

Background: DDI2 and DDI3 are two uncharacterized identical genes found in budding yeast.
Results: They encode novel cyanamide hydratases and are massively induced by cyanamide. Their deletion causes cellular sensitivity to cyanamide.
Conclusion: The two genes function in cyanamide detoxification and are tightly regulated.
Significance: This is the first attempt to understand the duplicated gene cluster in budding yeast.
Keywords: enzyme, gene regulation, metabolism, transcription, yeast, HD domain, cyanamide hydratase, gene duplication, hydratase, transcriptional regulation
Abstract
Two DNA damage-inducible genes in Saccharomyces cerevisiae, DDI2 and DDI3, are identical and encode putative HD domain-containing proteins, whose functions are currently unknown. Because Ddi2/3 also shows limited homology to a fungal cyanamide hydratase that converts cyanamide to urea, we tested the enzymatic activity of recombinant Ddi2. To this end, we developed a novel enzymatic assay and determined that the Km value of the recombinant Ddi2/3 for cyanamide is 17.3 ± 0.05 mm, and its activity requires conserved residues in the HD domain. Unlike most other DNA damage-inducible genes, DDI2/3 is only induced by a specific set of alkylating agents and surprisingly is strongly induced by cyanamide. To characterize the biological function of DDI2/3, we sequentially deleted both DDI genes and found that the double mutant was unable to metabolize cyanamide and became much more sensitive to growth inhibition by cyanamide, suggesting that the DDI2/3 genes protect host cells from cyanamide toxicity. Despite the physiological relevance of the cyanamide induction, DDI2/3 is not involved in its own transcriptional regulation. The significance of cyanamide hydratase activity and its induced expression is discussed.
Introduction
DDI2 and DDI3 are two Saccharomyces cerevisiae genes identified through a genome-wide microarray analysis of budding yeast gene expression in response to the typical DNA-damaging agent methyl methanesulfonate (MMS).5 In this study, two functionally unknown open reading frames (ORFs) YNL335W and YFL061W displayed the highest induction (>100-fold) after 0.1% MMS treatment (1), and thus were named DNA-damage inducible genes 2 and 3, respectively. DDI1 had previously been reported to be co-regulated with MAG1 (2–4) encoding a 3-methyladenine DNA glycosylase (5, 6), involved in DNA-damage checkpoint (7), and required for repression of protein secretion (8). It turns out that DDI2 and DDI3 are duplicated genes located on different chromosomes, with identical ORF sequences and only one nucleotide difference in their promoter (up to 1 kb) regions.
Unlike other previously characterized budding yeast DNA damage-inducible genes that are often induced by a variety of DNA-damaging agents regardless of whether they are involved in the repair of that type of DNA damage (9), DDI2 and DDI3 are only induced by MMS and selected DNA-alkylating agents (10), suggesting that they are regulated by a unique mechanism and that they function differently than most other DNA damage-inducible genes. A protein sequence alignment and analysis reveal that Ddi2/3 is a member of the HD domain metalloprotein superfamily (11) and is homologous to a reported cyanamide hydratase (EC 4.2.1.69) from the soil fungus Myrothecium verrucaria (12, 13).
Cyanamide (CN2H2) is used as fertilizer to provide nitrogen to soil. Cyanamide can be naturally converted to urea, a functional ingredient in nitrogen fertilizer, via hydrating water molecules in the air. Urea can then be broken down via further hydration to ammonia and carbon dioxide. Thus cyanamide is an environmentally “clean” fertilizer. Cyanamide is also used as a fungicide and herbicide due to its mild toxicity. Cyanamide can be biosynthesized from natural nitrogen by some hairy vetch species, such as Vicia villosa Roth, and is attributed to the growth suppression of many weeds. Crude extracts from hairy vetch leaves and stems containing 1.3 ppm of cyanamide inhibited radical growth of lettuce by 40% (14). Thus cyanamide was considered as an allelochemical responsible for strong allelopathic potential in hairy vetch (14–16). Cyanamide is also a raw compound in the pharmaceutical industry to make guanidine derivatives (17), and it is also used in treating alcoholic patients because it (or its metabolized products) can inhibit aldehyde dehydrogenase and thus inhibit ethanol metabolism (18).
Nitrile compounds are distributed widely in the environment, and most of them are toxic to higher eukaryotes. Many organisms have developed corresponding nitrile-metabolizing enzymes that participate in nitrile biodegradation and utilization. Based on the above analyses, we hypothesize that lower eukaryotic microorganisms may also contain such enzymes, including cyanamide hydratase for cyanamide detoxification and/or utilization. In this study, we characterized the enzyme activity of recombinant Ddi2/3 as well as its gene regulation and biological functions. We developed a novel cyanamide hydratase assay and demonstrated that Ddi2/3 is indeed a bona fide cyanamide hydratase. Indeed, the expression of DDI2 and DDI3 genes are greatly elevated by cyanamide treatment, and the inactivation of these genes results in compromised resistance to cyanamide. Hence, this study establishes a detoxification role of fungal cyanamide hydratase and justifies why the corresponding genes are under strict regulation.
EXPERIMENTAL PROCEDURES
Yeast Strains, Culture, and Manipulation
Yeast haploid strains used in this study are listed in Table 1. Targeted gene deletion mutants are isogenic derivatives of BY4741. Yeast cells were cultured in either YPD-rich medium or a synthetic dextrose (SD) minimal medium supplemented with required nutrients as described (19). A protocol modified from the standard LiAc method (20) was used for yeast transformation.
TABLE 1.
S. cerevisiae strains used in this study
| Strain name | Genotype | Source |
|---|---|---|
| DBY747 | MATa leu2-3,112 ura3-52 his3-Δ1 trp1-289 | D. Botstein |
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Consortium |
| WXY3147 | BY4741 with ddi2/3Δ∷HIS3 | This study |
| WXY3148 | BY4741 with ddi2/3Δ∷LEU2 | This study |
| WXY2929 | BY4741 with ddi2/3Δ∷HIS3 ddi2/3Δ∷LEU2 | This study |
| WXY3149 | BY4741 with ddi2Δ::NAT (chromosome VI deletion from 6104 to 15,552) | This study |
| WXY3143 | WXY3149 with ddi3Δ::HIS3 | This study |
| WXY3150 | BY4741 with ddi3Δ::NAT (chromosome XIV deletion from 8004 to 17,388) | This study |
| WXY3145 | WXY3150 with ddi2Δ::HIS3 | This study |
Northern Hybridization and β-Galactosidase (β-Gal) Activity Assay
DDI2/3 gene expression was assessed by Northern hybridization as described previously (1) using the DDI2/3 ORF as a probe and ACT1 as an internal control.
