed] [Google Scholar]
- 13. MacGregor A. W., Macri L. J., Schroeder S. W., Bazin S. L. (1994) Purification and characterisation of limit dextrinase inhibitors from barley. J. Cereal Sci. 20, 33–41 [Google Scholar]
- 14. MacGregor A. W., Donald L. J., MacGregor E. A., Duckworth H. W. (2003) Stoichiometry of the complex formed by barley limit dextrinase with its endogenous inhibitor. Determination by electrospray time-of-flight mass spectrometry. J. Cereal Sci. 37, 357–362 [Google Scholar]
- 15. Jensen J. M., Vester-Christensen M. B., Møller M. S., Bønsager B. C., Christensen H. E., Hachem M. A., Svensson B. (2011) Efficient secretory expression of functional barley limit dextrinase inhibitor by high cell-density fermentation of Pichia pastoris. Protein Expr. Purif. 79, 217–222 [DOI] [PubMed] [Google Scholar]
- 16. José-Estanyol M., Gomis-Rüth F. X., Puigdomènech P. (2004) The eight-cysteine motif, a versatile structure in plant proteins. Plant Physiol. Biochem. 42, 355–365 [DOI] [PubMed] [Google Scholar]
- 17. Svensson B., Fukuda K., Nielsen P. K., Bønsager B. C. (2004) Proteinaceous α-amylase inhibitors. Biochim. Biophys. Acta 1696, 145–156 [DOI] [PubMed] [Google Scholar]
- 18. Leslie A. G. (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 ESF-EAMCB Newsl. Protein Crystallogr., No. 26 [Google Scholar]
- 19. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Potterton E., Briggs P., Turkenburg M., Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 21. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rice P., Longden I., Bleasby A. (2000) EMBOSS: The European molecular biology open software suite. Trends Genet. 16, 276–277 [DOI] [PubMed] [Google Scholar]
- 25. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011) MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gouet P., Courcelle E., Stuart D., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]
- 27. Myszka D. G., He X., Dembo M., Morton T. A., Goldstein B. (1998) Extending the range of rate constants available from BIACORE: Interpreting mass transport-influenced binding data. Biophys. J. 75, 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Møller M. S., Abou Hachem M., Svensson B., Henriksen A. (2012) Structure of the starch-debranching enzyme barley limit dextrinase reveals homology of the N-terminal domain to CBM21. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 1008–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vester-Christensen M. B., Abou Hachem M., Svensson B., Henriksen A. (2010) Crystal structure of an essential enzyme in seed starch degradation: barley limit dextrinase in complex with cyclodextrins. J. Mol. Biol. 403, 739–750 [DOI] [PubMed] [Google Scholar]
- 30. Strobl S., Maskos K., Wiegand G., Huber R., Gomis-Rüth F. X., Glockshuber R. (1998) A novel strategy for inhibition of α-amylases: yellow meal worm α-amylase in complex with the Ragi bifunctional inhibitor at 2.5 Å resolution. Structure Fold Des. 6, 911–921 [DOI] [PubMed] [Google Scholar]
- 31. MacGregor E. A., Janecek S., Svensson B. (2001) Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim. Biophys. Acta 1546, 1–20 [DOI] [PubMed] [Google Scholar]
- 32. Li Q. F., Zhang G. Y., Dong Z. W., Yu H. X., Gu M. H., Sun S. S., Liu Q. Q. (2009) Characterization of expression of the OsPUL gene encoding a pullulanase-type debranching enzyme during seed development and germination in rice. Plant Physiol. Biochem. 47, 351–358 [DOI] [PubMed] [Google Scholar]
- 33. Morinaga K., Honda E., Morohashi Y., Matsushima H. (1997) Pullulanase in mung bean cotyledons. Purification, some properties and developmental pattern during and following germination. Physiol. Plant 101, 519–525 [Google Scholar]
- 34. Yamada J. (1981) Purification of oat debranching enzyme and occurrence of inactive debranching enzyme in cereals. Agric. Biol. Chem. 45, 1013–1015 [Google Scholar]
- 35. Dunn G., Manners D. J. (1975) Limit dextrinases from ungerminated oats (Avena sativa L.) and ungerminated rice (Oryza sativa L.). Carbohydr. Res. 39, 283–293 [DOI] [PubMed] [Google Scholar]
