

Background: Flavin-dependent lysine monooxygenases are involved in siderophore biosynthesis and are promising bacterial drug targets.
Results: Biochemical and structural characterization of lysine monooxygenase from Nocardia farcinica (NbtG) is presented.
Conclusion: An unprecedented domain conformation blocks the proper binding of NAD(P)H in the active site, which explains the high level of uncoupling observed in NbtG.
Significance: The structural and biochemical data should aid in drug design.
Keywords: flavin, flavoprotein, iron metabolism, siderophore, virulence factor, C4a-hydroperoxyflavin, N-hydroxylating monooxygenases, flavin-dependent monooxygenase, lysine monooxygenase
Abstract
N-Hydroxylating monooxygenases are involved in the biosynthesis of iron-chelating hydroxamate-containing siderophores that play a role in microbial virulence. These flavoenzymes catalyze the NADPH- and oxygen-dependent hydroxylation of amines such as those found on the side chains of lysine and ornithine. In this work we report the biochemical and structural characterization of Nocardia farcinica Lys monooxygenase (NbtG), which has similar biochemical properties to mycobacterial homologs. NbtG is also active on d-Lys, although it binds l-Lys with a higher affinity. Differently from the ornithine monooxygenases PvdA, SidA, and KtzI, NbtG can use both NADH and NADPH and is highly uncoupled, producing more superoxide and hydrogen peroxide than hydroxylated Lys. The crystal structure of NbtG solved at 2.4 Å resolution revealed an unexpected protein conformation with a 30° rotation of the NAD(P)H domain with respect to the flavin adenine dinucleotide (FAD) domain that precludes binding of the nicotinamide cofactor. This “occluded” structure may explain the biochemical properties of NbtG, specifically with regard to the substantial uncoupling and limited stabilization of the C4a-hydroperoxyflavin intermediate. Biological implications of these findings are discussed.
Introduction
Flavin-dependent N-hydroxylating monooxygenases (NMOs)5 are involved in the biosynthesis of hydroxamate-containing siderophores. These enzymes catalyze the NADPH- and oxygen-dependent hydroxylation of amine groups on the side chains of l-ornithine (l-Orn), l-lysine (l-Lys), and some primary aliphatic diamines (1–6). Many of these NMOs are linked to virulence in pathogenic bacteria. For instance, gene deletion experiments have shown that the l-Orn monooxygenases SidA from Aspergillus fumigatus and PvdA from Pseudomonas aeruginosa as well as the l-Lys monooxygenase MbtG from Mycobacterium tuberculosis are essential for functional siderophore biosynthesis and virulence (7–9). It is possible that these enzymes would make promising drug targets for various infectious diseases because of their requirement for the production of virulence-conferring siderophores and the lack of any human homologs.
Substantial mechanistic and structural data has been obtained in recent years on the l-Orn monooxygenases SidA, PvdA, and, more recently, on Kutzneria sp. 744 l-Orn monooxygenase (KtzI) (10–12). These enzymes are very selective for l-Orn and are highly coupled, producing little or no hydrogen peroxide during catalysis (1, 13) as they stabilize the C4a-hydroperoxyflavin (FADOOH) intermediate long enough to allow effective hydroxylation of l-Orn (14–16). A different strategy for oxygen consumption regulation was found in KtzI where flavin movement from an “out” to “in” conformation controls the oxidized/reduced state of the cofactor, similarly to what happens in Class A monooxygenases such as p-hydroxybenzoate hydroxylase. Despite this difference, the structures of SidA, PvdA and KtzI all share a similar overall fold to other Class B flavin monooxygenases and a similar binding conformation for NADP+, which is important for stabilization of the FADOOH (6, 10, 17, 18).
Comparison of the structures of SidA in complex with l-Orn and l-Lys revealed the mechanism of substrate selectivity where the Nϵ of l-Orn is in a favorable position for hydroxylation by the distal oxygen of the FADOOH. In contrast, l-Lys protrudes farther into the active site in a suboptimal position, which causes breakdown of the FADOOH resulting in production of high levels of hydrogen peroxide and low levels of hydroxylated l-Lys (10).
In contrast to the significant mechanistic and structural data of l-Orn monooxygenases, relatively little is known about l-Lys monooxygenases. IucD from Escherichia coli was the first l-Lys monooxygenase characterized. IucD shares similar mechanistic features to SidA and PvdA, as this enzyme has been shown to be highly coupled and specific for NADPH and l-Lys (3, 19). Recently, characterization of the l-Lys monooxygenase MbsG from M. smegmatis showed that this enzyme can use both NADH and NADPH and is highly uncoupled, making more superoxide and hydrogen peroxide than hydroxylated Lys (20, 21). Also, unlike other NMOs where substrate binding accelerates turnover, l-Lys binding decreases the activity of MbsG ∼2-fold by regulating the reaction with NAD(P)H (21). These results suggest that there is a higher degree of mechanistic variability among NMOs than previously thought.
The biosynthesis of the siderophore nocobactin in Nocardia farcinica requires functioning of NbtG, a l-Lys monooxygenase that shares >50% of sequence identity with MbsG (Scheme 1) (22). The Nocardia genus is comprised of parasitic bacteria, which can infect both plants and animals, including humans, and has a high mortality rate due to antibiotic resistance (22). Here, we present a thorough biochemical and structural characterization of NbtG. The enzyme is an effective l-Lys monooxygenase that is also able to hydroxylate d-Lys. NbtG is unable to stabilize the FADOOH intermediate, which results in production of hydrogen peroxide and superoxide. The structure of NbtG was solved by x-ray crystallography and the results show an unexpected and unprecedented protein conformation that can rationalize the biochemical properties of NbtG, specifically with regard to the substantial uncoupling and limited stabilization of the FADOOH.
SCHEME 1.

A, reaction catalyzed by NbtG. NbtG utilizes NADPH and molecular oxygen (shown in red) to hydroxylate the N6-atom of l-Lys. NADP+ and H2O are byproducts of the catalytic cycle. B, siderophore nocobactin produced by N. farcinica. N6-Hydroxy-l-lysines are incorporated into the backbone of nocobactin (oxygens inserted by NbtG are shown in red).
EXPERIMENTAL PROCEDURES
Materials
The synthetic gene encoding for NbtG from N. farcinica IFM 10152 was obtained from GenScript (Piscataway, NJ). PmeI and SgfI were from Promega (Madison, WI). E. coli TOP10 and BL21-T1R chemically competent cells were from Invitrogen. pVP56K was obtained from the Center for Eukaryotic Structural Genomics (University of Wisconsin, Madison, WI). DNA sequencing was performed at the DNA sequencing facility of the Virginia Bioinformatics Institute (Blacksburg, VA). Protein purification was performed on an ÄKTA Prime Plus FPLC (GE Healthcare). l-Lys, d-Lys, NADPH, NADP+, buffers, and salts were from Fisher. Amino acid analysis was performed using an Acquity Ultraperformance Liquid Chromatography system (Waters, Milford, MA). Oxygen consumption studies were performed on a Hansatech Oxygraph (Norfolk, UK). WST-1, used for superoxide quantification, was from Dojindo Molecular Technologies, Inc. (Rockville, MD). Alcohol dehydrogenase from Thermoanaerobium brockii was obtained from Sigma. Glucose oxidase was from MP Biomedical (Solon, OH). Rapid-reaction studies were performed on an Applied Photophysics SX20 stopped-flow spectrophotometer (Leatherhead, UK) housed in a Coy Glove Box (Grass Lake, MI).