To characterize the DDI2/3 promoter, the promoter region of DDI2/3 was amplified by PCR from genomic DNA by primers YFL061w-1 (5′-GGAAAATCCAAGCTTTCAAG-3′) and YFL061w-3 (5′-GCCCTGCAGCCTCATTGAAACTTACCT-3′; the restriction enzyme sequence used for cloning is underlined). The PCR product (containing −718 to +678 of DDI2/3) was then cloned as a HindIII-PstI (−711 to +457) fragment into YEp365R (21) to form YEpDDI2-lacZ. The resulting plasmid was transformed into yeast cells, and the confirmed transformants were maintained in an SD-Ura medium. The β-Gal assay was performed as described previously (22). Briefly, 0.5 ml of overnight yeast culture was subcultured in 2.5 ml of fresh SD-Ura medium, and incubation was continued for another 2 h until cell density reached approximately A600 nm = 0.2. A test chemical was added to predetermined concentrations, and cells were returned to culture for 3 h. 1 ml of the culture was used to determine cell density by measuring A600, and the remaining 2-ml culture was collected by centrifugation and used for the β-gal assay. Harvested cells were washed with phosphate-buffered saline (PBS) and suspended in 1 ml of buffer Z (60 mm Na2HPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgSO4, and 40 mm β-mercaptoethanol, pH 7.0). 50 μl of 0.1% SDS and chloroform were added, and the mixture was vortexed to permeabilize cells. After adding 0.2 ml of 4 mg/ml substrate ortho-nitrophenyl β-d-galactoside, the mixture was incubated at 30 °C for 20 min before stopping the reaction by adding 0.5 ml of 1 m NaCO3. The reaction was measured by spectrophotometry at 420 nm, and its activity is expressed in Miller units (23). Induction was calculated as a ratio of β-gal activity of the cells with and without treatment in the same experiment.
Expression of DDI2 in Escherichia coli and Recombinant Protein Purification
The DDI2/3 ORF was amplified from the yeast genome using primers YFL061w-2 (5′- GCCGAATTCATGTCACAGTACGGATTT-3′) and YFL061w-3 and inserted into plasmid pGEX-6P-1 (GE Healthcare) at the EcoRI and NotI sites. To express mutated Ddi2 proteins, site-specific mutations were created in the above pGEX-Ddi2 plasmid by a mega-primer approach (24) as described previously (25), with two common primers, pGEX-5′ and pGEX-3′, and two mutation-specific primers, Ddi2-H88AD89A (5′-CTGTTGTTGCAATAGCAGCAAGTAAGCAGGTG-3′; mutated sequences are in bold) and Ddi2-H137AD139A (5′-GGGCTGCAGAATGAGCCCCAAGGTGGTAATGTAGCCAGTCCCAGTCAAAGCCTGGGCACGAATGATGGC-3′). The entire ORFs of resulting plasmids were sequenced to confirm the mutant constructs.
The recombinant plasmid was transformed into E. coli strain BL21(DE3) for heterologous gene expression and recombinant protein production. The transformed cells were cultured in LB + 100 μg/ml ampicillin at 37 °C to A600 nm = 0.6 prior to induction by 0.1 mm isopropyl 1-thio-β-d-galactopyranoside over 16 h at room temperature. The cells were harvested by centrifugation and suspended in PBS (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, and 1.8 mm KH2PO4, pH 7.4). The cell lysate was collected after passing through a Cell Disrupter (TS Series Benchtop from Constant System Ltd.) at 35 p.s.i., and the cell debris was removed by spinning at 35,000 × g for 30 min. GST-fused Ddi2/3 in the supernatant fraction was collected by using glutathione-Sepharose 4B resin and eluted by adding an excess amount of reduced glutathione (10 mm). After purification, the GST tag was cleaved using PreScission protease (GE Healthcare) in a cleavage buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, and 4 mm DTT) at 4 °C for 16 h. The GST tag and PreScission protease, which is also GST-tagged but cannot be cleaved, was removed by running the digested product over glutathione-Sepharose 4B resin. Collected Ddi2/3 was dialyzed in a 50 mm HEPES-Na, pH 7.5, 100 mm NaCl buffer. During the purification process, protein samples were taken after each step and analyzed by SDS-PAGE. Western blot analysis was also performed to identify the GST fusion protein, in which a goat anti-GST (1:20,000, purchased from GE Healthcare) primary antibody and bovine anti-goat IgG-HRP (1:10,000, purchased from Santa Cruz Biotechnology) secondary antibody were used.
Cyanamide Hydratase Assay
Purified Ddi2/3 was incubated with various concentrations of cyanamide at room temperature (23 °C), and urea formation was monitored by adding excessive urease and glutamate dehydrogenase from an ammonia assay kit (Sigma catalog no. AA0100). Incorporation of ammonia is coupled with NADPH consumption in the formation of glutamate, and its molarity is equal to the consumption of NADPH, which has an absorbance peak at 340 nm. The reaction mixture for measuring kinetic parameters contains 0.62 μm urease (Sigma catalog no. U1500), 0.45 μm l-glutamate dehydrogenase, 3.4 mm α-ketoglutarate, and 0.23 mm NADPH. For each assay, 100 μl of Ddi2 containing 0.027 μm protein was incubated with the above reaction mix, and cyanamide solution was added last to start the reaction. Each reaction was monitored at different time intervals for at least 20 min to calculate the initial velocity. When cyanamide concentrations were below 5 mm, the cyanamide hydration reactions were continuously monitored for 15 min. When cyanamide concentrations were above 5 mm, real time monitoring of reactions for 15 min would be impossible because cyanamide increases the background in the ammonia assay, and the A340 nm soon drops to undetectable levels. In these situations, cyanamide hydration reactions and ammonia assays were separated. Briefly, the cyanamide hydration by Ddi2 was started in a 1.5-ml Eppendorf tube containing 0.027 μm Ddi2 and 0.62 μm urease, with cyanamide added last to start the reaction, making the total volume 1 ml. 50–200 μl of the reaction mixture was withdrawn every 7–10 min for the ammonia assay, and the total amount of ammonia produced in the original reaction was determined using the ammonia assay kit. To account for possible background ammonia production, a negative control reaction lacking recombinant Ddi2 was performed. All assays were repeated at least three times to calculate standard deviations. GST was used as a negative control in the enzymatic assay because it is readily available in the laboratory, and its molecular weight is comparable with that of Ddi2/3.
Yeast-targeted Gene Disruption
To make ddi2/3Δ disruption cassettes, the YFL061w-1/YFL061w-3 PCR product was cleaved by PvuII-NotI, and the 1.17-kb fragment containing a 0.5-kb promoter and the entire DDI2/3 ORF was cloned into SmaI-NotI of pBluescript. A 0.54-kb HpaI-BamHI fragment (−55 to +487) was then deleted from the resulting pBS-DDI2 and replaced by a BamHI linker, which was used to clone either 1.16-kb HIS3 from YDp-H or 1.6-kb LEU2 from YDp-L (26). The resulting ddi2/3Δ::HIS3 cassette was released by XbaI-XhoI digestion and ddi2/3Δ::LEU2 disruption cassette by BglII-XhoI digestion prior to yeast transformation. To achieve high efficiency and specificity of target gene disruption, the disruption cassettes were purified from agarose gel after electrophoresis. A single copy of the DDI2/3 gene was disrupted by either the ddi2Δ::HIS3 or ddi2Δ::LEU2 cassette by a one-step gene disruption method (27), followed by sequential disruption of the second copy with the opposite selectable marker. The double disruption lines were screened by their ability to grow on the SD minimal medium lacking both His and Leu and then further confirmed by genomic PCR with primers flanking the deleted region. To make chromosome deletions at the duplicated loci, chromosomes VI-specific primer pairs ChrVIf (5′-GTATTTCATTCAAGCGGTAACCGCTGTACGAGCAGTGACACATGGAGGCCCAGAATACC-3′; the sequence homologous to the template DNA is underlined) plus ChrVIr (5′-GCATTGTTCGTATTCAAGGAAACCGGGGGGCAAAATTTCCAGTATAGCGACCAGCATTC-3′) and XIV-specific primer pairs ChrXIVf (5′-ACGTTTCATTTTGGGTAACAACTGCTGTGCGAACAGTGAACATGGAGGCCCAGAATACC-3′) plus ChrXIVr (5′-GAGAAAATAAGCACTCAATCTTCAGACATATAAAAGGAGCAGTATAGCGACCAGCATTC-3′) were used to PCR-amplify a ClonNAT-resistant marker from plasmid pFA6a-natMX6 (28). The resulting disruption cassettes were used to delete the duplicated loci, and the desired transformants were confirmed by yeast genomic PCR with the above primers.
