

Background: CD47-SIRPα immunoregulatory pathway is an attractive clinical target.
Results: “Velcro” engineering enabled the creation of high affinity CD47 ectodomain that exquisitely targets myeloid cells to potentiate antibody-dependent cellular phagocytosis.
Conclusion: “Velcro” engineering is a powerful tool to enhance protein-protein interactions; Velcro-CD47 could be an adjuvant for cancer immunotherapy.
Significance: This study demonstrates a new general affinity maturation approach, which led to an appealing preclinical immunotherapeutic lead.
Keywords: cancer therapy, directed evolution, immunotherapy, phagocytosis, protein engineering, CD47, SIRPα, don't-eat-me signal
Abstract
CD47 is a cell surface protein that transmits an anti-phagocytic signal, known as the “don't-eat-me” signal, to macrophages upon engaging its receptor signal regulatory protein α (SIRPα). Molecules that antagonize the CD47-SIRPα interaction by binding to CD47, such as anti-CD47 antibodies and the engineered SIRPα variant CV1, have been shown to facilitate macrophage-mediated anti-tumor responses. However, these strategies targeting CD47 are handicapped by large antigen sinks in vivo and indiscriminate cell binding due to ubiquitous expression of CD47. These factors reduce bioavailability and increase the risk of toxicity. Here, we present an alternative strategy to antagonize the CD47-SIRPα pathway by engineering high affinity CD47 variants that target SIRPα, which has restricted tissue expression. CD47 proved to be refractive to conventional affinity maturation techniques targeting its binding interface with SIRPα. Therefore, we developed a novel engineering approach, whereby we augmented the existing contact interface via N-terminal peptide extension, coined “Velcro” engineering. The high affinity variant (Velcro-CD47) bound to the two most prominent human SIRPα alleles with greatly increased affinity relative to wild-type CD47 and potently antagonized CD47 binding to SIRPα on human macrophages. Velcro-CD47 synergizes with tumor-specific monoclonal antibodies to enhance macrophage phagocytosis of tumor cells in vitro, with similar potency as CV1. Finally, Velcro-CD47 interacts specifically with a subset of myeloid-derived cells in human blood, whereas CV1 binds all myeloid, lymphoid, and erythroid populations interrogated. This is consistent with the restricted expression of SIRPα compared with CD47. Herein, we have demonstrated that “Velcro” engineering is a powerful protein-engineering tool with potential applications to other systems and that Velcro-CD47 could be an alternative adjuvant to CD47-targeting agents for cancer immunotherapy.
Introduction
Macrophages are constantly faced with a “to-eat-or-not-to-eat” decision in maintaining tissue homeostasis, guarding against pathogens, and in some cases contributing to pathologic developments (1). For instance, tumor-associated macrophages infiltrate solid tumors, reside in the immunosuppressed microenvironment, and support tumor progression (2). Recent studies have demonstrated the therapeutic potential of harnessing the phagocytic ability of macrophages to combat cancer by targeting cell surface receptors on either macrophages or tumor cells (3–8).
One promising target is CD47, known as a “don't-eat-me” signal or a “marker-of-self” (4, 9–14). CD47 is a 50-kDa membrane glycoprotein expressed on the surface of virtually all cells in the body. It contains an N-terminal V-type immunoglobulin superfamily (IgSF)2 ectodomain (ECD), followed by five transmembrane helices and a short cytoplasmic tail (15). CD47 inhibits macrophage phagocytosis by engaging the receptor SIRPα (signal regulatory protein α). SIRPα belongs to the signal regulatory protein family and is expressed only on myeloid and neuronal cells (16). SIRPα is a highly polymorphic (17) transmembrane protein with three N-terminal extracellular IgSF domains and a long C-terminal intracellular domain consisting of two putative immunoreceptor tyrosine-based inhibition motifs (9, 18). The x-ray crystal structure of a complex of SIRPα domain 1 and CD47 ECD revealed an unexpected binding mode between the IgSF domains of the two proteins (19). The N-terminal IgSF-domain of SIRPα interacts with the CD47 IgSF domain via its complementarity determining region-like loops, resembling an antibody-antigen recognition mode. In contrast, the residues on CD47 that contact SIRPα are located on one face of the CD47 AGFCC′C″ β-sheet (19). A notable interaction hot spot residue is the N-terminal pyroglutamate, which is spontaneously cyclized from a glutamine residue upon the cleavage of the CD47 signal sequence (19). CD47 binding to SIRPα prompts the recruitment of SHP-1 and SHP-2 phosphatases (20), which in turn result in myosin-IIA dephosphorylation and inhibition of phagocytosis (10).
Although all normal cells express anti-phagocytic signals such as CD47 to maintain tissue homeostasis, cancer cells up-regulate CD47 as a means to evade destruction by the immune system (9, 13). There has been recent interest in blocking CD47-SIRPα interaction to restore macrophages to a pro-phagocytic state in cancer. For instance, the anti-CD47 monoclonal antibody (mAb) B6H12 blocks SIRPα binding to CD47, and it has shown pre-clinical efficacy in several hematologic malignancies and solid tumor models (4–7, 21). In addition, we have previously engineered a high affinity soluble variant of SIRPα IgSF domain 1, termed CV1 (22). CV1 exhibits similar efficacy as the anti-CD47 antibody in several cancer model systems, but with potentially enhanced tissue penetrance and decreased toxicity by the virtue of being a small monomeric protein (∼14 kDa) that lacks fragment crystallizable (Fc) effector functions. Both strategies exhibit remarkable synergy with existing tumor-specific mAbs to enhance macrophage-mediated, antibody-dependent cellular phagocytosis (ADCP) of tumor cells. However, the broad expression of CD47 causes both strategies to have a large antigen sink that reduces bioavailability and increases the potential for on-target toxicity to normal cells.
An alternative to targeting CD47 is to target SIRPα. Indeed, antagonistic anti-SIRPα antibodies that significantly enhance in vitro antibody-mediated killings of tumor cells by phagocytes have been generated to circumvent the potential issues associated with a large antigen sink (12). One potential limitation is that an antibody has poor tissue penetration into solid tumors due to its large size (23), and tissue penetration in the case of targeting SIRPα expressed on tumor-infiltrated macrophages is critical for therapeutic efficacy. Perhaps a smaller version of an anti-SIRPα blocking agent could have benefits in this regard.
In this study, we aimed to engineer a soluble high affinity variant of human CD47 ECD that binds human SIRPα to turn off the don't-eat-me signal and thereby promote tumor clearance by macrophages. Blocking SIRPα targets a much more defined cell population than blocking CD47. In addition, compared with anti-SIRPα antibodies (12), an engineered CD47-ECD may exhibit superior tissue penetrance, utilize the natural CD47-SIRPα-binding site so that resistance mechanisms are difficult to evolve, and be suitable for further chemical manipulation in imaging applications. To this end, we have developed a novel protein-engineering technique, coined “Velcro” engineering, which increases affinity of receptor-ligand interactions by extending an existing contact interface via peptide extension at the N terminus. This approach should be quite general for affinity maturation of receptor-ligand interactions that are targets for therapeutic development.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human SIRPα allele 1 domain 1 (a1d1), allele 2 domain 1 (a2d1), and CV1 were expressed as described previously (22). Briefly, SIRPα variants were cloned into a modified pMal-p2X expression vector (New England Biolabs), containing a 3C protease cleavage site (LEVLF(Q/G)P) after the maltose-binding protein tag and a C-terminal His8 tag, and were expressed in the periplasm of BL-21(DE3) Escherichia coli. Cells were induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside and incubated with shaking at room temperature overnight. Maltose-binding protein-fusion protein was purified by nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen) and then digested with 3C protease (produced in house) at 4 °C overnight to remove the maltose-binding protein tag. SIRPα proteins were further purified by Ni-NTA affinity chromatography, followed by size exclusion chromatography using a Superdex-75 column (GE Healthcare).