Cloning
To subclone the NbtG gene into the expression vector pVP56K, the pUC57:nbtG plasmid was incubated with PmeI and SgfI restriction endonucleases at 37 °C for 2 h. After digestion, the restriction endonucleases were inactivated by incubation at 65 °C for 30 min. The digested gene was then ligated into pVP56K, which was previously treated with PmeI and SgfI. The ligation reaction was then transformed into E. coli TOP10 cells. Five colonies were grown for plasmid purification and sequenced to determine proper ligation of nbtG into pVP56K.
Site-directed Mutagenesis
Mutagenesis was performed using the QuikChange (Agilent Technologies) method following the manufacturer's instructions. The wild-type NbtG gene, subcloned into the pVP56K plasmid, was used as the template. The mutations were confirmed by DNA sequencing at the Virginia Bioinformatics Core Sequencing Facility.
Protein Expression and Purification
A single colony of E. coli BL21-T1R transformed with pVP56K:nbtG was used to inoculate a 50-ml overnight LB culture flask (supplemented with 50 μg/ml kanamycin). After overnight incubation at 37 °C, 6 flasks containing 1 liter each of autoinduction media (23) (supplemented with 50 μg/ml kanamycin) were each inoculated with 10 ml of the overnight culture and grown at 37 °C with agitation at 250 rpm. When the optical density at 600 nm reached a value of ∼3, the temperature was lowered to 18 °C. After overnight incubation, the cells were harvested by centrifugation, and the cell pellet was stored at −80 °C. All subsequent steps were performed at 4 °C. Cell paste (50 g) was resuspended in 150 ml of buffer A (25 mm HEPES, 300 mm NaCl, 25 mm imidazole, 5% glycerol, pH 7.5) containing 1 mm phenylmethanesulfonyl fluoride, 100 μm FAD, and 25 μg/ml each of DNase, RNase, and lysozyme. This solution was stirred for 30 min. Cells were lysed by sonication using a Fisher Scientific Sonic Dismembrator Model 500 at 70% amplitude for 10 min (5 s pulses with 15 s delays). Next, the lysate was centrifuged at 35,000 × g for 1 h. The resulting supernatant was loaded at 5 ml/min onto 3 in-tandem 5-ml HisTrap FF crude columns (GE Healthcare) previously equilibrated with buffer A. After loading, the column was washed with buffer A followed by 100 ml of buffer A containing 75 mm imidazole. Bound His8-MBP-NbtG was eluted with 150 ml of buffer A containing 500 mm imidazole. Fractions were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and those containing His8-MBP-NbtG were combined. To liberate NbtG from the His8-MBP, 10 mg of tobacco etch virus protease was added, and the solution was then transferred to a 4,000 molecular weight cutoff dialysis bag and dialyzed in 2 liters of buffer A. The next day, the solution was loaded at 3 ml/min onto 3 in-tandem 5-ml HisTrap FF crude columns previously equilibrated with buffer A. In this step His8-MBP, His8-tobacco etch virus, and any uncleaved His8-MBP-NbtG remain bound to the column, whereas NbtG elutes in the flow-through. SDS-PAGE analysis of the flow-through showed highly pure NbtG; however, small amounts of His8-MBP-NbtG were still present. To remove the fusion from free NbtG, the sample was loaded at 3 ml/min onto 2 in-tandem 5-ml MBPTrap HP columns equilibrated in buffer B (25 mm HEPES, 500 mm NaCl, pH 7.5). The flow-through contained NbtG and was determined to be >95% pure by SDS-PAGE. The sample (∼20 ml) was dialyzed for 3 h in 1 liter of storage buffer (100 mm sodium phosphate, 50 mm NaCl, pH 7.5) then dialyzed in 1 liter of fresh storage buffer overnight. Dialyzed NbtG was concentrated to ∼300 μm, aliquoted (in ∼60 μl drops), and frozen in liquid nitrogen before storage at −80 °C. NbtG K184A seleno-l-methionine was produced using the metabolic inhibition method (24). Both seleno-l-methionine-labeled NbtG K184A and unlabeled enzymes were purified as described above.
Characterization of the Flavin Cofactor
A solution of NbtG (25 μm) was heated at 95 °C for 10 min. After incubation on ice for 5 min, the denatured protein was precipitated by centrifugation at 15,000 × g for 10 min. The supernatant containing the flavin cofactor was used for mass spectroscopy analysis as previously described (20, 21). The extinction coefficient of the bound FAD to NbtG was determined by the addition of 5% SDS and incubation at room temperature for 5 min. The spectrum of the liberated FAD was recorded, and an extinction coefficient at 452 nm of 12,600 m−1 cm−1 was calculated using the extinction coefficient for free FAD of 11,300 m−1 cm−1 .
NADP+ Binding
The binding of NADP+ to oxidized NbtG was monitored by recording the change in the flavin spectra at increasing concentrations of NADP+. Each solution consisted of 100 μl of 100 mm sodium phosphate, pH 7.5, NbtG (60 μm) and NADP+ (0–10 mm). The solutions were incubated on ice for 10 min before data collection. The spectra of NADP+ alone were subtracted from spectra of NADP+/NbtG at each NADP+ concentration. The absorbance differences showed the largest decrease at 395 nm. An extinction coefficient of 1,300 m−1cm−1 for this change in absorbance at 395 nm was determined and used to calculate the concentrations of NbtG-NADP+ complex.
Oxygen Consumption Assay
Oxygen consumption was monitored in 1-ml reactions of 100 mm sodium phosphate, pH 7.5. NAD(P)H was varied between 0.1 and 5 mm, l-Lys was varied between 0 and 5 mm, and d-Lys was varied between 0 and 100 mm. For assays where NAD(P)H was varied, no Lys, 2 mm l-Lys, or 10 mm d-Lys were present. In assays where Lys was varied, NAD(P)H was held constant at 2 mm. Assays were initiated with 1 μm NbtG and monitored on a Hansatech Oxygraph system at 25 °C under constant stirring.
Product Formation Assay
To determine if l-Lys was hydroxylated, NbtG (8 μm) was mixed with 5 mm NADPH and 1 mm l-Lys in 100 μl of 50 mm Tris-Cl, 100 mm NaCl, pH 8.0. At 0.5, 1, 2, and 3 min, the reaction was quenched with 200 μl of 100% acetonitrile and centrifuged at 13,000 rpm for 1 min. The supernatant was withdrawn and combined with 25 μl of 200 mm borate, pH 8.0. l-Lys was derivatized by adding 3.4 μl of FMOC-Cl, (150 mm) to 130 μl of the quenched reaction. This was allowed to incubate for 5 min at 25 °C. To remove any remaining derivatizing reagent, 158 μl of 1-aminoadamantane (53 mm) was added and incubated at room temperature for an additional 15 min. The sample was analyzed by reverse phase ultraperformance liquid chromatography by loading 2 μl onto a C18 AcQuity column (2.1 × 100 mm) equilibrated in 70% eluent A (0.1% trifluoroacetic acid in water) at 0.5 min/ml. The samples were eluted with a linear gradient from 30 to 100% eluent B (0.1% trifluoroacetic acid in acetonitrile) at 0.5 min/ml, and samples were monitored at 263 nm. The elution profiles of derivatized l-Lys and l-Lys-OH were consistent with previously published data (25).
Hydroxylated Lys was quantified by a variation of the Csaky iodine oxidation assay as previously described (20, 26). Each reaction consisted of 104 μl of 100 mm sodium phosphate, pH 7.5. NAD(P)H was varied between 0.1 and 10 mm, l-Lys was varied between 0.1 and 5 mm, and d-Lys was varied between 0 and 10 mm. In assays where NAD(P)H was varied, 2 mm l-Lys, or 10 mm d-Lys were used. For reactions with varying concentrations of d/l-Lys, 2 mm NAD(P)H was used. Reactions were initiated with 2 μm NbtG and allowed to proceed at 25 °C for 10 min while shaking at 750 rpm.