Yeast Sensitivity Assay
Wild type and confirmed ddi2/3Δ mutants were cultured overnight at 30 °C in 2 ml of YPD. Sterile double distilled H2O was used in adjusting cell density and making a series of 10-fold dilutions of yeast cultures, which were equally spotted on YPD and YPD containing different concentrations of testing chemicals. After the liquid was absorbed, the plates were incubated at 30 °C for 3 days or otherwise specified times before taking photographs.
Preparation of Yeast Whole Cell Extracts (WCEs)
Wild-type BY4741, its ddi2/3Δ single mutant strain, and its ddi2Δ ddi3Δ double mutant strain were cultured in a YPD medium and subcultured on the 2nd day until the A600 nm reached 0.3. The yeast cultures were treated with 25 mm cyanamide for 2 h at 30 °C, and the cells were collected by centrifugation and then washed with a lysis buffer containing 20 mm Tris-Cl, pH 8.0, 150 mm NaCl, and 0.5 mm EDTA. The pellet was weighed and resuspended in the lysis buffer at 1 ml/g, to which 1 mm PMSF and 500 μl of acid-washed glass beads were added. Yeast cells were lysed by vortexing 30 s followed by leaving on ice 30 s; after eight rounds of votexing, glass beads were removed by centrifugation, and the supernatant was centrifuged at 15,000 × g for 10 min at 4 °C. The clear supernatant was collected as WCE, and the total protein concentration was adjusted to 1 mg/ml after being measured by the Bradford assay using the Bio-Rad protein assay reagent.
Cyanamide Assay Using Yeast WCE
Cyanamide was added to 0.5 ml of yeast WCE to a final concentration of 2 mm, and the solution was incubated at 30 °C. The cyanamide concentration was monitored by using a colorimetric assay as described (29). Briefly, 100 μl of reaction mix was added to 500 μl of PBS, followed by adding 400 μl of 0.1 m sodium carbonate buffer, pH 10.4, and 200 μl of 4% sodium pentacyanoammine ferroate(II) (TCI, S0050) as the color reagent. Absorbance at 530 nm was measured to determine the remaining cyanamide concentration.
RESULTS
Induction of Two Duplicated Yeast Genes by MMS Treatment
We previously performed a study in which S. cerevisiae cells were treated with 0.1% MMS for 48 min, and the global transcriptional response was assessed by microarray analysis. During this microarray analysis, two genes, YNL335w and YFL061w, showed the highest induction (138- and 108-fold, respectively), and thus were designated DDI2 and DDI3, respectively. Interestingly, DDI2 and DDI3 are two identical genes located in a duplicated gene cluster region with identical ORFs and only one nucleotide difference in their promoter sequences of more than 1-kb; however, they are located on different chromosomes (see Fig. 2A and Saccharomyces Genome Database, Stanford University). Because essentially all subsequent assays cannot distinguish between the two genes and their products, we refer to them as DDI2/3 or simply DDI2 in cases where one gene or its product is isolated and characterized.
FIGURE 2.

Genomic and protein sequence analyses of Ddi2/3. A, DDI2 and DDI3 genes are located within a highly conserved duplicated region. DDI2 and DDI3 and their flanking 20-kb regions are thought to derive from gene duplication, in which the boxed regions are highly conserved in DNA sequence. B, amino acid sequence alignment of Ddi2/3 with CAH from M. verrucaria. Identical amino acid residues are highlighted. Signature conserved HD residues are marked with asterisks. Protein sequences were retrieved from the NCBI protein database (CAH: AAA33429.1, and Ddi2: P0CH63.1), and the alignment was done with ClustalW. C, phylogenetic tree illustrating the homology among available cyanamide hydratase domain-containing proteins: DEHA2A01694p (Debaryomyces hansenii CBS767), CaO19.9024 (Candida albicans SC5314), AO090103000133 (Aspergillus oryzae RIB40), AN6421.2 (A. nidulans FGSC A4), AO090102000308 (A. oryzae RIB40), AN5411.2 (Aspergillus nidulans FGSC A4), CAH (M. verrucaria) and FG11132.1 (Fusarium graminearum PH-1). Amino acid sequences of proteins were retrieved from the NCBI conserved domain database. MEGA5 was used to predict the phylogenetic tree based on maximum likelihood. The phylogeny was tested by using a bootstrap method, and the bootstrap replications were set at 1000.
To validate the microarray result, we performed Northern blot analysis to determine the transcriptional level of DDI2/3 after MMS treatment. The Northern blot result (Fig. 1A) showed that DDI2/3 is increased ∼110-fold after MMS treatment, consistent with the microarray data. Furthermore, we isolated DDI2 and its promoter and made a lacZ fusion construct. As expected, the β-gal assay also showed 150-fold induction of DDI2-lacZ by 0.01% MMS treatment for 3 h (Fig. 1B), indicating that the DDI2-lacZ fusion reporter faithfully represents the native DDI2/3 promoter and that the induction is most likely due to enhanced transcriptional initiation.
FIGURE 1.

DDI2/3 expression in response to DNA-damaging agents. A, Northern hybridization showing that after MMS treatment, the DDI2/3 transcript level is drastically increased in DBY747 cells. Yeast cells were treated with 0.1% MMS for 48 min prior to RNA isolation and Northern hybridization. Each lane contains 15 μg of total RNA. The blot was stripped and re-hybridized with an ACT1 probe. B–H, induction of DDI2-lacZ by different DNA-damaging agents. The β-gal assays were processed as described under “Experimental Procedures,” and the results after various DNA-damaging agents are shown as fold induction relative to untreated. All data represent the average of at least three experiments with standard deviations.
DDI2/3 Is Only Highly Induced by Sn2-type Alkylating Agents
Because all of the well documented DNA damage-inducible genes in budding yeast seem to respond to a wide spectrum of DNA-damaging agents (9), we examined the induction of DDI2-lacZ after treating yeast cells with representative DNA-damaging agents. To our surprise, the expression of DDI2-lacZ was only highly induced by Sn2-type alkylating agents, including MMS and dimethyl sulfate, up to 350- and 150-fold, respectively (Fig. 1, B and C). These compounds alkylate predominantly at nitrogens rather than oxygens in DNA bases. The Sn1-type alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine, which efficiently alkylates oxygens, weakly induces the expression of DDI2-lacZ (Fig. 1D), probably due to its overlapping activities with Sn2-type alkylating agents. Other DNA-damaging agents, such as ethyl methanesulfonate, γ-radiation, or hydroxyurea, only mildly induced the expression of DDI2-lacZ by no more than 8-fold (Fig. 1, E–G). Furthermore, UV irradiation, a well known DNA-damaging agent, did not induce DDI2-lacZ expression at all (Fig. 1H). The above observations collectively indicate that DDI2/3 are not typical DNA damage-inducible genes and that they may be involved in a cellular metabolic response related to Sn2-type alkylation stress.