Human CD47 IgSF domain (residues 1–117) and high affinity Velcro-CD47 variants, both with a C15G mutation (19) and C-terminal His8 tag, were cloned into pAcGP67a using Gibson assembly (24) to remove the N-terminal scar from restriction enzyme digestion of the plasmid, ensuring a free N terminus. The plasmids were transfected into Trichoplusia ni (High Five) cells (Invitrogen) using the BaculoGold baculovirus expression system (BD Biosciences) for secretion and purified by Ni-NTA and size exclusion chromatography with a Superdex-75 column.
Biotinylated CD47 and SIRPα variants were expressed with a C-terminal biotin acceptor peptide tag (GLNDIFEAQKIEWHE) and purified as described above. The purified proteins were biotinylated in vitro with BirA ligase and then re-purified from the reaction mixture by size exclusion chromatography.
For profiling human peripheral blood, CV1 A17C and N3612 F14C were expressed and purified as described above to allow site-specific conjugation via maleimide linking chemistry. The proteins were conjugated to Alexa Fluorophore 647 (A647) maleimide (Life Technologies, Inc.) according to the manufacturer's protocol and re-purified from the reaction mixture by size exclusion chromatography.
For in vitro phagocytosis assays, endotoxin was removed using Triton X-114 as described previously (22), and endotoxin removal was confirmed using the ToxinSensor Chromogenic LAL endotoxin assay kit (Genscript).
Yeast Display and Construction of the CD47 Extension Library
The human CD47 IgSF domain, with a C15G mutation (25), was displayed on the surface of Saccharomyces cerevisiae strain EBY100 as an N-terminal fusion to Aga2 using the pYAL vector (26), leaving a free N terminus.
To construct the CD47 extension library, the mutagenized CD47 DNA constructs from N3L0, N3L2, and N3L4 molecule designs were mixed and combined with linearized pYAL vector and EBY100 yeast. The N3L0 molecule design extends the N terminus by three additional residues and randomizes Gln-1, Leu-3, Gly-52, Ala-53, and Leu-54. The N3L2 molecule design extends the N terminus by three additional residues, extends the FG loop region by two additional residues, and randomizes Gln-1, Leu-3, Gly-52, Ala-53, and Leu-54. The N3L4 molecule design extends the N terminus by three additional residues, extends the FG loop region by four additional residues, and randomizes Gln-1, Leu-3, Gly-52, Ala-53, and Leu-54.
The NNK codon was used at all of the positions randomized and/or extended. Electroporation, rescue, and expansion of the yeast library were performed as described previously (27). Final library contained ∼3 × 108 yeast transformants.
Selection of the CD47 Extension Library
The selections of the yeast library were performed as described previously with some modifications (22). Briefly, the initial selections (rounds 1–3) were conducted using a magnetically activated cell sorter (MACS). For round 1, 1.0 × 109 cells were selected with paramagnetic streptavidin microbeads (Miltenyi Biotec) that were pre-coated with 400 nm biotinylated SIRPα a2d1. For rounds 2 and 3, 1.0 × 108 yeasts were stained and selected with 100 nm monomeric biotinylated SIRPα a2d1. To normalize apparent affinity by protein expression on the cell surface, library selection was performed using two-color fluorescence activated cell sorting (FACS) in the subsequent rounds. After incubation with target protein during each subsequent round, the yeast library was washed twice and co-labeled with streptavidin-Alexa Fluor 647 (SIRPα binding) and anti-c-Myc-Alexa Fluor 488 (CD47 variant expression) for 10 min at 4 °C. Alexa647+Alexa488+ yeast were purified using a FACS Jazz cell sorter (BD Biosciences). For round 4, the yeast library was stained using 10 nm monomeric biotinylated SIRPα a2d1 for 1 h at 4 °C. For round 5, the yeast library was incubated with 200 nm biotinylated SIRPα a1d1 for 1 h at room temperature, washed twice with PBE (PBS, pH 7.4, + 0.5% (w/v) BSA + 2 mm EDTA, pH 8.0), and competed with 1 μm unbiotinylated SIRPα a1d1 for 4 h at room temperature. For round 6, the yeast library was stained with 1 nm SIRPα a1d1 for 1 h at 4 °C.
After the sixth round of selection, 100 μl of the yeast library, which was expanded in SD-CAA medium overnight, was collected to extract library DNA using the ZymoprepTM Yeast Plasmid Miniprep II kit (Zymo Research), according to the manufacturer's instructions. The extracted DNA was transformed into DH5α E. coli and plated to sequence individual colonies.
Modeling SIRPα-N3612 Complex
The N-terminal extension (“Velcro”) of N3612 was built onto the structure of the SIRPα-CD47 complex downloaded from the RCSB Protein Data Bank (Protein Data Bank code 2JJS) (19) using PyMOL Version 1.7.0.5 (Schrödinger, LLC) and Coot (28). The structure was then energy-minimized for 1000 cycles using the crystallography and NMR system (CNS) (29, 30).
Surface Plasmon Resonance
The binding affinity and kinetics were measured using a BIAcore T100 (GE Healthcare). Protein concentrations were quantified by 280 nm absorbance using a Nanodrop2000 spectrometer (Thermo Scientific). Biotinylated SIRPα a1d1 and a2d1 was immobilized on a BIAcore SA sensor chip (GE Healthcare) in different flow channels with an Rmax ∼150 and ∼130 response units, respectively. An unrelated biotinylated protein was captured in another flow channel with a similar response unit value to control for nonspecific binding. Experiments were carried out at 25 °C, and measurements were made with serial dilutions of CD47 variants in HBS-P+ buffer (GE Healthcare) supplemented with 0.1% (w/v) BSA. All data were analyzed with the BIAcore T100 evaluation software version 2.0 with a 1:1 Langmuir binding model.
Cell-based SIRPα Blocking Assay
Biotinylated wild-type CD47 was incubated with A647-conjugated streptavidin for 15 min at room temperature to form CD47 tetramers. Labeled human (for human cells) or mouse (for mouse cells) CD47 tetramers at 100 nm were combined with serial dilutions of unlabeled CD47 variants and simultaneously added to stain undifferentiated THP-1, human macrophages, or NSG (NOD scid γ) splenocytes. Cells were incubated for 1 h at 4 °C and then washed with PBE to remove unbound proteins. Samples were analyzed on an Accuri C6 flow cytometer (BD Biosciences). Data represent the mean fluorescence intensity normalized to maximal binding for each class of reagents, and points were fit to sigmoidal dose-response curves using Prism 5 (GraphPad).
In Vitro Macrophage Phagocytosis
Macrophage derivation and phagocytosis experiments were conducted as described previously (22). Briefly, human macrophages were isolated from leukocyte reduction system chambers (Stanford Blood Center) using anti-CD14 whole blood magnetic beads (Miltenyi Biotec) and purified with autoMACS Pro Separator (Miltenyi Biotec). The cells were cultured in Iscove's modified Dulbecco's medium + GlutaMAX (Life Technologies, Inc.) supplemented with 10% human antibody serum (Invitrogen) for 7–10 days. Assays were performed by incubation of macrophages with GFP+ tumor cells at a 1:2 ratio in serum-free medium at 37 °C for 2 h. Phagocytosis was analyzed using an LSRFortessa cell analyzer with a high throughput sampler (BD Biosciences) and evaluated as the percentage of GFP+ macrophages using FlowJo Version 9.4.10 (Tree Star, Inc.). The data were normalized to the maximal response by each independent donor against each cell line and were fit to sigmoidal dose-response curves using Prism5 (GraphPad).