Hydrogen Peroxide Formation Assay
Hydrogen peroxide formed during the reaction was measured using the Pierce hydrogen peroxide detection kit as described previously (20, 21). Each reaction consisted of 104 μl of 100 mm sodium phosphate, pH 7.5. NAD(P)H was varied between 0.1 and 10 mm in the absence of d/l-Lys or with l-Lys or d-Lys held constant at 2 mm and 10 mm, respectively. Reactions were initiated with 2 μm NbtG and allowed to proceed at 25 °C for 10 min under constant orbital shaking at 750 rpm.
Superoxide Formation Assay
Superoxide formed during the reaction was measured by coupling the reaction with WST-1 as described previously (20, 21). Each reaction consisted of 200 μl of 100 mm sodium phosphate, pH 7.5. NAD(P)H was varied between 0.1 and 10 mm in the absence of d/l-Lys or with l-Lys or d-Lys held constant at 2 and 10 mm, respectively. Reactions were initiated with 2 μm NbtG and allowed to proceed at 25 °C for 10 min while shaking at 750 rpm.
Flavin Reduction
Flavin reduction was monitored at 25 °C in 100 mm sodium phosphate, pH 7.5, in a stopped-flow spectrophotometer in single mixing mode. The stopped-flow spectrophotometer system and buffers were made anaerobic as described previously (21). Oxidized NbtG (15 μm after mixing) was mixed with various concentrations of NADPH (0.025–5 mm after mixing). The reaction was monitored with a photodiode array spectrophotometer until flavin reduction was complete. This was repeated with l-Lys or d-Lys at 2 mm and 10 mm (after mixing), respectively.
Flavin Oxidation
Flavin oxidation was monitored at 25 °C in 100 mm sodium phosphate, pH 7.5, in a stopped-flow spectrophotometer in double mixing mode. In the first mixing step, 30 μm NbtG (after mixing) was allowed to react with a stoichiometric concentration of NADPH (30 μm after mixing) for 500 s. In the second mixing step, reduced NbtG was reacted with various concentrations of molecular oxygen (100–600 μm after mixing). The reaction was monitored using a photodiode array spectrophotometer until flavin oxidation was complete. Oxygen saturated buffer (1.2 mm) was obtained by bubbling 100% oxygen for 30 min at 25 °C. Various oxygen concentrations were obtained by mixing with appropriate volumes of anaerobic buffer. Experiments with l-Lys or d-Lys contained 2 and 10 mm, respectively. To determine if excess NADP+ was required to stabilized the FADOOH intermediate, NADP+ was added to the second mix (0–10 mm after mixing) during oxidation.
Synthesis of Deuterated NADPH
R-4H2-NADPH was synthesized by the method of Jeong and Gready (27) with some minor modifications as previously described (28). Briefly, 25 mg of NADP+ (5.6 mm final concentration), 480 μl of 2-propanol-d8 (1 m final concentration), and 50 units of alcohol dehydrogenase (T. brockii) were added to 6 ml of 25 mm Tris-Cl, pH 9.0. The reaction was allowed to proceed for 30 min at 40 °C with light agitation until the A260/A340 ratio reached a value of ∼2.5.
Data Analysis
The kinetic data were fit using KaleidaGraph (Synergy Software, Reading, PA). Steady-state kinetic data were fit to the Michaelis-Menten equation. Data that exhibited substrate inhibition were fit to Equation 1 to obtain the turnover number (kcat), the Michaelis constant (Km), and the substrate inhibition constant (Kis). The decrease in absorbance at 452 nm was fit to a double exponential decay equation (Equation 2), and the resulting kobs values were plotted as a function of NADPH concentration. These data were fit using Equation 3 to determine the maximum rate of flavin reduction (kred) and KD values. The rate of flavin oxidation was determined by fitting the increase in absorbance at 452 nm to a single exponential rise equation (Equation 4). The resulting kobs values were plotted as a function of oxygen concentration and fit to a linear equation to determine the bimolecular rate constant for flavin oxidation (kox).
 |
 |
Crystallization and Structure Determination
Purified samples of NbtG (6 mg/ml) in 25 mm HEPES/NaOH, pH 7.5, 500 mm NaCl, and 5 mm NADP+ in the presence of either l-Lys or d-Lys (various concentrations ranging from 5 to 100 mm) were used for crystallization screenings. As no crystals were obtained in these initial trials, surface mutagenesis experiments were carried out. Using the surface entropy reduction prediction server (SERp Server) (29), Lys-184, Lys-200, and Lys-202 were selected and mutated to Ala. After initial screening, promising conditions were found only with the K184A variant. As shown by the crystal structure, Lys-184 is on a loop, which is fully exposed to the solvent at >20 Å distance from the flavin ring. Moreover, the kinetic properties were very similar to those of the wild type (data not shown). Yellow crystals of the K184A NbtG variant grew in 3–4 days at 20 °C using the hanging drop vapor diffusion method by mixing 1 μl of protein solution with an equal volume of a reservoir solution containing 10% w/v PEG 6,000, 5% v/v 2,4-methylpentanediol, and 0.1 m HEPES/NaOH, pH 7.5. Optimization was achieved by the micro-seeding technique using a seed stock generated by vortexing crystals for 90 s in a seed bead tube. Crystallization experiments were then performed by adding 0.3 μl of a 100-fold diluted seed stock solution into a drop with a 2:1 ratio of protein to reservoir solution. Similar procedures were followed to crystallize a seleno-l-methionine-labeled sample of K184A NbtG.
For x-ray data collection, NbtG crystals were soaked in a cryo solution containing 16% w/v PEG 6,000, 14% v/v 2,4-methylpentanediol, 0.1 m HEPES/NaOH, pH 7.5, 5 mm NADP+, d/l-Lys (at the concentration used for crystallization), 20% v/v glycerol, and flash-cooled in liquid nitrogen. Many crystals were screened for x-ray diffraction at the beamlines ID23-EH1 (European Synchrotron Radiation Facility, Grenoble, France), X06SA (Swiss Light Source, Villigen, Switzerland), and X29 (National Synchrotron Light Source, Upton, NY). Data processing and scaling were performed using MOSFLM (30) and programs of the CCP4 package (31). Final statistics of the collected datasets are reported in Table 1. The NbtG structure was solved by single-wavelength anomalous diffraction on the selenium-edge using the program SHELX (32). Because the crystals were highly isomorphous, it was possible to merge two datasets at 2.7 and 2.9 Å resolution to increase redundancy. The eight selenium sites (including that of the N-terminal methionine, which is left in the recombinant protein after His-tag cleavage) were identified and used for phasing with the program PHENIX (33). The quality of the initial electron density map was poor (figure of merit = 0.27), but a significant improvement was obtained after PHENIX density modification (averaging among the 4 molecules present in the asymmetric unit). This allowed us to perform autobuilding, which yielded good statistics (model-map CC = 62%, Rf = 36%, free Rf = 41%), although a large portion of the protein needed to be manually traced by COOT (34). Refinement was carried out using a native data set at 2.4 Å resolution with the program REFMAC5 (35). Figures were produced by CCP4mg (36).
TABLE 1.