DDI2/3 Encodes a Member of the HD Domain Family of Proteins
DDI2/3 encodes a putative 225 amino acid polypeptide containing an HD domain (Fig. 2B). The HD domain represents a large superfamily of metal-dependent phosphohydrolases, which includes phosphatases, phosphodiesterases, dNTPases, and redox enzymes (11, 30). No polypeptide sequences in the worm, mouse, or human genome database show significant similarity to Ddi2/3. However, close homologs of Ddi2/3 are identified in some Ascomycota fungi. Interestingly, an amino acid sequence alignment (Fig. 2B) revealed 36% (81/225) sequence identity with a previously characterized cyanamide hydratase (CAH) from M. verrucaria (12); both contain the HD domain (Fig. 2B). To date, only nine predicted proteins from fungi contain such a domain (Fig. 2C), all belonging to Ascomycota. Among them, only CAH has been demonstrated to have a cyanamide hydratase activity (12, 31), and Ddi2/3 and CAH appear to be distantly related (Fig. 2, B and C). The above analyses prompted us to ask whether DDI2/3 also exhibits cyanamide hydratase catalytic activity.
Recombinant Ddi2 Exhibits Cyanamide Hydratase Activity
To date, there is only one reported characterization of cyanamide hydratase activity from M. verrucaria cell extract, with a measured Km of 27 mm and high substrate specificity (12). To test our hypothesis that DDI2/3 encodes a cyanamide hydratase and to further characterize this class of enzymes, we cloned the DDI2/3 ORF into plasmid pGEX6P-1 as a GST fusion. Following overexpression, purification, and PreScission protease cleavage to remove the GST tag, the resulting recombinant Ddi2/3 contains eight additional amino acid residues (Gly-Pro-Leu-Gly-Ser-Pro-Glu-Phe) at the N terminus, with a calculated molecular mass of 25.8 kDa. The cleaved Ddi2 protein was purified to apparent homogeneity as judged by SDS-PAGE, and the corresponding Western blot shows that it is free of detectable GST contamination (Fig. 3A).
FIGURE 3.

Enzymatic characterization of the recombinant Ddi2 protein. A, SDS-polyacrylamide gel image and the corresponding anti-GST Western blot to show the purification of the recombinant Ddi2 protein. Lane M, Precision Plus ProteinTM unstained standards (from Bio-Rad); lane 1, purified GST-Ddi2; lane 2, GST-Ddi2 incubated with PreScission Protease for 16 h; lane 3, recombinant Ddi2 after removal of the GST tag, which was used in the enzymatic assay. B, colorimetric assays of cyanamide. The experiment protocol was modified from Refs. 29, 33. Linear regression was programmed by using SigmaPlot12. R2 = 0.99. C, time courses of urea formation of Ddi2 and site-specific double mutants. Each reaction contained 44.8 mm cyanamide and 0.027 μm wild-type Ddi2 or 0.39 μm mutant Ddi2. ●, wild-type Ddi2; ▾, Ddi2-H88AD89A; and ■, Ddi2-H137AD139A. Note that Ddi2-H137AD139A was not purified to the same extent as the WT and Ddi2-H88AD89A proteins. D, Michaelis-Menten curve of Ddi2 to cyanamide. 0.7 μg (27 nm) of Ddi2 was applied in the assay. The urea formation rates from 0.3 to 88 mm cyanamide were measured. Nonlinear regression was determined by the SigmaPlot 12 program with the Michaelis-Menten equation. E, time course monitoring of urea formation by Ddi2 and His6-tagged Ddi2 proteins. ○, Ddi2 protein purified from GST expression system, in which the GST tag was cleaved and removed. ●, C-terminal His6-tagged Ddi2, expressed from a pET28 vector. Applied proteins were 20 μg; cyanamide concentration was 0.15 mm. Linear regressions were programmed by using SigmaPlot12. R2 values of both trend lines are 0.997. F, SDS-polyacrylamide gel image showing the purified wild-type Ddi2 and its mutant derivative. Lane M, Precision Plus ProteinTM unstained standards; lane 1, purified Ddi2; lane 2, purified Ddi2-H88AD89A. Note that the purification resulted in a single intense band corresponding to Ddi2 plus additional minor bands.
The initial CAH enzymatic activity was determined by measuring the consumption of cyanamide after incubation with CAH for 15–60 min, followed by a colorimetric assay at 530 nm (32). It was reported that A530 nm absorption is linear with cyanamide concentrations only up to 200 μm (33), but in our hands the linear relationship can be extended up to 5 mm cyanamide (Fig. 3B). Enzymatic activity is presented as a reaction rate, which needs to be measured with substrate concentration as high as five times Km to guarantee that the enzyme is saturated and the reaction rate approaches Vmax to ensure an accurate Km value of the enzyme. Because the established Km of CAH is 27 mm and Ddi2 is expected to have similar properties, the reported colorimetric assay is unlikely to provide accurate data on enzymatic reaction rate to high cyanamide concentrations. Here, we developed a novel method for measuring cyanamide hydratase activity based on the determination of the product urea concentration. In principle, excessive urease in the cyanamide hydratase reaction allows immediate hydrolysis of the produced urea to ammonia, which can be accurately quantified by coupling the urea hydrolysis by adding α-ketoglutarate and glutamate dehydrogenase and monitoring the consumption of the NADPH cofactor (available as a commercial kit from Sigma) (34, 35). The time courses of urea formation can be plotted, as shown in Fig. 3C. Initial velocities of the cyanamide hydration reaction to various concentrations of substrate were measured, and an initial velocity versus substrate concentration curve for Ddi2 was plotted using cyanamide concentrations ranging from 0.3 to 88 mm for 0.027 μm Ddi2. The kinetic curve was fitted by the program SigmaPlot12 with the Michaelis-Menten equation, in which the R2 of regression fitting reached 0.9997, and Ddi2 fits a one-site saturation model for cyanamide. In summary, the recombinant Ddi2 exhibits a Km = 17.3 ± 0.5 mm, Vmax = 15.9 ± 0.16 nmol·min−1, and kcat = 589 nmol of cyanamide consumed·min−1·nmol−1 enzyme (Fig. 3D). To rule out the possibility that the N-terminal extra amino acid residues in the recombinant Ddi2 influence its activity, we also performed the CAH assay with purified His6-tagged recombinant Ddi2, and similar activities were obtained (Fig. 3E).
Ddi2 HD Domain Is Required for Cyanamide Hydratase Activity
The HD domain is named due to the most conserved histidine and aspartate residues in the region and features a characteristic … H … HD…. D … pattern (11). Mutations in the conserved HD residues drastically affect the enzymatic activity (36).
Because the Ddi2 protein belongs to the HD domain superfamily but no structural analysis of this subfamily has been reported, we wished to ask whether the conserved HD metal-binding residues are required for cyanamide hydratase activity. The sequence alignment of all nine putative cyanamide hydratase domain-containing proteins reveals 19 identical amino acids, among which five are HD residues, including Ddi2-His-55, His-88, Asp-89, His-137, and Asp-139. We created two Ddi2 double mutations, Ddi2-H88A,D89A and Ddi2-H137A,D139A, that presumably compromise metal binding and impair cyanamide hydratase activity, and we tested their enzymatic activities. During recombinant protein preparation, we noticed that the Ddi2-H137A,H139A mutation severely affected protein yield and solubility, whereas the Ddi2-H88A,D89A protein was readily expressed and purified (Fig. 3F). As shown in Fig. 3C, both mutated proteins completely lost their cyanamide hydratase activity, indicating that the conserved HD domain and presumed metal-binding site are essential for the above enzymatic activity. It also suggests that the required metal ions can be incorporated into recombinantly expressed Ddi2. Furthermore, this observation also rules out the possibility that the cyanamide hydratase activity was due to potential contaminant from bacterial cells.