Human Immune Cell Profiling
The CD47 and SIRPα expression profile on various blood cell populations was determined by multicolor flow cytometry. A human buffy coat was obtained from Stanford Blood Center and treated with ACK lysing buffer (Life Technologies, Inc.) to remove red blood cells (RBCs). Cells (1 × 106/well, 100 μl) were stained with markers for B cells (CD19+CD20+), CD4-T cells (CD3+CD4+), CD8-T cells (CD3+CD8+), NK cells (CD3−CD14−CD19−CD16+CD56+), monocytes (FSChiCD14+), neutrophils (SSChiCCR3−CD16+), eosinophils (SSChiCCR3+CD16−), and basophils (CD3−CD14−CD19−CD56−SSCloCD123hiCCR3+) in PBS containing 2% human serum and 0.1 mg/ml mouse serum IgG as described previously (31). RBCs (CD235a+) were stained directly from freshly isolated heparinized human whole blood (Stanford Blood Center). Dead cells were excluded by propidium iodide (Life Technologies, Inc.) staining, and single cells were selected based on SSC and FSC properties. The fluorescence minus one method was used to determine the threshold of negative versus positive staining. The samples were co-stained with anti-CD47 (high affinity SIRPα CV1) or anti-SIRPα (high affinity CD47 N3612, 15-414, REA144, SE5A5) agents. CV1-A647 and N3612-A647 were used at 250 nm. Anti-SIRPα clone 15-414 (eBioscience), clone REA144 (Miltenyi), and clone SE5A5 (BioLegend) were used at 1:50 dilution. All analyses were performed on LSRFortessa cell analyzer with high throughput sampler (BD Biosciences), and data were analyzed with FlowJo software (Tree Star, Inc.).
RESULTS
“Velcro” Engineering of Human CD47 to Generate High Affinity CD47 Variants
The use of wild-type (WT) human CD47-ECD as a human SIRPα antagonist is limited by the relatively low affinity interaction (∼0.5 μm) between CD47 and SIRPα (Fig. 5B) (19). To address this limitation, we used in vitro evolution via yeast surface display (27) to increase the affinity of CD47 for SIRPα, thereby increasing the potency of CD47 as a SIRPα antagonist to modulate macrophage recognition and elimination of cancer cells (Fig. 1A). WT CD47-ECD has a free, unpaired cysteine at position 15 that may contribute to undesirable disulfide aggregates in solution or when displayed on the surface of yeast. Therefore, we mutated Cys-15 to Gly (19), for all experiments described herein, and refer to CD47 C15G as WT CD47 for simplicity. Functional WT CD47-ECD was displayed on the surface of S. cerevisiae as an N-terminal fusion to Aga2, leaving a free N terminus to ensure the formation of the required pyroglutamate residue (data not shown). Several mutant libraries were subsequently created by structure-guided, site-directed mutagenesis of the contact interface as well as hydrophobic residues that directly support the contact interface (Fig. 2A). Successive rounds of selections with increasing stringency (Fig. 2B) resulted in the convergence of the libraries to only three unique clones (Fig. 2C). However, affinity estimation via on-yeast binding titration curves indicate that these CD47 variants had the same EC50 values as WT CD47 but with a significantly higher level of maximal binding to SIRPα on yeast (Fig. 2D). This suggests that the selection process had enriched for clones with increased and/or more stable yeast surface expression rather than higher affinity for SIRPα. Two additional libraries were created using a more conservative “soft” randomization-directed mutagenesis approach or a random mutagenesis by error-prone PCR. However, neither approach resulted in CD47 variants with high affinity binding to SIRPα (data not shown). We speculate that this may be because SIRPα has a rather recessed binding site that accommodates the bullet-like tip of CD47, rendering CD47 a suboptimal interface for engineering. Moreover, CD47 binds to SIRPα with structurally rigid β-sheets that may be less amenable to affinity modulation than loops protruding from the tips of the sheets, as seen in SIRPα.
FIGURE 5.

Biophysical characterization of Velcro-CD47. A, amino acid sequence alignment of known human SIRPα-binding domain alleles, showing that there are only two variations, a1d1 or a2d1, at the CD47-contact interface. Red text marks the amino acid sequence of V1 (a2d1), the most prominent human SIRPα allele, and blue text marks that of V2 (a1d1), the second most prominent human SIRPα allele. The black boxes indicate the residues that interact with CD47, and the pale orange shade indicates residues that are different from the V1 sequence. B, representative surface plasmon resonance sensorgrams of WT CD47 and high affinity variant N3612 binding to immobilized SIRPα a1d1 (upper panel) and a2d1 (lower panel). RU, response unit. C, dose-response curves of SIRPα antagonism on panel i, undifferentiated THP-1; panel ii, primary human macrophages; and panel iii, NSG splenocytes with WT human CD47 (blue) and high affinity CD47 variant N3612 (red). Cells were stained with titrating concentrations of SIRPα blocking agents in competition with 100 nm Alexa Fluor 647-conjugated WT human CD47 tetramer for THP-1 cells and human macrophages or WT mouse CD47 tetramer for NSG splenocytes. Data represent mean fluorescence intensity normalized to maximal binding for each cell type ± S.E. of triplicate wells. Data are representative of two independent experiments.
FIGURE 1.
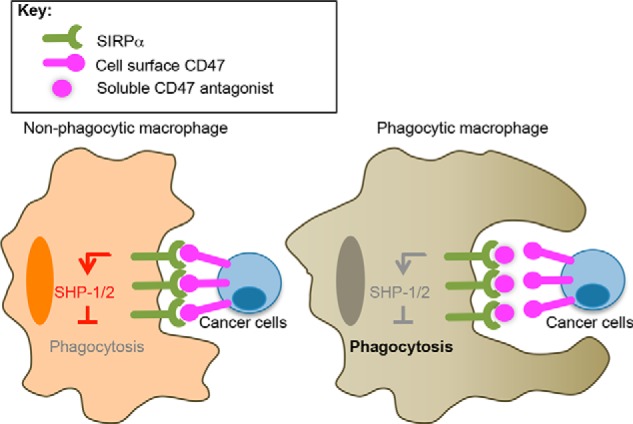
Potentiation of macrophage phagocytosis of tumor cells by SIRPα blockade. A schematic of SIRPα blockade by soluble high affinity CD47 is shown. Left, CD47 expression on cancer cells activates SIRPα on macrophages, recruiting SHP-1 and -2 tyrosine phosphatases and preventing macrophage phagocytosis of cancer cells. Right, soluble high affinity CD47 prevents CD47 on the cancer cell surface from engaging SIRPα on macrophages, thereby acting as a competitive antagonist.
FIGURE 2.

Directed evolution of CD47 using classical library creation strategies. A, CD47 library designs. Left, crystal structure of SIRPα (green) and CD47 (magenta) is shown in cartoon representation. Amino acid positions randomized in the libraries are highlighted and shown in sphere representation. CD47 is divided into three different patches as follows: core (cyan), contact region 1 (gray), and contact region 2 (orange). Randomizing combinations of these three patches created multiple CD47 libraries that were selected independently for the first three rounds but mixed to select as a single library on the last round of selection. Middle right, tables of randomized positions with possible amino acid variations and the corresponding degenerate DNA codons (noted in the parentheses). a.a., amino acid. B, schematics of the criteria at each round of CD47 library selections. MACS, magnetic activated cell sorting; FACS, fluorescence activated cell sorting; and SA, streptavidin. C, summary of sequences of selected CD47 variants. The position of mutated residues and their corresponding sequence in WT is denoted at the top of the table. D, binding titration curves of SIRPα a2d1 on yeast expressing WT CD47 (black) or selected variant A9 (yellow), B4 (blue), or C1 (green). Cells were stained with serial dilutions of biotinylated SIRPα a2d1 and analyzed by flow cytometry. Data represent raw (top) or normalized (bottom) mean fluorescence intensity ± S.E. Data are representative of two independent experiments. a.u., arbitrary units.