Data collection and refinement statistics for the NbtG structure
SeMet, seleno-l-methionine.
| SeMet (crystal 1) | SeMet (crystal 2) | Native | |
|---|---|---|---|
| PDB code | 4d7e | ||
| Space group | P21 | P21 | P21 |
| a (Å) | 80.3 | 80.4 | 80.6 |
| b (Å) | 121.5 | 122.1 | 121.9 |
| c (Å) | 92.2 | 92.6 | 91.9 |
| β (°) | 97.7 | 97.6 | 97.7 |
| Wavelength (Å) | 0.98 | 0.98 | 1.07 |
| Resolution (Å) | 2.7 | 2.9 | 2.4 |
| Rsyma,b (%) | 10.1 (96.8) | 10.8 (99.5) | 7.6 (98.4) |
| CC1/2b,c | 0.997 (0.423) | 0.997 (0.627) | 0.999 (0.563) |
| Completenessb (%) | 85.4 (79.9) | 100.0 (99.9) | 99.2 (98.5) |
| Anomalous completenessb (%) | 82.3 (78.3) | 99.6 (99.0) | |
| Redundancy | 6.1 (6.4) | 7.1 (7.1) | 6.5 (6.3) |
| Anomalous redundancy | 3.0 (3.1) | 3.6 (3.6) | |
| Unique reflections | 40862 | 39427 | 68,260 |
| I/σb | 13.4 (1.4) | 14.2 (1.8) | 12.3 (1.0) |
| Protein/FAD atoms (N) | 11,431/53 × 4 | ||
| Water atoms (N) | 239 | ||
| Average B value (Å2) | 43.5 | ||
| Rcrystd (%) | 20.7 | ||
| Rfreed (%) | 28.2 | ||
| Root mean square bond length (Å) | 0.013 | ||
| Root mean square bond angles (°) | 1.7 |
a Rsym = Σ|Ii − 〈I〉|/ΣIi, where Ii is the intensity of ith observation, and 〈I〉 is the mean intensity of the reflection.
b Values in parentheses are for reflections in the highest resolution shell.
c A cut-off criterion for resolution limits was applied on the basis of the mean intensity correlation coefficient of half-subsets of each dataset (CC1/2) (45).
d Rcryst = Σ|Fobs − Fcalc|/Σ|Fobs|, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively. Rcryst and Rfree were calculated using the working and test sets, respectively.
RESULTS
Purification of Wild-type NbtG and Variants
Wild-type NbtG and the K184A isoform used for crystallography were expressed as a fusion to His8-MBP in pVP56K. This was critical to obtaining soluble active enzymes, as expression with only a His8 tag produced insoluble protein. Overall, a yield of ∼30 mg of purified NbtG was obtained per liter of growth media with a flavin incorporation of ∼75%. High resolution MALDI-TOF of the extracted flavin from the active site of NbtG yielded an m/z value of 784.15, which is consistent with FAD (data not shown). The flavin spectrum of NbtG displayed characteristic absorbance maxima at 380 and 452 nm for oxidized FAD (Fig. 1). The extinction coefficient at 452 nm was 12,600 m−1cm−1. The same results were obtained for the NbtG P238R and K64A variants.
FIGURE 1.
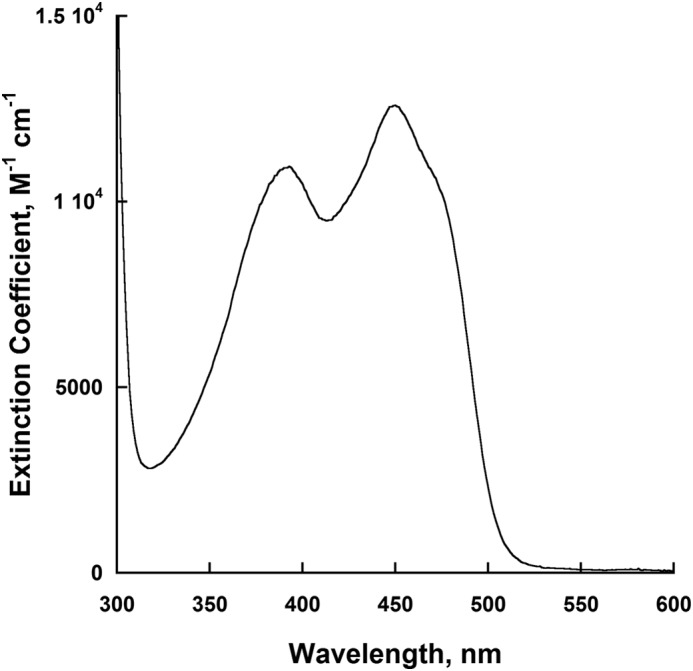
UV-visible spectrum of the flavin bound to NbtG, which has absorbance peaks at 380 nm and 452 nm.
Oxygen Consumption
The activity of NbtG was monitored by measuring the rate of oxygen consumption with either NADPH or NADH (Table 2). NbtG is highly active in the absence of l-Lys, having ∼2-fold higher activity with NADPH as compared with NADH. Under these conditions, NbtG functions as an oxidase, as no hydroxylation occurs. With NADPH, in the presence of l-Lys a ∼2-fold increase in the kcat value was determined, and a ∼5-fold increase was determined when d-Lys was used as substrate. When NADH was used as the coenzyme, the kcat values increased ∼4- and ∼8-fold in the presence of l-Lys and d-Lys, respectively. In the presence of l-Lys the kcat/KNADPH was only 2-fold higher than for kcat/KNADH. In the presence of d-Lys the kcat/KNADPH value was ∼4-fold higher. Thus, NbtG does not have significant coenzyme selectivity, in contrast to other Class B flavin monooxygenases, which are all highly selective for NADPH (1, 37–39).
TABLE 2.
Activity of NbtG monitored by oxygen consumption
Conditions were 100 mm sodium phosphate, pH 7.5, 25 °C. l-Lys and d-Lys were present at saturating concentrations (2 mm for l-Lys and 10 mm for d-Lys).
| Parameter | No Lys | l-Lys | d-Lys |
|---|---|---|---|
| NADPH | |||
| kcat (s−1) | 0.58 ± 0.02 | 1.08 ± 0.01 | 2.9 ± 0.1 |
| KNADPH (mm) | 0.20 ± 0.02 | 0.14 ± 0.01 | 0.51 ± 0.06 |
| kcat/KNADPH (m−1s−1) | 2900 ± 300 | 7700 ± 500 | 5600 ± 700 |
| NADH | |||
| kcat (s−1) | 0.32 ± 0.01 | 1.29 ± 0.04 | 2.5 ± 0.1 |
| KNADH (mm) | 0.36 ± 0.05 | 0.40 ± 0.04 | 1.6 ± 0.2 |
| kcat/KNADH (m−1s−1) | 900 ± 100 | 3300 ± 400 | 1500 ± 200 |
Lysine Hydroxylation
NbtG displayed significant activity as measured by the oxygen consumption assay. To determine if hydroxylation of l-Lys was taking place, NbtG was reacted with NADPH in excess and 1 mm l-Lys, and the reaction was monitored by ultraperformance liquid chromatography after derivatization of the amino acid as described under “Experimental Procedures.” The results clearly show that NbtG is able to produce l-Lys-OH (Fig. 2). To quantitate the amount of l-Lys-OH produced, the Csaky iodine oxidation assay was performed. The reaction was initially done using NADPH as the reducing substrate (Table 3). Under fully coupled conditions, one equivalent of hydroxylated d/l-Lys should be produced for every 1 eq of oxygen consumed. Very similar kcat values were determined for hydroxylation of d/l-Lys; however, the values were ∼3–7-fold lower than the kcat values obtained in the oxygen consumption assay. These results suggest that NbtG is highly uncoupled, producing reactive oxygen species in addition to hydroxylated d/l-Lys. Substrate inhibition was observed when NADPH was varied in the presence of saturating levels of either d- or l-Lys, with a lower KI(NADPH) value with l-Lys. The kcat/Km value for l-Lys was ∼6-fold higher than for d-Lys, which originates mainly from a lower Km value for l-Lys. Weak substrate inhibition was observed with l-Lys, which is a common feature observed in other NMOs. With d-Lys, no substrate inhibition was observed, most likely because of the lower affinity. Very similar results were observed when NADH was used as the coenzyme (Table 3).