- 36. Zhu Z. P., Hylton C. M., Rossner U., Smith A. M. (1998) Characterization of starch-debranching enzymes in pea embryos. Plant Physiol. 118, 581–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ludwig I., Ziegler P., Beck E. (1984) Purification and properties of spinach leaf debranching enzyme. Plant Physiol. 74, 856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li B., Servaites J. C., Geiger D. R. (1992) Characterization and subcellular localization of debranching enzyme and endoamylase from leaves of sugar beet. Plant Physiol. 98, 1277–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Repellin A., Båga M., Chibbar R. N. (2008) In vitro pullulanase activity of wheat (Triticum aestivum L.) limit-dextrinase type starch debranching enzyme is modulated by redox conditions. J. Cereal Sci. 47, 302–309 [Google Scholar]
- 40. MacGregor A. W., Macri L. J., Bazin S. L., Sadler G. W. (1995) Limit dextrinase inhibitor in barley and malt and its possible role in malting and brewing. Proceedings of 25th Congress, European Brewery Convention, Brussels, pp. 185–192, Oxford University Press, Oxford, UK [Google Scholar]
- 41. Payan F. (2004) Structural basis for the inhibition of mammalian and insect α-amylases by plant protein inhibitors. Biochim. Biophys. Acta 1696, 171–180 [DOI] [PubMed] [Google Scholar]
- 42. Strobl S., Mühlhahn P., Bernstein R., Wiltscheck R., Maskos K., Wunderlich M., Huber R., Glockshuber R., Holak T. A. (1995) Determination of the 3-dimensional structure of the bifunctional α-amylase/trypsin inhibitor from ragi seeds by NMR-spectroscopy. Biochemistry 34, 8281–8293 [DOI] [PubMed] [Google Scholar]
- 43. Gourinath S., Alam N., Srinivasan A., Betzel C., Singh T. P. (2000) Structure of the bifunctional inhibitor of trypsin and α-amylase from ragi seeds at 2.2 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 56, 287–293 [DOI] [PubMed] [Google Scholar]
- 44. MacGregor E. A., Bazin S. L., Ens E. W., Lahnstein J., Macri L. J., Shirley N. J., MacGregor A. W. (2000) Structural models of limit dextrinase inhibitors from barley. J. Cereal Sci. 31, 79–90 [Google Scholar]
- 45. Stahl Y., Alexander R. D., Coates S., Bryce J. H., Jenkinson H. R., Morris P. C. (2007) The barley limit dextrinase inhibitor: Gene expression, protein location and interaction with 14-3-3 protein. Plant Sci. 172, 452–461 [Google Scholar]
- 46. Stites W. (1997) Protein-protein interactions: Interface structure, binding thermodynamics, and mutational analysis. Chem. Rev. 97, 1233–1250 [DOI] [PubMed] [Google Scholar]
- 47. Oneda H., Lee S., Inouye K. (2004) Inhibitory effect of 0.19 α-amylase inhibitor from wheat kernel on the activity of porcine pancreas α-amylase and its thermal stability. J. Biochem. 135, 421–427 [DOI] [PubMed] [Google Scholar]
- 48. Sheinerman F. B., Norel R., Honig B. (2000) Electrostatic aspects of protein-protein interactions. Curr. Opin. Struct. Biol. 10, 153–159 [DOI] [PubMed] [Google Scholar]
- 49. Schroeder S., MacGregor A. (1998) Synthesis of limit dextrinase in germinated barley kernels and aleurone tissues. J. Am. Soc. Brew. Chem. 56, 32–37 [Google Scholar]
- 50. Finnie C., Andersen B., Shahpiri A., Svensson B. (2011) Proteomes of the barley aleurone layer: A model system for plant signalling and protein secretion. Proteomics 11, 1595–1605 [DOI] [PubMed] [Google Scholar]
- 51. Longstaff M. A., Bryce J. H. (1993) Development of limit dextrinase in germinated barley (Hordeum vulgare L.): Evidence of proteolytic activation. Plant Physiol. 101, 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Østergaard O., Finnie C., Laugesen S., Roepstorff P., Svennson B. (2004) Proteome analysis of barley seeds: Identification of major proteins from two-dimensional gels (pl 4–7). Proteomics 4, 2437–2447 [DOI] [PubMed] [Google Scholar]
- 53. Carbonero P., García-Olmedo F. (1999) A multigene family of trypsin/α-amylase inhibitors from cereals. Seed Proteins, 1st Ed., pp. 617–633, Springer, Dordrecht, The Netherlands [Google Scholar]
- 54. Weiss M. (2001) Global indicators of x-ray data quality. J. Appl. Crystallogr. 34, 130–135 [Google Scholar]
- 55. Behnke C. A., Yee V. C., Trong I. L., Pedersen L. C., Stenkamp R. E., Kim S. S., Reeck G. R., Teller D. C. (1998) Structural determinants of the bifunctional corn Hageman factor inhibitor: X-ray crystal structure at 1.95 angstrom resolution. Biochemistry 37, 15277–15288 [DOI] [PubMed] [Google Scholar]
- 56. Oda Y., Matsunaga T., Fukuyama K., Miyazaki T., Morimoto T. (1997) Tertiary and quaternary structures of 0.19 α-amylase inhibitor from wheat kernel determined by X-ray analysis at 2.06 angstrom resolution. Biochemistry 36, 13503–13511 [DOI] [PubMed] [Google Scholar]
- 57. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]