The lack of success using traditional interface targeting engineering methods is a rather common problem in evolving protein-protein interactions and can be attributed to factors such as interface shape and the nature of the contact residues within the binding interface, as well as other unknown factors. Recent work by Chen et al. (32) has shown a positive correlation between binding affinity and the buried surface area at protein-protein interfaces. We therefore explored the possibility of “artificially” increasing the buried surface area between CD47 and SIRPα as a means of increasing affinity. We applied two methods to increase the interaction surface by inspection of the CD47-SIRPα co-crystal structure (19). One approach, which we term “Velcro” engineering, was to extend the N terminus of CD47, which may thread itself through a tunnel-like depression formed by residues from Tyr-50 to Thr-67 on SIRPα (Fig. 3A, black arrow). The other approach was to insert additional residues to extend the FG loop region at position 52, which may allow CD47 to reach up and form additional contacts with SIRPα (Fig. 3A, red arrow). This method resembles antibody complementarity-determining region-loop engineering to gain affinity (33, 34).
FIGURE 3.

“Velcro” engineering of human CD47. A, schematic of strategy to increase CD47 affinity for SIRPα by N-terminal and loop extension. The co-crystal structure of SIRPα (green in surface representation) and CD47 (magenta in cartoon representation) is highlighted with the N-terminal pyroglutamate (blue) and the loop at positions 52–54 (cyan) regions in sphere representation. The arrows represent the direction of intended amino acid extension. B, extension library design. Residues colored in pale orange are native to CD47 and are included in randomization. Residues colored in blue indicate the three additional amino acids that were added for N-terminal extension, and residues colored in cyan represent the 0, 2, or 4 additional amino acids that were added for loop extension. NxLy represents the types of molecule present in the extension library, where x and y indicate the number of amino acid extensions at the N terminus and at the FG loop region, respectively. The randomized DNA of N3L0, N3L2, and N3L4 was made separately and then mixed to transform a single yeast library. AA, amino acid.
To construct the extension library, we added three amino acids to the N terminus of CD47 and either 0, 2, or 4 amino acids to the tip of the CD47 FG loop. The additional residues were randomized via degenerate NNK codons, allowing all 20 amino acids to be sampled at each position. In addition, we randomized Gln-1, Leu-3, Gly-52, Ala-53, and Leu-54 via degenerate NNK codons as these residues are in close proximity to the CD47-SIRPα binding interface and regions where the various amino acid extensions were installed (Fig. 3B).
The resulting library was subjected to six rounds of selection using the two most prominent human SIRPα alleles (17), allele 1 domain 1 (a1d1) and allele 2 domain 1 (a2d1) as bait (Fig. 4A). With each successive round of selection, we decreased the bait concentration to increase selection stringency and alternated the SIRPα allele used for selections to evolve a CD47 variant that cross-reacts with both alleles. In the final rounds of selection, we used a competition-based selection strategy (35), where rebinding of labeled SIRPα was prevented by the addition of a large excess of “cold” or unlabeled SIRPα to enrich for variants with slow dissociation rates. This selection strategy resulted in a progressive increase in binding affinity and dissociation half-life for both SIRPα alleles (Fig. 4B). Twenty four individual clones were sequenced after the last round of selection, and 17 unique CD47 sequences were identified (Fig. 4C). The amino acid sequence was numbered such that the three additional residues at the N terminus were labeled −3, −2, and −1 for the first, second, and third residues, respectively, and the native N terminus remained position number 1.
FIGURE 4.

Affinity evolution of CD47 extension library. A, schematics of the selection criteria at each round of the extension library selections. MACS, magnetic activated cell sorting; FACS, fluorescence-activated cell sorting; and SA, streptavidin. B, histogram overlays assessing SIRPα a1d1 (left) and a2d1 (right) staining of the library at each round. C, summary of sequences of engineered CD47 variants. The position of mutated residues and their corresponding sequence in WT is denoted at the top of the table. Positions −3, −2, and −1 denote the “Velcro” residues added to the N terminus. Red text indicates the consensus mutations. D, model of SIRPα-N3612 complex. The structure was modeled using the WT complex structure with PyMOL and Coot and then energy-minimized for 1000 cycles using crystallography and NMR system. SIRPα (green) is shown in surface representation. CD47 (magenta) is shown in ribbon representation, and the mutations (cyan) present in N3612, including the “Velcro”, are shown in stick representation.
Although the library was still diverse after six rounds of selection, an emerging consensus sequence was apparent. Tryptophan and glutamine residues are absolutely conserved at positions −3 and −2, respectively. Furthermore, there is at least one proline residue at positions −1 or 1. We speculate that this proline residue serves as a molecular kink that stabilizes the “Velcro” peptide in the groove on SIRPα and/or as a structural mimetic for the pyroglutamate (36, 37) that is necessary for the CD47-SIRPα interaction. Moreover, we found that the internal loop region preferred its original length and that it is indifferent to mutations because no clear consensus sequence emerged (Fig. 4C). These findings suggest that the gain in affinity is a result of the N-terminal extension, and we coined this approach “Velcro” engineering because the three extended amino acids at the N terminus presumably act like a hook, which we have modeled extending into the cleft adjacent to Glu-54 of SIRPα (Fig. 4D). Unfortunately we have not been able to crystallize the “Velcro” complex, but it is plausible that the N-terminal extension would simply fill the hydrophobic cavity immediately adjacent to the CD47 N terminus.
Velcro-CD47 Binds with High Affinity to Human SIRPα Alleles
Although SIRPα is highly polymorphic (17), amino acid sequence alignment of the known human SIRPα alleles shows that there are only two unique sequences, represented by alleles a1d1 and a2d1 at the CD47 binding interface (Fig. 5A). WT CD47-ECD binds with similar affinities to all known human SIRPα alleles (25). Therefore, we expect that an engineered CD47 variant that binds with high affinity to both SIRPα a1d1 and a2d1 will also bind with high affinity to all other human SIRPα alleles, as long as the WT binding interface is preserved. To assess the enhancement of binding affinity of the Velcro-CD47s compared with WT CD47, we used surface plasmon resonance to measure their differential binding properties for human SIRPα a1d1 and a2d1. Biotinylated SIRPα alleles were immobilized on the surface of a sensor chip, and soluble CD47 variants were passed over as analytes. Consistent with previous reports, WT CD47 binds SIRPα a1d1 and a2d1 with an affinity of 290 and 480 nm, respectively (Fig. 5B) (19, 25). In contrast, a representative high affinity CD47 variant N3612 binds SIRPα a1d1 and a2d1 with an affinity of 370 pm and 2.5 nm, respectively, corresponding to a 200–800-fold increase in affinity for both alleles (Fig. 5B). The increase in affinity is primarily due to the significantly slower off-rate of N3612 (Table 1), as expected based on the selection strategy used.
TABLE 1.