FIGURE 2.

Production of hydroxylated lysine (l-Lys-OH) by NbtG. A, scheme of the derivatization reaction of l-Lys with FMOC-Cl. FMOC-Cl can react with the amino groups of either l-Lys or l-Lys-OH. B, ultraperformance liquid chromatography chromatogram (263 nm) showing the elution of l-Lys and l-Lys-OH derivatized with FMOC-Cl. The inset shows a zoomed-in view of the l-Lys peak decreasing and the l-Lys-OH peak increasing between 0 and 3 min.
TABLE 3.
Activity of NbtG monitored by Lys hydroxylation
Conditions were 100 mm sodium phosphate, pH 7.5, 25 °C. The final concentration of l-Lys was 2 mm and of d-Lys was 10 mm. Uncoupling was calculated using the expression (1 − kcat(N-OH)/kcat(O2)) × 100, where kcat(N-OH) is the kcat calculated measuring hydroxylated d/l-Lys, and kcat(O2) is kcat from the oxygen consumption assay.
| Parameter | NADPH |
NADH |
||
|---|---|---|---|---|
| l-Lys | d-Lys | l-Lys | d-Lys | |
| kcat (s−1) | 0.32 ± 0.02 | 0.41 ± 0.03 | 0.62 ± 0.08 | 0.31 ± 0.02 |
| KNAD(P)H (mm) | 0.7 ± 0.2 | 0.5 ± 0.1 | 0.6 ± 0.1 | 3.8 ± 2.4 |
| KI (NAD(P)H) (mm) | 2.4 ± 0.6 | 35 ± 24 | 5.7 ± 1.7 | 2.1 ± 1.5 |
| kcat/KNAD(P)H (m−1 s−1) | 460 ± 130 | 520 ± 120 | 1100 ± 300 | 80 ± 50 |
| KLys (mm) | 0.35 ± 0.05 | 2.6 ± 0.3 | 0.9 ± 0.3 | 4.2 ± 0.6 |
| KI(Lys) (mm) | 15 ± 3 | 6.3 ± 2.5 | ||
| kcat/Km(Lys) (m−1 s−1) | 910 ± 140 | 100 ± 20 | 580 ± 230 | 70 ± 10 |
| Uncoupling (%) | 70 | 86 | 52 | 92 |
Reactive Oxygen Species Production
By comparing the kcat values measured in the oxygen consumption assay (in the presence of either l- or d-Lys) with the kcat values measured in the hydroxylation assay, it was determined that there was a substantial degree of uncoupling (less than one hydroxylated d/l-Lys per oxygen consumed) in the reaction catalyzed by NbtG. In general, coupling was higher with l-Lys than with d-Lys and did not significantly vary if NADPH or NADH were used (Table 3).
With enzymatic assays highlighting such a substantial uncoupling, we next asked the question as to the type of product generated, as flavoenzymes can produce both hydrogen peroxide and superoxide upon reacting with oxygen. The kinetic analysis indicated that the oxidase activity of NbtG partly produces superoxide and hydrogen peroxide, which were detected using both NADPH and NADH either in the absence or presence of d/l-Lys (Table 4). In summary, NbtG is rather inefficient in Lys hydroxylation because flavin reoxidation can efficiently occur through the direct generation of hydrogen peroxide and/or superoxide. This finding warranted a more in-depth kinetic and structural analysis to understand the features underlying such a functional property.
TABLE 4.
Oxygen reactive species produced during oxidation of NbtG
Conditions were 100 mm sodium phosphate, pH 7.5, 25 °C. The final concentration of l-Lys was 2 mm and of d-Lys 10 mm.
| Assay |
kcat (s−1) |
||
|---|---|---|---|
| No Lys | 2 mm l-Lys | 10 mm d-Lys | |
| Hydrogen peroxide | |||
| NADPH | 0.3 ± 0.1 | 0.20 ± 0.04 | 0.40 ± 0.05 |
| NADH | 0.27 ± 0.02 | 0.32 ± 0.05 | 0.40 ± 0.04 |
| Superoxide | |||
| NADPH | 0.30 ± 0.02 | 0.30 ± 0.02 | 0.23 ± 0.01 |
| NADH | 0.22 ± 0.02 | 0.170 ± 0.004 | 0.34 ± 0.02 |
Flavin Reduction
Flavin reduction was monitored as a function of NADPH in both the presence and absence of l-Lys (Fig. 3). The reaction was monitored by measuring the decrease in absorbance at 452 nm in the stopped-flow spectrophotometer. Two kinetic phases were observed in this reduction of NbtG. The first phase was ∼10-fold faster than the second, and both were dependent on NADPH concentration. The slow phase might originate from a small population of less active enzyme in our sample. Binding of either l-Lys or d-Lys did not change the rate of flavin reduction or the binding affinity for NADPH (Table 5).
FIGURE 3.
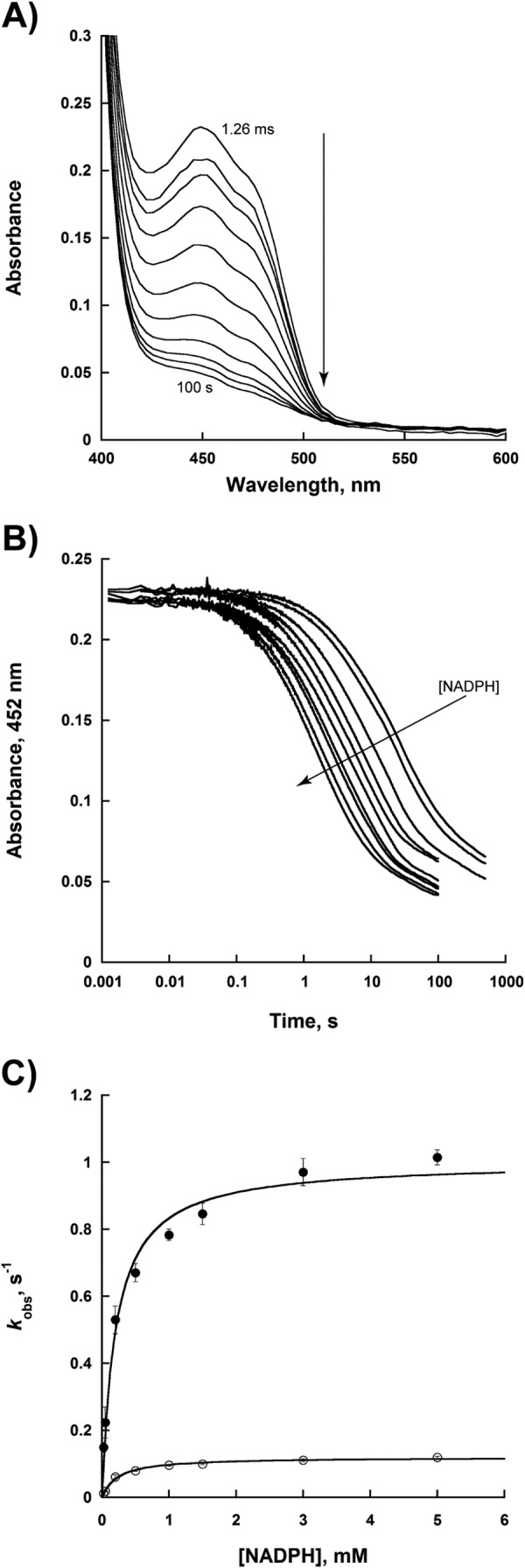
Flavin reduction with NADPH. A, spectra of reduction with 3 mm NADPH monitored over 100 s. B, change in the flavin absorbance at 452 nm at various concentrations of NADPH (0.025–5 mm). The changes in absorbance at 452 nm were fit to Equation 2 to obtain the kobs values of the two phases. C, dependence of the kobs values as a function of NADPH for the fast phase (●) and slow phase (○). The data were fit to Equation 3.