DDI2/3 Transcription Is Significantly Induced by Cyanamide
Because DDI2/3 encodes a cyanamide-metabolizing enzyme and its basal expression is at an extremely low level, but can be induced by certain alkylating agents, we suspected that it may also be induced by cyanamide. Using a reporter gene assay, we found that as low as 0.01% MMS can induce DDI2 over 150-fold, and treatment with 0.03% MMS achieves the highest induction of over 350-fold (Fig. 1B). Under the same experimental conditions, cyanamide can lead to even higher induction than MMS. For example, 10 mm cyanamide reached induction of over 600-fold for DDI2/3 (Fig. 4A). Hence, DDI2/3 is a cyanamide-inducible gene.
FIGURE 4.
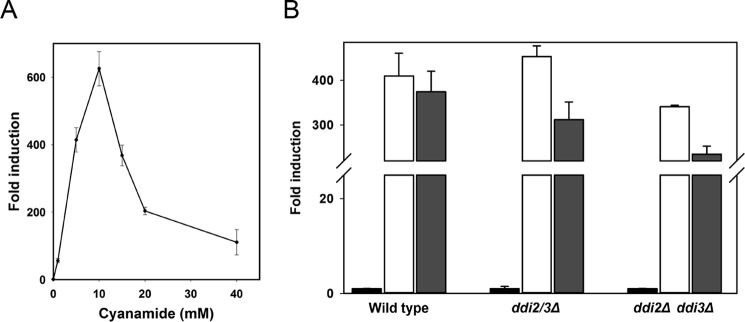
DDI2/3 is induced by cyanamide but is not involved in its own transcriptional regulation. A, fold induction of DDI2/3-lacZ by cyanamide. Experimental conditions are as described in Fig. 1, B–H. B, deletion of both DDI2 and DDI3 genes does not affect DDI2/3-lacZ induction by cyanamide or MMS. Black bars, no treatment; open bars, treated with 0.03% MMS; gray bars, treated with 10 mm cyanamide. Experimental conditions are as described in Fig. 1, B–H.
DDI2/3 Confers Resistance to Cyanamide and MMS
DDI2 and DDI3 reside in a duplicated chromosomal region in which three genes (including DDI2/3) and their flanking sequences are nearly identical (Fig. 2A). Because of this, the yeast gene deletion collection does not have a reliable single mutant, let alone the ddi2Δ ddi3Δ double mutant. To characterize the biological function(s) of DDI2/3, we created two ddi2/3 disruption cassettes with different selectable markers (Fig. 5A) and performed a sequential gene deletion procedure. As shown in Fig. 5B, the ddi2/3Δ::HIS3 and ddi2/3Δ::LEU2 single mutants contain a wild-type copy and a disrupted copy, although the PCR method cannot distinguish which chromosomal copy was disrupted. Nevertheless, in the ddi2/3Δ::HIS3 ddi2/3Δ::LEU2 double mutant, both DDI2 and DDI3 genes were disrupted, and the strain no longer contains a wild-type allele. The growth of the ddi2/3Δ single and double mutants was tested by a serial dilution assay in the presence of MMS or cyanamide. As shown in Fig. 5C, the ddi2Δ ddi3Δ double mutant displays increased sensitivity to cyanamide compared with the parental wild-type strain, suggesting that DDI2/3 protect cells from cyanamide toxicity. Surprisingly, the ddi2/3Δ single mutant displays a level of sensitivity similar to the double mutant, suggesting that although DDI2 and DDI3 are identical genes they are not simply redundant in function. Similarly, both single and double mutants display comparable levels of sensitivity to MMS. There are at least two possibilities to explain the above observations. 1) One of the two genes is functional and another is not. 2) The differences between single and double mutants can only be observed under certain conditions. To distinguish between the two possibilities, we created chromosomal deletion strains by targeting each of the two repeats, followed by deleting the single remaining DDI2 or DDI3 gene. As seen in Fig. 5D, deletion of either single chromosomal region alone is sufficient to confer cyanamide or MMS sensitivity regardless of further DDI2 or DDI3 deletion, which effectively rules out the first possibility. To test the second possibility, we thought that perhaps the agent concentration is the most influential parameter. Indeed, at higher cyanamide concentrations, the double mutant becomes apparently more sensitive than the isogenic single mutant (Fig. 5E).
FIGURE 5.

Characterization of the ddi2/3 deletion mutants. A, schematic diagrams of DDI2/3 gene deletion constructs. B, yeast genomic PCR confirms the disruption of DDI2 and/or DDI3 genes. Lane M, 1-kb DNA ladders (from Invitrogen); lane 1, BY4741; lane 2, WXY3148 (ddi2/3Δ::LEU2); lane 3, WXY3147 (ddi2/3Δ::HIS3); lane 4, WXY2929 (ddi2/3Δ::LEU2 ddi2/3Δ::HIS3). Primers are DDI2–4 and DDI2–5, flanking the DDI2/DDI3 ORFs. C, serial dilution assay of ddi2/3Δ cells to test sensitivity to cyanamide (Cy) or MMS. Overnight yeast cultures were normalized, and diluted cells were equally spotted on YPD and cyanamide- or MMS-containing YPD plates. D, serial dilution assay of the mutants with disrupted duplicated chromosomal regions and/or ddi2/3 mutations showing killing by cyanamide or MMS. E, serial dilution assay of ddi2/3Δ cells showing sensitivity to cyanamide at higher concentrations. Plates shown in C–E were incubated at 30 °C for 3 days before being photographed.
Endogenous Cyanamide Hydratase Activity Encoded by DDI2/3
The above observations collectively suggest that DDI2/3 genes encode a cyanamide hydratase and are highly induced by cyanamide. To critically test this hypothesis, we optimized experimental conditions to obtain the yeast WCE and adapted an assay as described to monitor the reduction of cyanamide as a quantitative measurement of the cyanamide hydratase activity. As shown in Fig. 6, the cyanamide concentration in the reaction mixture remained stable over 24 h in the absence of WCE, and addition of WCE from wild-type cells without prior cyanamide treatment did not alter the cyanamide concentration. Addition of WCE from cyanamide-induced cells resulted in the reduction of cyanamide concentration by one-third in 5 h and more than 5-fold in 24 h, indicating that the cyanamide hydratase activity in yeast cells is inducible by cyanamide. In contrast, the WCE from the cyanamide-treated ddi2Δ ddi3Δ double mutant was unable to reduce cyanamide concentration even over 24 h. Hence, we conclude from the above observations that DDI2/3 is solely responsible for cyanamide metabolism in vivo and most likely encodes cyanamide hydratase. Interestingly, WCE from the cyanamide-treated ddi2/3Δ single mutant displayed an intermediate cyanamide hydratase activity in comparison with those of wild-type and the double mutant, which appears to be consistent with the observed cyanamide sensitivity data as shown in Fig. 5E.
FIGURE 6.