Binding constants determined by surface plasmon resonance
| SIRPα allele | CD47 | kON | kOFF | KD | t½ |
|---|---|---|---|---|---|
| m−1 s−1 | s−1 | m | min | ||
| a1d1 | WT | 6.1 × 105 | 3.7 × 10−1 | 2.9 × 10−7 | 0.031 |
| N3612 | 1.5 × 105 | 5.7 × 10−4 | 3.7 × 10−10 | 20 | |
| a2d1 | WT | 3.0 × 105 | 4.8 × 10−1 | 4.8 × 10−7 | 0.024 |
| N3612 | 1.3 × 106 | 3.2 × 10−3 | 2.5 × 10−9 | 3.6 |
Velcro-CD47 Potently Antagonizes the SIRPα-CD47 Interaction on Human SIRPα- but Not Mouse SIRPα-expressing Cells
We evaluated the ability of soluble high affinity CD47-ECD variants to antagonize cell surface SIRPα using an in vitro competition assay. Monomeric N3612 potently inhibits tetrameric WT human CD47 binding to human SIRPα expressed on the surface of undifferentiated human THP-1 leukemia cells and primary human macrophages, with an IC50 of ∼12 and 65 nm, respectively (Fig. 5C, panels i and ii). In contrast, monomeric WT human CD47 is a weak antagonist of the SIRPα-CD47 axis (Fig. 5C, panels i and ii), as expected given the low affinity of WT human CD47 for SIRPα. This indicates that affinity enhancement through “Velcro” engineering is a viable strategy to engineer CD47-based antagonists of cell surface SIRPα.
We also determined the ability of WT human CD47 and N3612 to antagonize the mouse SIPRα-CD47 interaction using splenocytes prepared from NSG (NOD scid γ) mice. NSG mice express the same SIPRα allele as the NOD mouse, which is known to support the engraftment of human hematopoietic stem cells due to enhanced binding to human CD47 compared with other mouse strains (17). Although both WT human CD47 and N3612 antagonize the mouse SIPRα-CD47 interaction, the potency of N3612 is substantially less than that of WT CD47, with an IC50 of ∼ 3 μm and 40 nm, respectively (Fig. 5C). Given the weak potency, we do not expect N3612 to have therapeutic activity in mice. Future studies using human SIRPα transgenic mice (38) are necessary to determine the ability of N3612 to potentiate ADCP and tumor clearance in vivo.
Velcro-CD47 Enhances Monoclonal Antibody-mediated ADCP
Next, we assessed the ability of N3612 to potentiate macrophage phagocytosis of tumor cells in vitro in comparison with a high affinity variant of SIRPα, termed CV1, which exhibits this property. Primary human macrophages were incubated with target tumor cells and various combinations of tumor-specific mAbs and CD47 or SIRPα antagonists, and phagocytosis was quantified by flow cytometry (22). Similar to the CD47 antagonist CV1 (22), treatment with the SIPRα antagonist N3612 alone does not induce phagocytosis of cancer cells by macrophages compared with a PBS control (Fig. 6). This is expected given the absence of a pro-phagocytic signal to initiate macrophage phagocytosis of tumor cells, such as an antibody Fc domain (12, 22). Treatment with the anti-tumor mAbs rituximab, trastuzumab, and cetuximab significantly increased macrophage phagocytosis of Raji, SKBR3, and DLD-1 tumor cells, respectively, compared with no treatment or treatment with CD47 or SIRPα antagonists alone (Fig. 6). Both CV1 and N3612 synergize with the mAbs to increase phagocytosis of cancer cells (Fig. 6, A–C), in agreement with previous reports (22). Similar to CV1, N3612 significantly increases the maximal potency of macrophage-mediated ADCP, whereas WT CD47 has little to no effect on ADCP, likely due to its weak affinity for SIRPα (Fig. 6, D and E). Collectively, our data indicate that SIPRα antagonist N3612 is an effective alternative to CD47 antagonists to potentiate ADCP.
FIGURE 6.

High affinity Velcro-CD47 variant enhances mAb ADCP. A–C, phagocytosis of GFP+ Raji lymphoma (A), SKBR3 breast cancer (B), and DLD-1 colon cancer (C) cells by donor-derived human macrophages with or without 10 μg/ml respective tumor-specific mAbs in the presence of PBS (blue), WT CD47 (maroon), N3612 (gray), and CV1 (green). D and E, phagocytosis of GFP+ SKBR3 breast cancer (D) and DLD-1 colon cancer (E) cells with titrating amounts of respective tumor-specific mAbs in the presence of the same protein treatments as in A–C. The proteins were used at 1 μm. A–E, all phagocytosis was performed with human macrophages derived from a minimum of two independent blood donors; data are representative of at least two independent experiments. Data represent the percentage of phagocytosis normalized to the maximal response by each donor for each cell line ± S.E. of duplicate wells. ns, not significant; **, p < 0.01; ***, p < 0.001, by one-way analysis of variance test.
Velcro-CD47 Specifically Targets a Subset of Myeloid-derived Cells in Human Blood
CD47 is expressed on nearly all cell types in the body, whereas SIRPα expression is mainly limited to cells of the myeloid lineage and of the central nervous system. This widespread distribution of CD47 raises potential safety issues with the use of CD47 targeted therapeutics (12). We used ex vivo profiling of human peripheral blood mononuclear cells to predict the in vivo antigen sink and other potential toxicity concerns due to indiscriminate target cell binding. To facilitate the profiling, we reformatted N3612 as a diagnostic agent by installing a cysteine mutation at the loop distal to the SIRPα-binding site, F14C (Fig. 7A). N3612-F14C was site-specifically coupled to A647 using maleimide chemistry and behaved as a monodisperse peak over a Superdex-75 size exclusion column (Fig. 7B). The purified N3612 F14C-A647 was active and bound to both SIRPα alleles with similar affinity as biotinylated N3612 without the F14C mutation (Fig. 7, C and D). We also used a similar approach to conjugate the fluorophore to the high affinity SIRPα variant CV1 (data not shown). Subsequently, cell types of the immune system in the blood were identified using multicolor FACS analysis. After selecting live singlet cells, the gating strategy for lymphoid lineages (Fig. 8A) is as follows. 1) For T cells, the CD3+ fraction was subdivided into CD4+ and CD8+ fractions to differentiate helper and cytotoxic T cells, respectively. 2) CD19+CD20+ marked the B cell subpopulation. 3) Lin−CD16+CD56+, where Lin− markers included CD3, CD14, and CD19, was used to detect NK cells. In addition, we identified FSChiSSCmedCD14+, SSChiCCR3−CD16+, SSChiCCR3+CD16−, and Lin−CCR3+CD123+ as blood monocytes, neutrophils, eosinophils, and basophils, respectively (Fig. 8, B and C). Finally, we used CD235a+ marker to label red blood cells (Fig. 8D). These samples were simultaneously co-stained with CV1-A647, N3612-A647, or three commercially available anti-SIRPα-APC mAbs.
FIGURE 7.

Reformatting high affinity CD47 variant N3612 into a diagnostic agent. A, cartoon representation of x-ray crystallographic structure of the SIRPα (green)-CD47 (magenta) complex. The cysteine mutation (F14C) installed to allow site-specific fluorophore conjugation is highlighted in blue and shown in sphere representation. B, representative size exclusion chromatogram trace of N3612-A647. C and D, titration curves of biotinylated WT CD47 (blue), biotinylated N3612 (pink), and N3612 F14C conjugated to Alexa Fluor 647 (gray) binding to yeast expressing SIRPα a1d1 (C) and yeast expressing SIRPα a2d1 (D). Data represent mean fluorescence intensity normalized to maximal binding for each cell type ± S.E. Data are representative of two independent experiments. a.u., arbitrary units; mAu, milli-absorbance units.
FIGURE 8.