TABLE 5.
Rapid reaction kinetic parameters for NbtG with NADPH
Conditions were 100 mm sodium phosphate, pH 7.5, 25 °C.
| Parameter | Fast phase | Slow phase |
|---|---|---|
| No Lys | ||
| kred, s−1 | 1.00 ± 0.03 | 0.120 ± 0.002 |
| KD, mm | 0.20 ± 0.03 | 0.23 ± 0.03 |
| kred/KD, m−1s−1 | 5000 ± 760 | 520 ± 70 |
| l-Lys | ||
| kred, s−1 | 0.92 ± 0.03 | 0.106 ± 0.003 |
| KD, mm | 0.26 ± 0.04 | 0.28 ± 0.04 |
| kred/KD, m−1s−1 | 3540 ± 560 | 380 ± 60 |
| d-Lys | ||
| kred, s−1 | 1.00 ± 0.02 | 0.120 ± 0.001 |
| KD, mm | 0.20 ± 0.01 | 0.190 ± 0.006 |
| kred/KD, m−1s−1 | 5000 ± 270 | 620 ± 20 |
Kinetic Isotope Effects
The hydride transfer step was probed using deuterium kinetic isotope effects. Kinetic isotope effects were calculated by measuring the ratio of the kinetic values determined with NADPH and to those determined with R-[4-2H]NADPH. Under steady-state conditions a Dkcat value of 1.58 ± 0.03 and a Dkcat/Km value of 1.0 ± 0.1 were obtained. Under rapid reaction conditions, a kinetic isotope effect for flavin reduction was observed for both phases with Dkred values being 1.5 ± 0.1 and 1.4 ± 0.1 for the fast and slow phase, respectively. A Dkred/KD value of 0.7 ± 0.2 was measured for the fast phase, and a Dkred/KD value of 1.0 ± 0.3 was measured for the slow phase. These data are consistent with the hydride transfer step being only partially rate-limiting. Solvent kinetic isotope effects were measured to determine if a proton transfer occurs in the rate-limiting step in the reaction catalyzed by NbtG. A D2Okcat value of 2.43 ± 0.03 was determined, which indicates that the proton transfer step is significantly rate-limiting.
Flavin Oxidation
Flavin oxidation was monitored as a function of molecular oxygen concentration. NbtG oxidation occurs in a single step process without the stabilization of a FADOOH intermediate. In the absence of d/l-Lys, a rate of 3,100 ± 200 m−1 s−1 was determined. In the presence of l-Lys, this value increased to 3,900 ± 100 m−1 s−1, whereas the rate was 5,400 ± 400 m−1 s−1 in the presence of d-Lys (Fig. 4). The effect of excess NADP+ on flavin oxidation was also determined. The addition of 0.5–10 mm NADP+ did not change the rate of flavin oxidation, nor was there evidence of the spectra for the FADOOH intermediate (Fig. 4C).
FIGURE 4.
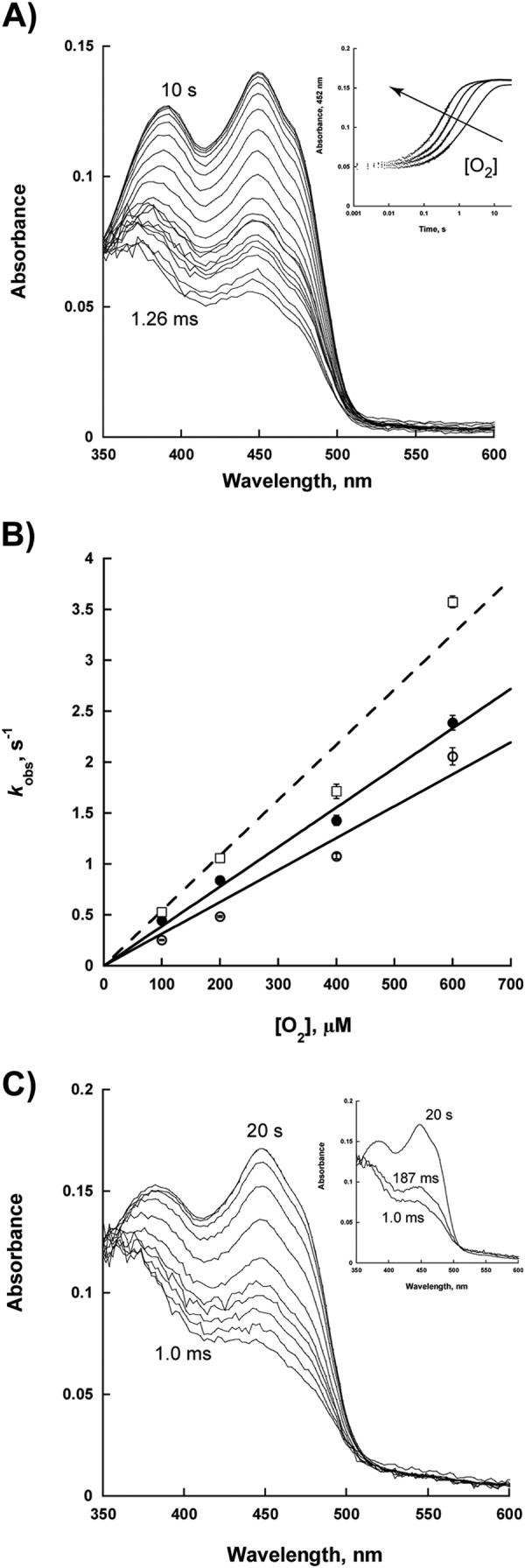
Flavin oxidation. A, spectra changes of the reaction of reduced NbtG with 270 μm O2. The inset shows the changes in absorbance at 452 nm as a function of oxygen concentration (100, 200, 400, and 600 μm O2). The data were fit to Equation 4. B, kobs values as a function of O2 without Lys (open circles), with 2 mm l-Lys (solid circles), and with 10 mm d-Lys (open squares, dashed line). C, spectra of NbtG flavin oxidation with 3 mm NADP+. The spectra of flavin oxidation were subtracted from the spectrum of 3 mm NADP+ alone. The inset shows the deconvoluted flavin spectra. The data were fit to a two-step process to determine if a transient C4a-hydroperoxyflavin is present. Pro-K (Applied Photophysics) was used to deconvolute the flavin spectra.
NADP+ Binding
It has been determined that NADP+ is essential for stabilization of the FADOOH intermediate in members of Class B flavin-dependent monooxygenases. In PvdA and SidA, NADP+ has similar affinity to NADPH. Using the spectral perturbation methods, a KD value of 2.0 ± 0.5 mm was determined for NADP+ binding to oxidized NbtG (Fig. 5). Thus, for NbtG, the KD for NADP+ is ∼10- fold higher than the KD for NADPH (Table 5).
FIGURE 5.