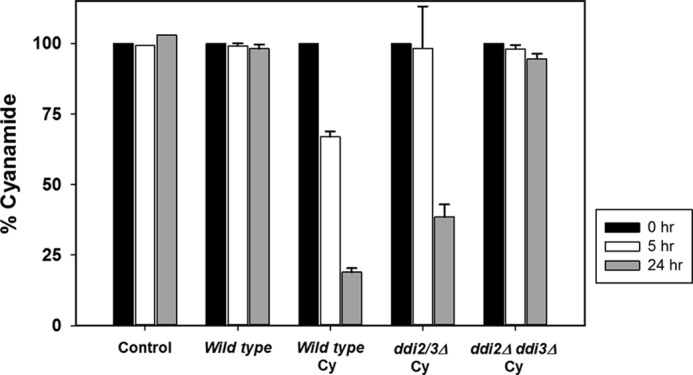
Influence of yeast WCE on cyanamide metabolism. 2 mm cyanamide was either incubated in the reaction mixture alone (Control) or in the presence of WCE from 1) wild-type cells without prior cyanamide treatment; 2) wild-type cells induced by cyanamide (Cy); and 3) the ddi2Δ ddi3Δ mutant cells induced by cyanamide. The cyanamide concentration was measured by a colorimetric assay at A530 nm. The cyanamide concentration at time 0 (filled bars) is taken as 100%. Open bars, after a 5-h incubation; and gray bars, after a 24-h incubation. The results are the average of three independent experiments with standard deviation.
Ddi2/3 Is Not Required for Its Own Induction
The fact that DDI2/3 is highly induced by cyanamide and encodes a cyanamide hydratase raises the possibility that DDI2/3 may serve as a sensor in its own induction pathway. To test whether Ddi2/3 is involved in such a regulatory circuit, we examined DDI2-lacZ induction by cyanamide or MMS in the ddi2Δ ddi3Δ double mutant. As seen in Fig. 4B, deletion of both DDI2 and DDI3 genes does not undermine DDI2-lacZ induction, indicating that Ddi2/3 is not involved in the transcriptional regulation of its own gene.
DISCUSSION
In this study, we revealed the biochemical activity and biological functions of two previously uncharacterized genes from the budding yeast S. cerevisiae. DDI2 and DDI3 were previously named by our laboratory because they are highly induced by the DNA-damaging agent MMS but had unassigned functions. The two genes turn out to be identical in their coding and promoter regions and are obviously co-regulated. Based on their protein sequence alignment and analysis, we developed a novel enzymatic assay and confirmed that the DDI2/3 genes encode a protein with CAH activity.
Cyanamide hydratase enzymes have not been characterized to the level of many other enzymes, and to date there is only one report on CAH enzymatic activity based on proteins extracted from the fungus M. verrucaria (12). This study reports for the first time real time monitoring of a recombinant cyanamide hydratase reaction, and our recorded Km value for Ddi2/3 (17 mm) is comparable with that of the reported native M. verrucaria CAH (27 mm). This relatively low substrate affinity indicates that either cyanamide is not a physiological substrate or that Ddi2/3 and CAH are not effective enzymes to hydrolyze cyanamide into urea. However, in addition to the above enzymatic assay, several pieces of evidence are consistent with Ddi2/3 as a bona fide cyanamide hydratase. First, because cyanamide is synthesized by vetch species, the presence of such an enzyme may be vital to the survival of soil fungi. Second, the expression of DDI2/3 is highly induced by cyanamide, which may be explained by the relatively poor efficiency of the enzyme for the reaction (kcat/Km = 5.67 × 102 m−1 s−1). Third, we have demonstrated that deletion of DDI2 and DDI3 genes sensitize cells to as low as 2 mm cyanamide in the medium, suggesting that they protect cells from cyanamide toxicity. Fourth, yeast cells lacking DDI2/3 genes or wild-type cells without prior cyanamide induction were unable to metabolize cyanamide, whereas wild-type cells treated with cyanamide are able to metabolize cyanamide, indicating a cyanamide-inducible hydratase activity in vivo. Finally, microorganisms with putative cyanamide hydratase activity may be able to utilize cyanamide as a carbon and/or nitrogen source. To this end, it is of great interest to notice that the budding yeast DUR1,2 gene encodes two ureases that effectively convert urea to ammonia and CO2 (35).
DDI2/3 encodes a member of the HD domain family of proteins, and its HD domain is required for the cyanamide hydratase activity. Well studied members of this family include dGTPase, tRNA nucleotidyltransferase, and 5′-deoxyribonucleotidase YfbR in E. coli, dNTP triphosphohydrolase in Thermus thermophilus, and phosphodiesterase in S. cerevisiae (36–41). These enzymes are involved in nucleic acid metabolism, signal transduction, and possibly other functions in bacteria, archaea, and eukaryotes. Structural analyses of HD family members reveal that conserved HD residues are metal-binding ligands (42), and crystallographic studies on some functional HD domain-containing phosphohydrolases reveal that the conserved HD residues bind to metal ions such as nickel, zinc, or iron, and substrates interact with the nearby amino acids (43, 44). It appears that Ddi2 purified from bacteria cells contains zinc (data not shown), indicating that it is indeed a bona fide member of this superfamily.
This study also raises at least two questions to be addressed by future investigations. First, why is DDI2/3 also induced by Sn2-type DNA-methylating agents? We are entertaining two possibilities as follows. (i) Methylating agents like MMS or their derivatives share similar molecular structure with cyanamide and hence may serve as a cyanamide analog. However, MMS does not appear to be a small molecular inhibitor of Ddi2/3 enzymatic activity (data not shown), although deletion of both DDI2 and DDI3 genes does result in an increased sensitivity to MMS. Nevertheless, because Ddi2/3 is not involved in the signal transduction leading to DDI2/3 induction by cyanamide or MMS, the definite answer to this question has to wait until the above signal transduction cascade is established. (ii) Cyanamide is involved in DNA metabolism. Cyanamide has also been regarded as an inhibitor of aldehyde dehydrogenase and carbonic anhydrase (45), but to date it has not been reported to exhibit a DNA-damaging effect. Interestingly enough, carbonic anhydrase catalyzes cyanamide hydration to urea, which in turns serves as its inhibitor (45). Budding yeast NCE103 encodes a putative carbonic anhydrase (46), but apparently its cyanamide metabolism activity is relatively low (if any) compared with Ddi2/3 in cells exposed to cyanamide. However, it has been reported that pyrimidine ribonucleotides can be synthesized from cyanamide along with other chemicals under prebiotically plausible conditions (47), leaving this possibility open. Second, why are the DDI2 and DDI3 genes located in the core of a duplicated chromosomal region? The Saccharomyces genome database reveals that the haploid laboratory strain contains up to 30% genomic duplications (48). However, the chromosome VI and XIV regions where DDI2 and DDI3 reside are the only area (except the rDNA cluster) in which the nucleotide sequences are highly conserved with three genes and their flanking regions nearly identical in sequence. Among these three pairs of genes, SNO2/3 and SNZ2/3 are found to be regulated by thiamine (49, 50), and their adjacent THI5 and THI2 genes are related to thiamine (vitamin B1) synthesis (51), suggesting that the duplicated regions contain a gene cluster related to thiamine metabolism. Thiamine is a very important physiological molecule because it serves as a cofactor (in the form of thiamine diphosphate) for several enzymes involved primarily in carbohydrate catabolism. Thiamine is composed of pyrimidine and thiazole rings linked by a methylene bridge, whereas cyanamide can be utilized in pyrimidine synthesis (52). Hence, it is plausible to speculate that cyanamide could be utilized by Ddi2/3 in pyrimidine rings and further in thiamine synthesis. Future studies are needed to determine whether and how Ddi2/3 is involved in thiamine metabolism. It is also of great interest to understand whether maintaining such duplicated gene clusters with high degrees of nucleotide sequence identity confer a selective advantage for yeast growth. To this end, we have created the first dual gene deletion in this region and also made chromosomal deletions of each duplicated gene cluster, which will facilitate future investigations. As an initial step toward this goal, we noticed that deletion of a single DDI2/3 gene or particularly either chromosome gene cluster sensitizes cells to cyanamide and MMS to an extent comparable with that of the double gene deletion, indicating that the cluster duplication indeed contributes to cellular protection against environmental stresses.