Human cell immunophenotyping and reactivity with CD47/SIRPα antagonists via flow cytometry. A, gating strategy to identify B cell, CD4-T cell, CD8-T cell, and NK cell subpopulations. B cells = CD19+CD20+; CD4 T cells = CD3+CD4+; CD8 T cells = CD3+CD8+; NK cells = CD3−CD14−CD19−CD16+CD56+. B, gating strategy to identify monocytes. Monocytes = FSChiCD14+. C, gating strategy to identify neutrophils, eosinophils, and basophils. Neutrophils = CD3−CD14−CD19−CD56−SSChiCCR3−; eosinophils = CD3−CD14−CD19−CD56−SSChiCCR3+; basophils = CD3−CD14−CD19−CD56−SSCloCD123hiCCR3+. D, gating strategy to identify red blood cells. RBC = CD235a+. E, heat map representation of percentage of human peripheral blood cells positively stained with the indicated agent/clone targeting either SIRPα (N3612, 15-414, SE5A5, and REA144) or CD47 (CV1). FMO, fluorescence minus one. N3612 and CV1 proteins were used at 250 nm, and the anti-SIRPα antibodies were used at 1:50 dilution.
CV1-A647 strongly stained all cell types in the blood, in agreement with the widespread expression of CD47 (Fig. 8E, supplemental Fig. S1, and Table 2). In contrast, N3612-A647 staining profile was consistent with anti-SIPRα antibody clone 15-414. They labeled only monocytes and, to a lesser extent, neutrophils, as expected given the restricted distribution of SIRPα compared with CD47 (Fig. 8E, supplemental Fig. S1, and Table 2). The binding profile of the other anti-SIPRα antibody clones, SE5A5 and REA144, showed higher staining for many different cell subpopulations, likely due to increased levels of non-SIRPα-specific binding (Fig. 8E, supplemental Fig. S1, and Table 2). One notable difference in the anti-CD47 (CV1 and other anti-CD47 antibodies) and the anti-SIRPα (N3612) approaches is that the former targets red blood cells (RBCs) and may induce RBC clearance and associated toxicity. Because of the enhanced specificity and reduced indiscriminate cell binding, we anticipate that the N3612 could be an alternative, less toxic adjuvant to anti-CD47 mAb or CV1 for cancer immunotherapy.
TABLE 2.
Percentage of human immune cells positively stained with indicated agent
| Clone/Target | FMO | N3612/SIRPα | 15-414/SIRPα | SE5A5/SIRPα | REA144/SIRPα | CV1/CD47 | |
|---|---|---|---|---|---|---|---|
| Lymphoid | B cell | 0 | 1.8 | 1.5 | 7.4 | 10 | 100 |
| CD4 T cell | 0 | 2.2 | 0.4 | 6.2 | 7.5 | 100 | |
| CD8 T cell | 0 | 2.8 | 2.1 | 5.9 | 5.9 | 100 | |
| NK cell | 0 | 0 | 9.3 | 2.0 | 2.4 | 98 | |
| Erythroid | RBC | 0 | 0.07 | 0.1 | 0.06 | 0.1 | 100 |
| Myeloid | Basophil | 0.5 | 12 | 18 | 22 | 28 | 100 |
| Eosinophil | 0 | 4.6 | 5.8 | 95 | 95 | 97 | |
| Neutrophil | 0.02 | 23 | 21 | 100 | 100 | 100 | |
| Monocyte | 0 | 100 | 99 | 100 | 100 | 100 |
DISCUSSION
The CD47-SIRPα immunoregulatory axis is increasingly recognized as an attractive clinical target for cancer immunotherapy (8, 21, 39). Current approaches to antagonize the CD47-SIRPα interaction have principally targeted CD47 (4, 5, 22), which is widely distributed, raising concerns with bioavailability and potential on-target toxicity to normal cells. Alternative agents such as anti-SIRPα antibodies have also been developed and show promise as substitutes to anti-CD47 treatment (12). Nevertheless, antibodies have poor tissue penetration, especially into solid tumors due to their large molecular weight (23). Here, we report a high affinity variant of human CD47-ECD (N3612) that is a small protein capable of greater tissue penetrance without the potential toxicity associated with Fc effector functions and that potently binds to SIRPα, which has limited tissue distribution.
Insights into the Mechanism of SIRPα Activation
One potential pitfall of our approach using a soluble high affinity CD47 variant as a competitive SIRPα antagonist was that CD47 is the natural agonist of SIRPα. If high receptor occupancy were sufficient to induce SIRPα signaling, we would observe inhibition of phagocytosis. Instead, our results indicate that a single CD47 binding event inhibits SIRPα signaling, which results in the potentiation of phagocytosis (Fig. 6). This implies that there must be other mechanisms necessary for induction of SIRPα activation. One possibility is receptor clustering or dimerization. This model is supported by experiments showing treatments of soluble CD47-Fc, and higher avidity fusion proteins are capable of reducing phagocytosis of porcine cells by human macrophages in vitro (40) and dampening the production of several inflammatory cytokines by SIRPα+ dendritic cells in the Crohn disease model in vivo (41). Another possible mechanism of SIRPα activation is mechano-transduction by virtue of a vectorial force exerted during a cell-cell interaction (42). This model is consistent with the observations that CD47 expression on the target cells is critical for their evasion from macrophage phagocytosis (40) and that signal activation of SIRPα has only been directly observed by co-culturing macrophages with red blood cells (9, 10, 43) or beads bearing recombinant CD47 (10, 44). Collectively, then, activation of SIRPα by CD47 in vivo probably requires some degree of clustering as well as formation of a cell-cell junction.
Velcro-CD47 in Clinically Relevant Applications
Our results show that Velcro-CD47 N3612 broadly enhances the effectiveness of tumor-specific antibodies (Fig. 6) and specifically targets the monocyte subpopulation (Fig. 8). This warrants further evaluation of its efficacy and safety as an immunotherapeutic agent for cancer in pre-clinical and clinical models.
Furthermore, the ability to image tissue macrophages is of immense clinical and research interest because maladaptive macrophages participate in a number of inflammatory and autoimmune diseases such as inflammatory bowel disease (45), rheumatoid arthritis (46), obesity (47), and diabetes (48). Current methods mostly involve using targeted or nontargeted nanomaterials, but the metabolic processing of many of these nanoparticles is not fully understood (49, 50). Nano-medicine also faces challenging manufacturing processes, requiring a large number of optimization trials to develop a procedure with consistency and reproducibility (50).
Velcro-CD47 N3612 presents a potential alternative macrophage-tracking agent. We show that N3612 only stains the blood monocytes and neutrophils (Fig. 8) and that it can be site-specifically conjugated and purified with ease, resulting in a collection of uniform imaging agents (Fig. 7). Furthermore, as a small ∼15-kDa protein, N3612 may be easily removed by the renal excretory system (51). These properties make Velcro-CD47 N3612 an appealing imaging agent for macrophages.
“Velcro” Engineering as a New Tool to Enhance Protein-Protein Interactions
Immunoreceptor-ligand interactions are currently active targets for therapeutic intervention, such as the “checkpoint” inhibitors anti-PD1 and anti-CTLA4 (52–54). These interactions are typically of low affinity, making a soluble receptor or ligand ECD antagonist approach ineffective in most cases. Although antagonistic antibodies can fulfill most of the desired functional requirements for therapeutic antagonism, the natural ligand or receptor ECDs do offer several advantages. First, the small ECDs are likely to have better tissue penetration for solid tumor applications. Second, patients are less likely to develop resistance through acquired mutations because this would also disrupt the natural receptor-ligand interaction in vivo. An unfortunate reality is that conventional interface-targeted affinity maturation approaches are unsuccessful for a variety of reasons, including the structural context of the interaction that is not tolerant to changes.