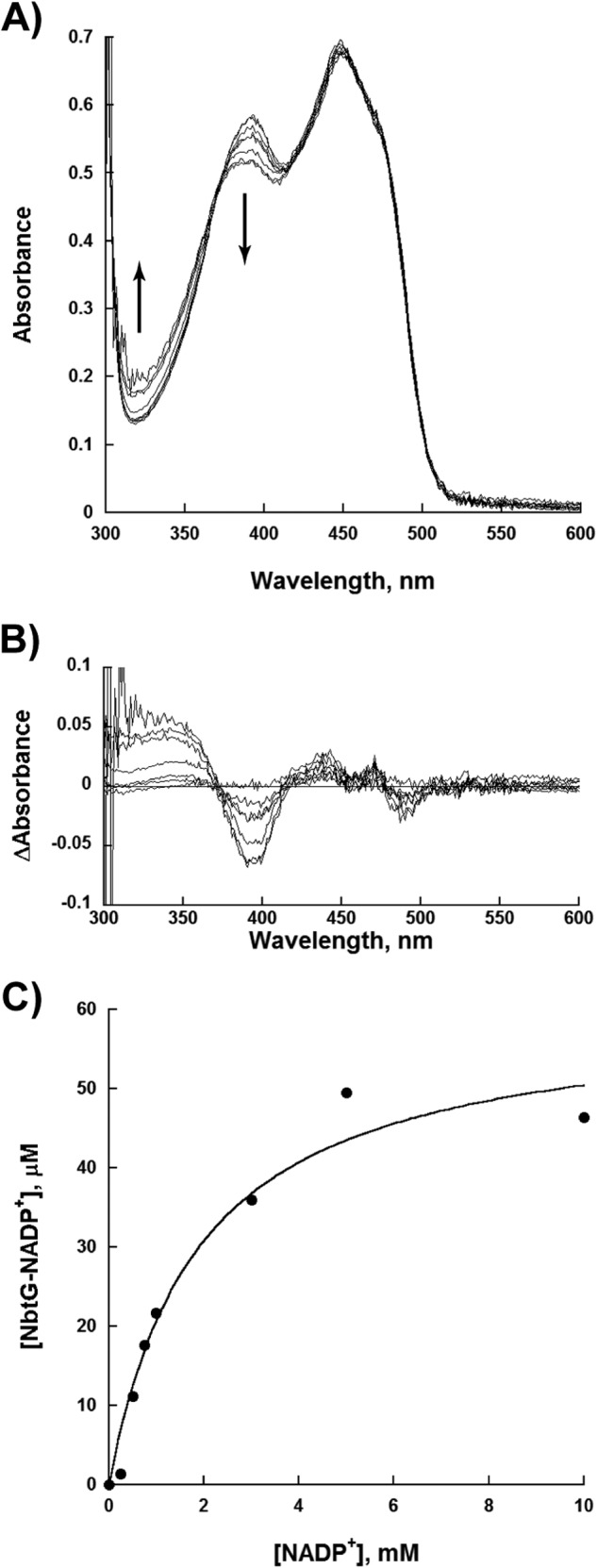
NADP+ binding. A, flavin spectra changes as a function of increasing concentrations of NADP+ (0–10 mm). The spectral changes show an increase in absorbance at 340 nm and a decrease in absorbance at 395 nm. B, spectra differences after subtracting the spectrum of NbtG with 0 mm NADP+. C, determination of the KD value of NADP+. The concentration of the NbtG-NADP+ complex was plotted as a function of NADP+ to determine a KD value.
Three-dimensional Structural Analysis of NbtG
The crystal structure of NbtG was solved by single-wavelength anomalous diffraction at the selenium edge and refined at 2.4 Å resolution (Table 1). The four NbtG molecules present in the asymmetric unit form a compact tetramer with the same arrangement observed in the l-Orn monooxygenases SidA (10) and PvdA (11) (their monomeric and dimeric structures, respectively, form a tetramer through application of crystallographic symmetry operators); in KtzI a similar but not identical tetramer was observed (12). The NbtG subunit folds into a two-domain topology similar to that of other NMOs: the FAD binding domain (residues 1–178 and 337–429), including the non-covalently bound FAD cofactor (Fig. 6B), and the NADPH binding domain (residues 179–336). Nearly all 429 residues of the protein could be modeled for each of the four polypeptide chains present in the asymmetric unit except for monomer D, in which ⅓ of the molecule (i.e. most of the NADP+ domain) lacks clear electron density (Fig. 6A).
FIGURE 6.
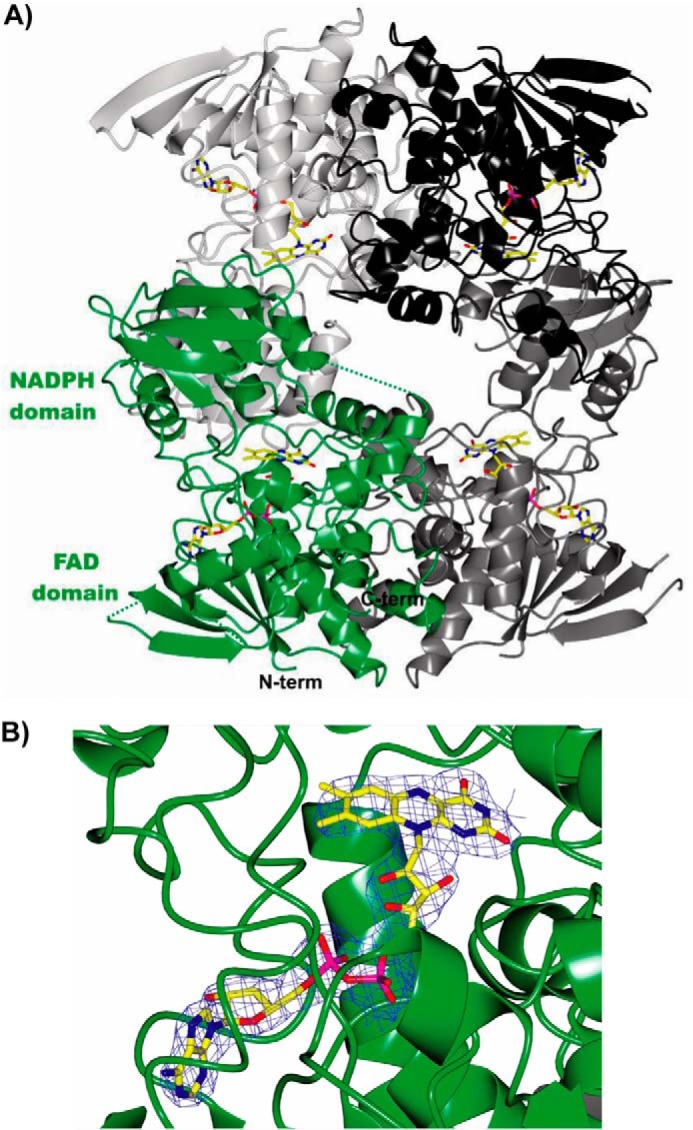
Crystal structure of NbtG. A, ribbon diagram of the crystallographic tetramer of NbtG observed in the asymmetric unit. Monomers A, B, C, and D of the tetramer are represented in green, light gray, dark gray, and black, respectively. The FAD cofactor is drawn as sticks for each monomer, with carbon, oxygen, nitrogen, and phosphorus atoms colored in yellow, red, blue, and magenta, respectively. For the sake of clarity, flexible regions that lack electron density are indicated only for monomer A as green dashed lines (residues 143–149, 158–160, 241–247; the C-terminal 414–429 residues could not be included in the final model). B, close-up view (same orientation as A) of FAD bound to monomer A with its refined weighted 2Fo − Fc electron density contoured at 1.2 σ.