Acknowledgments
We thank Michelle Hanna for technical assistance and proofreading the manuscript. We also thank Dr. Mark Patchett, Massey University, Palmerston North, New Zealand, for valuable suggestions regarding the enzyme assay.
This work was supported in part by the Natural Sciences and Engineering Research Council of Canada Discovery Grants RGPIN-2014-04580 and SAM 262138-2011 (to W. X.).
- MMS
- methyl methanesulfonate
- WCE
- whole cell extract
- CAH
- cyanamide hydratase.
REFERENCES
- 1. Fu Y., Zhu Y., Zhang K., Yeung M., Durocher D., Xiao W. (2008) Rad6-Rad18 mediates a eukaryotic SOS response by ubiquitinating the 9-1-1 checkpoint clamp. Cell 133, 601–611 [DOI] [PubMed] [Google Scholar]
- 2. Liu Y., Dai H., Xiao W. (1997) UAS(MAG1), a yeast cis-acting element that regulates the expression of MAG1, is located within the protein coding region of DDI1. Mol. Gen. Genet. 255, 533–542 [DOI] [PubMed] [Google Scholar]
- 3. Zhu Y., Xiao W. (1998) Differential regulation of two closely clustered yeast genes, MAG1 and DDI1, by cell-cycle checkpoints. Nucleic Acids Res. 26, 5402–5408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhu Y., Xiao W. (2004) Pdr3 is required for DNA damage induction of MAG1 and DDI1 via a bi-directional promoter element. Nucleic Acids Res. 32, 5066–5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen J., Derfler B., Maskati A., Samson L. (1989) Cloning a eukaryotic DNA glycosylase repair gene by the suppression of a DNA repair defect in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 86, 7961–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen J., Derfler B., Samson L. (1990) Saccharomyces cerevisiae 3-methyladenine DNA glycosylase has homology to the AlkA glycosylase of E. coli and is induced in response to DNA alkylation damage. EMBO J. 9, 4569–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clarke D. J., Mondesert G., Segal M., Bertolaet B. L., Jensen S., Wolff M., Henze M., Reed S. I. (2001) Dosage suppressors of pds1 implicate ubiquitin-associated domains in checkpoint control. Mol. Cell. Biol. 21, 1997–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White R. E., Dickinson J. R., Semple C. A., Powell D. J., Berry C. (2011) The retroviral proteinase active site and the N terminus of Ddi1 are required for repression of protein secretion. FEBS Lett. 585, 139–142 [DOI] [PubMed] [Google Scholar]
- 9. Fu Y., Pastushok L., Xiao W. (2008) DNA damage-induced gene expression in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 32, 908–926 [DOI] [PubMed] [Google Scholar]
- 10. Fu Y. (2008) The Regulatory Network Controlling DNA Damage Responses in Saccharomyces cerevisiae. Ph.D. thesis, University of Saskatchewan [Google Scholar]
- 11. Aravind L., Koonin E. V. (1998) The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 23, 469–472 [DOI] [PubMed] [Google Scholar]
- 12. Maier-Greiner U. H., Obermaier-Skrobranek B. M., Estermaier L. M., Kammerloher W., Freund C., Wülfing C., Burkert U. I., Matern D. H., Breuer M., Eulitz M. (1991) Isolation and properties of a nitrile hydratase from the soil fungus Myrothecium verrucaria that is highly specific for the fertilizer cyanamide and cloning of its gene. Proc. Natl. Acad. Sci. U.S.A. 88, 4260–4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stransky H., Amberger A. (1973) Isolation and properties of a cyanamide hydratase (Ec-Group4,2,1) from Myrothecium verrucaria Alb and Schw. Zeitschrift Fur Pflanzenphysiol. 70, 74–87 [Google Scholar]
- 14. Kamo T., Hiradate S., Fujii Y. (2003) First isolation of natural cyanamide as a possible allelochemical from hairy vetch Vicia villosa. J. Chem. Ecol. 29, 275–283 [DOI] [PubMed] [Google Scholar]
- 15. Kamo T., Endo M., Sato M., Kasahara R., Yamaya H., Hiradate S., Fujii Y., Hirai N., Hirota M. (2008) Limited distribution of natural cyanamide in higher plants: occurrence in Vicia villosa subsp varia, V. cracca, and Robinia pseudo-acacia. Phytochemistry 69, 1166–1172 [DOI] [PubMed] [Google Scholar]
- 16. Kamo T., Sato M., Kato K., Hiradate S., Nakajima E., Fujii Y., Hirota M. (2006) Quantification of cyanamide contents in herbaceous plants. Biosci. Biotechnol. Biochem. 70, 2310–2312 [DOI] [PubMed] [Google Scholar]
- 17. Katla V. R., Syed R., Kuruva C. S., Kuntrapakam H. K., Chamarthi N. R. (2013) Synthesis of novel phosphorylated guanidine derivatives from cyanamide and their anti-inflammatory activity. Chem. Pharm. Bull. 61, 25–32 [DOI] [PubMed] [Google Scholar]
- 18. DeMaster E. G., Redfern B., Nagasawa H. T. (1998) Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem. Pharmacol. 55, 2007–2015 [DOI] [PubMed] [Google Scholar]
- 19. Sherman F., Fink G. R., Hicks J. (1983) Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 20. Ito H., Fukuda Y., Murata K., Kimura A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Myers A. M., Tzagoloff A., Kinney D. M., Lusty C. J. (1986) Yeast shuttle and integrative vectors with multiple cloning sites suitable for construction of lacZ fusions. Gene 45, 299–310 [DOI] [PubMed] [Google Scholar]
- 22. Jia X., Zhu Y., Xiao W. (2002) A stable and sensitive genotoxic testing system based on DNA damage induced gene expression in Saccharomyces cerevisiae. Mutat. Res. 519, 83–92 [DOI] [PubMed] [Google Scholar]
- 23. Guarente L. (1983) Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 101, 181–191 [DOI] [PubMed] [Google Scholar]
- 24. Ke S. H., Madison E. L. (1997) Rapid and efficient site-directed mutagenesis by single-tube ‘megaprimer’ PCR method. Nucleic Acids Res. 25, 3371–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pastushok L., Moraes T. F., Ellison M. J., Xiao W. (2005) A single Mms2 “key” residue insertion into a Ubc13 pocket determines the interface specificity of a human Lys63 ubiquitin conjugation complex. J. Biol. Chem. 280, 17891–17900 [DOI] [PubMed] [Google Scholar]
- 26. Berben G., Dumont J., Gilliquet V., Bolle P. A., Hilger F. (1991) The YDp plasmids: a uniform set of vectors bearing versatile gene disruption cassettes for Saccharomyces cerevisiae. Yeast 7, 475–477 [DOI] [PubMed] [Google Scholar]
- 27. Rothstein R. J. (1983) One-step gene disruption in yeast. Methods Enzymol. 101, 202–211 [DOI] [PubMed] [Google Scholar]