Random mutagenesis and site-directed mutagenesis of interface contact residues and/or residues that do not directly contact but contribute to the structural integrity of a protein-protein interface account for the success of a vast majority of protein engineering approaches to increase binding affinity for ligand-receptor interactions (22, 55–58). However, this was not the case with CD47 (Fig. 2). The concept of an “affinity ceiling” for a protein-protein interaction has been documented for antibodies and T cell receptors in vivo (59–61), and we speculate that there is an analogous notion in vitro. Therefore, although a sizable number of protein-protein interfaces are amenable to in vitro manipulations to evolve artificially high affinity, some, such as CD47, are refractive to common affinity maturation techniques.
To address this limitation, we developed a new engineering approach to increase the affinity of CD47 for SIRPα by installing additional contact residues on its N terminus to increase affinity through expanding the existing interface, termed “Velcro” engineering. Using this approach, we extended the N terminus of CD47 by a small “Velcro” peptide of three amino acids in length (Fig. 3). The “Velcro” strategy is highly efficient and has allowed us to generate a 200–800-fold higher affinity clone in a single library (Figs. 4 and 5). This methodology may be applicable to alternative receptor-ligand pairs that are difficult to affinity mature using techniques such as site-directed or random mutagenesis. It can also be a complementary approach to other systems that are amenable to classical affinity maturation techniques. Our current finding expands the repertoire of protein engineering tools that can be used to increase the affinity of protein-protein and/or protein-peptide interactions.
Supplementary Material
Acknowledgments
We thank K. Jude, V. Luca, C. Janda, M. Birnbaum, A. M. Levin, D. Waghray, S. Fischer, N. Goriatcheva, T. Storm, L. Jerabek, N. B. Fernhoff, A. K. Volkmer, and J. P. Volkmer for technical assistance, discussions, and reagents.
This work was supported, in whole or in part, by National Institutes of Health Grant R01CA177684 (to K. C. G.). This work was also supported by Siebel Stem Cell Institute (to C. H.), by Stanford Molecular and Cellular Immunobiology National Institutes of Health Training Grant 5T32 AI072905 (to J. T. S.), and by the Virginia and D. K. Ludwig Fund for Cancer Research (to I. L. W.).

This article contains supplemental Fig. S1.
- IgSF
- immunoglobulin superfamily
- a1d1
- allele 1 domain 1
- a2d1
- allele 2 domain 1
- ADCP
- antibody-dependent cellular phagocytosis
- A647
- Alexa Fluor 647
- ECD
- extracellular domain (or ectodomain)
- Fc
- fragment crystallizable
- MACS
- magnetic-activated cell sorting
- PDB
- Protein Data Bank
- RBC
- red blood cells
- SIRPα
- signal regulatory protein α
- NK
- natural killer
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Wynn T. A., Chawla A., Pollard J. W. (2013) Macrophage biology in development, homeostasis and disease. Nature 496, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lewis C. E., Pollard J. W. (2006) Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66, 605–612 [DOI] [PubMed] [Google Scholar]
- 3. Beatty G. L., Chiorean E. G., Fishman M. P., Saboury B., Teitelbaum U. R., Sun W., Huhn R. D., Song W., Li D., Sharp L. L., Torigian D. A., O'Dwyer P. J., Vonderheide R. H. (2011) CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chao M. P., Alizadeh A. A., Tang C., Jan M., Weissman-Tsukamoto R., Zhao F., Park C. Y., Weissman I. L., Majeti R. (2011) Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 71, 1374–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chao M. P., Alizadeh A. A., Tang C., Myklebust J. H., Varghese B., Gill S., Jan M., Cha A. C., Chan C. K., Tan B. T., Park C. Y., Zhao F., Kohrt H. E., Malumbres R., Briones J., et al. (2010) Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 142, 699–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chao M. P., Tang C., Pachynski R. K., Chin R., Majeti R., Weissman I. L. (2011) Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood 118, 4890–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Edris B., Weiskopf K., Volkmer A. K., Volkmer J. P., Willingham S. B., Contreras-Trujillo H., Liu J., Majeti R., West R. B., Fletcher J. A., Beck A. H., Weissman I. L., van de Rijn M. (2012) Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. U.S.A. 109, 6656–6661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Houot R., Kohrt H. E., Marabelle A., Levy R. (2011) Targeting immune effector cells to promote antibody-induced cytotoxicity in cancer immunotherapy. Trends Immunol. 32, 510–516 [DOI] [PubMed] [Google Scholar]
- 9. Oldenborg P. A., Zheleznyak A., Fang Y. F., Lagenaur C. F., Gresham H. D., Lindberg F. P. (2000) Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 [DOI] [PubMed] [Google Scholar]
- 10. Tsai R. K., Discher D. E. (2008) Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 180, 989–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oldenborg P. A., Gresham H. D., Lindberg F. P. (2001) CD47-signal regulatory protein α (SIRPα) regulates Fcγ and complement receptor-mediated phagocytosis. J. Exp. Med. 193, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao X. W., van Beek E. M., Schornagel K., Van der Maaden H., Van Houdt M., Otten M. A., Finetti P., Van Egmond M., Matozaki T., Kraal G., Birnbaum D., van Elsas A., Kuijpers T. W., Bertucci F., van den Berg T. K. (2011) CD47-signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc. Natl. Acad. Sci. U.S.A. 108, 18342–18347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jaiswal S., Jamieson C. H., Pang W. W., Park C. Y., Chao M. P., Majeti R., Traver D., van Rooijen N., Weissman I. L. (2009) CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Majeti R., Chao M. P., Alizadeh A. A., Pang W. W., Jaiswal S., Gibbs K. D., Jr., van Rooijen N., Weissman I. L. (2009) CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown E. J., Frazier W. A. (2001) Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 11, 130–135 [DOI] [PubMed] [Google Scholar]
- 16. Adams S., van der Laan L. J., Vernon-Wilson E., Renardel de Lavalette C., Döpp E. A., Dijkstra C. D., Simmons D. L., van den Berg T. K. (1998) Signal regulatory protein is selectively expressed by myeloid and neuronal cells. J. Immunol. 161, 1853–1859 [PubMed] [Google Scholar]
- 17. Takenaka K., Prasolava T. K., Wang J. C., Mortin-Toth S. M., Khalouei S., Gan O. I., Dick J. E., Danska J. S. (2007) Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 8, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 18. Liu Y., Tong Q., Zhou Y., Lee H. W., Yang J. J., Bühring H. J., Chen Y. T., Ha B., Chen C. X., Yang Y., Zen K. (2007) Functional elements on SIRPα IgV domain mediate cell surface binding to CD47. J. Mol. Biol. 365, 680–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hatherley D., Graham S. C., Turner J., Harlos K., Stuart D. I., Barclay A. N. (2008) Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol. Cell 31, 266–277 [DOI] [PubMed] [Google Scholar]
- 20. Veillette A., Thibaudeau E., Latour S. (1998) High expression of inhibitory receptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 in macrophages. J. Biol. Chem. 273, 22719–22728 [DOI] [PubMed] [Google Scholar]
- 21. Willingham S. B., Volkmer J. P., Gentles A. J., Sahoo D., Dalerba P., Mitra S. S., Wang J., Contreras-Trujillo H., Martin R., Cohen J. D., Lovelace P., Scheeren F. A., Chao M. P., Weiskopf K., Tang C., et al. (2012) The CD47-signal regulatory protein α (SIRPα) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. U.S.A. 109, 6662–6667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weiskopf K., Ring A. M., Ho C. C., Volkmer J. P., Levin A. M., Volkmer A. K., Ozkan E., Fernhoff N. B., van de Rijn M., Weissman I. L., Garcia K. C. (2013) Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chames P., Van Regenmortel M., Weiss E., Baty D. (2009) Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 157, 220–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A., 3rd, Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 25. Hatherley D., Lea S. M., Johnson S., Barclay A. N. (2014) Polymorphisms in the human inhibitory signal regulatory protein α do not affect binding to its ligand CD47. J. Biol. Chem. 289, 10024–10028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Birnbaum M. E., Mendoza J. L., Sethi D. K., Dong S., Glanville J., Dobbins J., Ozkan E., Davis M. M., Wucherpfennig K. W., Garcia K. C. (2014) Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boder E. T., Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 [DOI] [PubMed] [Google Scholar]
- 28. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 30. Brunger A. T. (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 31. Mori Y., Iwasaki H., Kohno K., Yoshimoto G., Kikushige Y., Okeda A., Uike N., Niiro H., Takenaka K., Nagafuji K., Miyamoto T., Harada M., Takatsu K., Akashi K. (2009) Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J. Exp. Med. 206, 183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J., Sawyer N., Regan L. (2013) Protein-protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 22, 510–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lamminmäki U., Paupério S., Westerlund-Karlsson A., Karvinen J., Virtanen P. L., Lövgren T., Saviranta P. (1999) Expanding the conformational diversity by random insertions to CDRH2 results in improved anti-estradiol antibodies. J. Mol. Biol. 291, 589–602 [DOI] [PubMed] [Google Scholar]
- 34. Lee C. V., Liang W. C., Dennis M. S., Eigenbrot C., Sidhu S. S., Fuh G. (2004) High affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J. Mol. Biol. 340, 1073–1093 [DOI] [PubMed] [Google Scholar]
- 35. Boder E. T., Wittrup K. D. (1998) Optimal screening of surface-displayed polypeptide libraries. Biotechnol. Prog. 14, 55–62 [DOI] [PubMed] [Google Scholar]
- 36. Miyauchi S., Gopal E., Babu E., Srinivas S. R., Kubo Y., Umapathy N. S., Thakkar S. V., Ganapathy V., Prasad P. D. (2010) Sodium-coupled electrogenic transport of pyroglutamate (5-oxoproline) via SLC5A8, a monocarboxylate transporter. Biochim. Biophys. Acta 1798, 1164–1171 [DOI] [PubMed] [Google Scholar]
- 37. Ganapathy V., Roesel R. A., Howard J. C., Leibach F. H. (1983) Interaction of proline, 5-oxoproline, and pipecolic acid for renal transport in the rabbit. J. Biol. Chem. 258, 2266–2272 [PubMed] [Google Scholar]
- 38. Strowig T., Rongvaux A., Rathinam C., Takizawa H., Borsotti C., Philbrick W., Eynon E. E., Manz M. G., Flavell R. A. (2011) Transgenic expression of human signal regulatory protein α in Rag2−/− γ (c)−/− mice improves engraftment of human hematopoietic cells in humanized mice. Proc. Natl. Acad. Sci. U.S.A. 108, 13218–13223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barclay A. N., Van den Berg T. K. (2014) The interaction between signal regulatory protein α (SIRPα) and CD47: structure, function, and therapeutic target. Annu. Rev. Immunol. 32, 25–50 [DOI] [PubMed] [Google Scholar]
- 40. Ide K., Wang H., Tahara H., Liu J., Wang X., Asahara T., Sykes M., Yang Y. G., Ohdan H. (2007) Role for CD47-SIRPα signaling in xenograft rejection by macrophages. Proc. Natl. Acad. Sci. U.S.A. 104, 5062–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baba N., Van V. Q., Wakahara K., Rubio M., Fortin G., Panzini B., Soucy G., Wassef R., Richard C., Tamaz R., Lahaie R., Bernard E. J., Caussignac Y., Leduc R., Lougnarath R., et al. (2013) CD47 fusion protein targets CD172a+ cells in Crohn's disease and dampens the production of IL-1β and TNF. J. Exp. Med. 210, 1251–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van der Merwe P. A., Dushek O. (2011) Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55 [DOI] [PubMed] [Google Scholar]
- 43. Wang H., VerHalen J., Madariaga M. L., Xiang S., Wang S., Lan P., Oldenborg P. A., Sykes M., Yang Y. G. (2007) Attenuation of phagocytosis of xenogeneic cells by manipulating CD47. Blood 109, 836–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodriguez P. L., Harada T., Christian D. A., Pantano D. A., Tsai R. K., Discher D. E. (2013) Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science 339, 971–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bain C. C., Mowat A. M. (2011) Intestinal macrophages–specialised adaptation to a unique environment. Eur. J. Immunol. 41, 2494–2498 [DOI] [PubMed] [Google Scholar]
- 46. Ji H., Ohmura K., Mahmood U., Lee D. M., Hofhuis F. M., Boackle S. A., Takahashi K., Holers V. M., Walport M., Gerard C., Ezekowitz A., Carroll M. C., Brenner M., Weissleder R., Verbeek J. S., et al. (2002) Arthritis critically dependent on innate immune system players. Immunity 16, 157–168 [DOI] [PubMed] [Google Scholar]
- 47. Lumeng C. N., Bodzin J. L., Saltiel A. R. (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bhargava P., Lee C. H. (2012) Role and function of macrophages in the metabolic syndrome. Biochem. J. 442, 253–262 [DOI] [PubMed] [Google Scholar]
- 49. Weissleder R., Nahrendorf M., Pittet M. J. (2014) Imaging macrophages with nanoparticles. Nat. Mater. 13, 125–138 [DOI] [PubMed] [Google Scholar]
- 50. Desai N. (2012) Challenges in development of nanoparticle-based therapeutics. AAPS J. 14, 282–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maack T., Johnson V., Kau S. T., Figueiredo J., Sigulem D. (1979) Renal filtration, transport, and metabolism of low-molecular-weight proteins: a review. Kidney Int. 16, 251–270 [DOI] [PubMed] [Google Scholar]
- 52. Momtaz P., Postow M. A. (2014) Immunologic checkpoints in cancer therapy: focus on the programmed death-1 (PD-1) receptor pathway. Pharmgenomics Pers. Med. 7, 357–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peggs K. S., Quezada S. A., Korman A. J., Allison J. P. (2006) Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr. Opin. Immunol. 18, 206–213 [DOI] [PubMed] [Google Scholar]
- 54. Shin D. S., Ribas A. (2015) The evolution of checkpoint blockade as a cancer therapy: what's here, what's next? Curr. Opin. Immunol. 33C, 23–35 [DOI] [PubMed] [Google Scholar]
- 55. Levin A. M., Bates D. L., Ring A. M., Krieg C., Lin J. T., Su L., Moraga I., Raeber M. E., Bowman G. R., Novick P., Pande V. S., Fathman C. G., Boyman O., Garcia K. C. (2012) Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine.’ Nature 484, 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Junttila I. S., Creusot R. J., Moraga I., Bates D. L., Wong M. T., Alonso M. N., Suhoski M. M., Lupardus P., Meier-Schellersheim M., Engleman E. G., Utz P. J., Fathman C. G., Paul W. E., Garcia K. C. (2012) Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat. Chem. Biol. 8, 990–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ring A. M., Manglik A., Kruse A. C., Enos M. D., Weis W. I., Garcia K. C., Kobilka B. K. (2013) Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pepper L. R., Cho Y. K., Boder E. T., Shusta E. V. (2008) A decade of yeast surface display technology: where are we now? Comb. Chem. High Throughput Screen. 11, 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Caoili S. E. (2012) On the meaning of affinity limits in B-cell epitope prediction for antipeptide antibody-mediated immunity. Adv. Bioinformatics 2012:346765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Poulsen T. R., Jensen A., Haurum J. S., Andersen P. S. (2011) Limits for antibody affinity maturation and repertoire diversification in hypervaccinated humans. J. Immunol. 187, 4229–4235 [DOI] [PubMed] [Google Scholar]
- 61. Foote J., Eisen H. N. (2000) Breaking the affinity ceiling for antibodies and T cell receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 10679–10681 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.