- 28. Goldstein A. L., McCusker J. H. (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541–1553 [DOI] [PubMed] [Google Scholar]
- 29. Weeks J., Koshiyama K., Maier-Greiner U., Schaeffner T., Anderson O. (2000) Wheat transformation using cyanamide as a new selective agent. Crop Sci. 40, 1749–1754 [Google Scholar]
- 30. Thaller M. C., Schippa S., Rossolini G. M. (1998) Conserved sequence motifs among bacterial, eukaryotic, and archaeal phosphatases that define a new phosphohydrolase superfamily. Protein Sci. 7, 1647–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marchler-Bauer A., Derbyshire M. K., Gonzales N. R., Lu S., Chitsaz F., Geer L. Y., Geer R. C., He J., Gwadz M., Hurwitz D. I., Lanczycki C. J., Lu F., Marchler G. H., Song J. S., Thanki N., et al. (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res. 43, D222–D226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steller W. A., Frankel B., Morgan P. W. (1965) Defoliant residues, determination of cyanamide residues on ginned cottonseed. J. Agric. Food Chem. 10.1021/Jf60140a011 [DOI] [Google Scholar]
- 33. Zhang X. H., Zhong W. Q., Widholm J. M. (2005) Expression of a fungal cyanamide hydratase in transgenic soybean detoxifies cyanamide in tissue culture and in planta to provide cyanamide resistance. J. Plant Physiol. 162, 1064–1073 [DOI] [PubMed] [Google Scholar]
- 34. Mondzac A., Ehrlich G. E., Seegmiller J. E. (1965) An enzymatic determination of ammonia in biological fluids. J. Lab. Clin. Med. 66, 526–531 [PubMed] [Google Scholar]
- 35. Olson J. A., Anfinsen C. B. (1953) Kinetic and equilibrium studies on crystalline l-glutamic acid dehydrogenase. J. Biol. Chem. 202, 841–856 [PubMed] [Google Scholar]
- 36. Zimmerman M. D., Proudfoot M., Yakunin A., Minor W. (2008) Structural insight into the mechanism of substrate specificity and catalytic activity of an HD-domain phosphohydrolase: The 5′-deoxyribonucleotidase YfbR from Escherichia coli. J. Mol. Biol. 378, 215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kondo N., Kuramitsu S., Masui R. (2004) Biochemical characterization of TT1383 from Thermus thermophilus identifies a novel dNTP triphosphohydrolase activity stimulated by dATP and dTTP. J. Biochem. 136, 221–231 [DOI] [PubMed] [Google Scholar]
- 38. Yakunin A. F., Proudfoot M., Kuznetsova E., Savchenko A., Brown G., Arrowsmith C. H., Edwards A. M. (2004) The HD domain of the Escherichia coli tRNA nucleotidyltransferase has 2′,3′-cyclic phosphodiesterase, 2′-nucleotidase, and phosphatase activities. J. Biol. Chem. 279, 36819–36827 [DOI] [PubMed] [Google Scholar]
- 39. Rao F., Qi Y., Murugan E., Pasunooti S., Ji Q. (2010) 2′,3′-cAMP hydrolysis by metal-dependent phosphodiesterases containing DHH, EAL, and HD domains is non-specific: Implications for PDE screening. Biochem. Biophys. Res. Commun. 398, 500–505 [DOI] [PubMed] [Google Scholar]
- 40. Nagata M., Kaito C., Sekimizu K. (2008) Phosphodiesterase activity of CvfA is required for virulence in Staphylococcus aureus. J. Biol. Chem. 283, 2176–2184 [DOI] [PubMed] [Google Scholar]
- 41. Quirk S., Bessman M. J. (1991) dGTP triphosphohydrolase, a unique enzyme confined to members of the family Enterobacteriaceae. J. Bacteriol. 173, 6665–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brown P. M., Caradoc-Davies T. T., Dickson J. M., Cooper G. J., Loomes K. M., Baker E. N. (2006) Crystal structure of a substrate complex of myo-inositol oxygenase, a di-iron oxygenase with a key role in inositol metabolism. Proc. Natl. Acad. Sci. U.S.A. 103, 15032–15037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kondo N., Nakagawa N., Ebihara A., Chen L., Liu Z.-J., Wang B.-C., Yokoyama S., Kuramitsu S., Masui R. (2007) Structure of dNTP-inducible dNTP triphosphohydrolase: insight into broad specificity for dNTPs and triphosphohydrolase-type hydrolysis. Acta Crystallogr. D Biol. Crystallogr. 63, 230–239 [DOI] [PubMed] [Google Scholar]
- 44. Brown P. M., Caradoc-Davies T. T., Dickson J. M., Cooper G. J., Loomes K. M., Baker E. N. (2006) Purification, crystallization, and preliminary crystallographic analysis of mouse myo-inositol oxygenase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 811–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Briganti F., Mangani S., Scozzafava A., Vernaglione G., Supuran C. T. (1999) Carbonic anhydrase catalyzes cyanamide hydration to urea: is it mimicking the physiological reaction? J. Biol. Inorg. Chem. 4, 528–536 [DOI] [PubMed] [Google Scholar]
- 46. Götz R., Gnann A., Zimmermann F. K. (1999) Deletion of the carbonic anhydrase-like gene NCE103 of the yeast Saccharomyces cerevisiae causes an oxygen-sensitive growth defect. Yeast 15, 855–864 [DOI] [PubMed] [Google Scholar]
- 47. Powner M. W., Gerland B., Sutherland J. D. (2009) Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 459, 239–242 [DOI] [PubMed] [Google Scholar]
- 48. Coissac E., Maillier E., Netter P. (1997) A comparative study of duplications in bacteria and eukaryotes: the importance of telomeres. Mol. Biol. Evol. 14, 1062–1074 [DOI] [PubMed] [Google Scholar]
- 49. Padilla P. A., Fuge E. K., Crawford M. E., Errett A., Werner-Washburne M. (1998) The highly conserved, coregulated SNOand SNZ gene families in Saccharomyces cerevisiae respond to nutrient limitation. J. Bacteriol. 180, 5718–5726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rodríguez-Navarro S., Llorente B., Rodríguez-Manzaneque M. T., Ramne A., Uber G., Marchesan D., Dujon B., Herrero E., Sunnerhagen P., Pérez-Ortín J. E. (2002) Functional analysis of yeast gene families involved in metabolism of vitamins B1 and B6. Yeast 19, 1261–1276 [DOI] [PubMed] [Google Scholar]
- 51. Wightman R., Meacock P. A. (2003) The THI5 gene family of Saccharomyces cerevisiae: distribution of homologues among the hemiascomycetes and functional redundancy in the aerobic biosynthesis of thiamin from pyridoxine. Microbiology 149, 1447–1460 [DOI] [PubMed] [Google Scholar]
- 52. Hulme R., Zamora O. D., Mota E. J., Pastén M. A., Contreras-Rojas R., Miranda R., Valencia-Hernández I., Correa-Basurto J., Trujillo-Ferrara J., Delgado F. (2008) Cyanamide: a convenient building block to synthesize 4-aryl-2-cyanoimino-3,4-dihydro-1H-pyrimidine systems via a multicomponent reaction. Tetrahedron 10.1016/j.tet.2008.01.087 [DOI] [Google Scholar]