The structures of the homologous SidA (10) and PvdA (11) NMOs were elucidated in complex with l-Orn and NADP+, whereas despite extensive screening of NbtG crystals grown in the presence of either NADP+/NAD+ or NADPH/NADH and/or d/l-Lys, no electron density for any ligand except for the FAD cofactor could be detected. NbtG shares about 20% sequence identity and the same domain topology with SidA, PvdA, and KtzI. However, comparison of NbtG structure with those of SidA, PvdA, and KtzI reveals a large change in domain orientation. More specifically, the program DynDom (40) indicates that the NADPH domain of NbtG is rotated by 33° with respect to the same domain of SidA using the superimposed FAD domains as reference. Such a rotation occurs through an axis passing through the residues (175–178 and 335–337 in NbtG) linking the two protein domains (Fig. 7A). This finding provides a rationale for the unsuccessful attempts to obtain the NADP+ and d/l-Lys bound to NbtG in this crystalline form. The domain rotation found in the NbtG structure makes the nicotinamide binding site in front of the FAD ring become occluded by an α-helical segment, preventing NADP(H) binding (Figs. 7B and 8). Likewise, structural comparison shows that the residues responsible for binding the l-Orn backbone group in SidA are conserved in NbtG (Lys-64, Asn-252, Ser-402), but their relative positions are modified (Fig. 8). In essence, the domain rotation featured by the NbtG structure drastically alters the active site geometry and accessibility. Differences in the NAD(P)+ binding site may also explain the lack of NAD+/NADP+ specificity found in NbtG. In SidA the position of the NADP+ adenine-ribose moiety is stabilized by a network of interactions that also involve the phosphate. In particular, the ribose 3′-OH is H-bonded to both the side chain and the peptidic nitrogen of Ser-254, whereas the phosphate interacts with Ser-325 and with Arg-279 (37). In NbtG, Arg-279 is substituted by Pro-238, and Ser-325 is substituted by Asp-277, whose longer side chain is likely to interact with the ribose rather than with the phosphate. All of these NbtG residues are also conserved in MbsG, which is consistent with the similar cofactor specificity properties of the mycobacterial enzyme.
FIGURE 7.
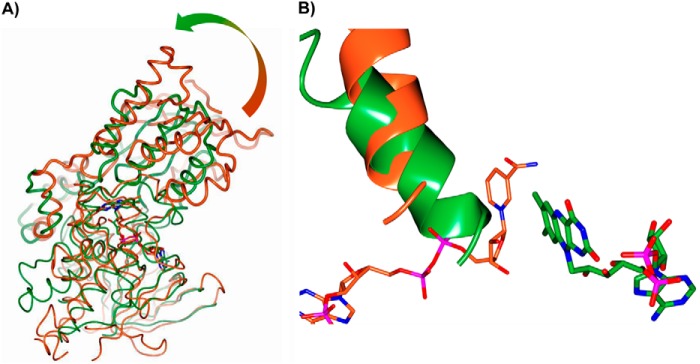
Comparison of SidA (PDB code 4b64; NADP+ complex) and NbtG. A, the large change in the orientations of the NADPH binding domains is visualized by superimposing NbtG (green) and SidA (orange) structures using their respective FAD binding domains (root mean square deviation = 2.5 Å for the equivalent Cα atoms). The shaded arrow outlines the additional 33° rotation, which is needed for the optimal superposition of the NADP+ binding domains (highlighted as bold worms). B, close-up view of the flavin site in NbtG (green) and SidA (orange). Because of the domain rotation, the nicotinamide-ribose site in NbtG is physically occluded by an α-helix (residues 215–229) of the NADP+ binding domain.
FIGURE 8.
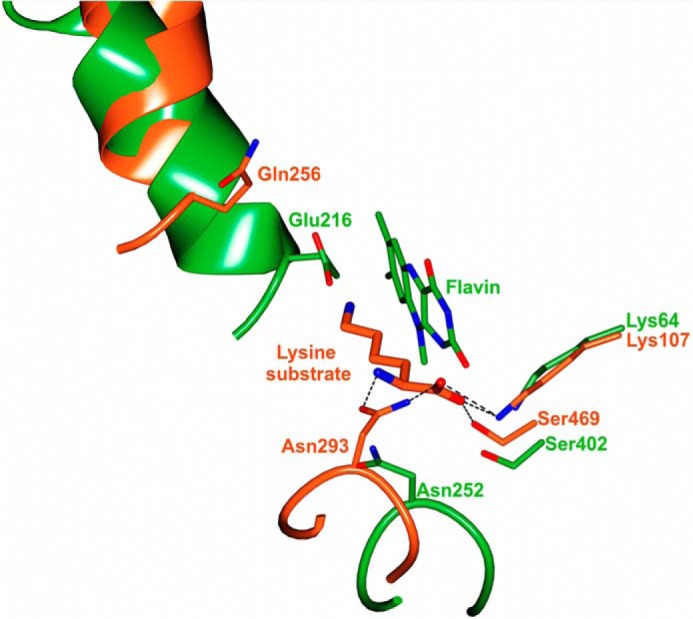
Comparison of NbtG (green) and SidA (orange) active sites. For the sake of clarity, only the flavin ring of NbtG is drawn. Equivalent residues surrounding the flavin site of the two enzymes and the position of the l-Lys substrate bound to the SidA active site are drawn (dashed lines indicate hydrogen bonds, which stabilize substrate binding). Loop 276–279 of NbtG and loop 321–324 of SidA are shown to highlight the different conformation.
Steady-state Kinetic Properties of P238R
To determine if replacing Pro at position 238 to Arg resulted in a higher affinity and specificity for NADPH, the NbtG P238R variant was created. The steady-state kinetic parameters were determined by monitoring oxygen consumption and lysine hydroxylation. Oxygen consumption data show that in the presence of l-Lys the kcat value is only ∼2-fold lower, and the KNADPH value is ∼5-fold higher than wild type; however, no activity was observed with NADH (Table 6). Similar results were obtained when lysine hydroxylation was monitored. The levels of uncoupling are very similar to those calculated for the wild-type enzyme.
TABLE 6.
Steady-state kinetic parameters for NbtG P238R
Conditions were 100 mm sodium phosphate, pH 7.5, with 2 mm d/l-Lys. Uncoupling was calculated using the expression (1 − kcat(N-OH)/kcat(O2)) × 100, where kcat(N-OH) is the kcat calculated measuring hydroxylated l-Lys, and kcat(O2) is kcat from the oxygen consumption assay.
| Parameter | O2 consumption | l-Lys hydroxylation |
|---|---|---|
| kcat (s−1) | 0.60 ± 0.04 | 0.146 ± 0.009 |
| KNADPH (mm) | 0.8 ± 0.2 | 0.7 ± 0.2 |
| kcat/KNADPH (m−1s−1) | 790 ± 170 | 210 ± 60 |
| KI(NADPH) (mm) | 1.6 ± 0.4 | |
| Uncoupling (%) | 76 |
Steady-state Kinetic Properties of K64A
Although the structure of NbtG did not contain electron density for Lys, the amino acids involved in l-Orn binding in SidA, PvdA, and KtzI are conserved in NbtG (Fig. 8). We probed the role of K64 in l- and d-Lys binding by mutating it to Ala. The steady-state lysine hydroxylation data clearly show that the major effect is on the Km value for l- and d-Lys. The Kl-Lys value increased ∼40-fold, whereas the Kd-Lys value increased ∼15–20-fold. In contrast, the kcat values decreased only 3–13-fold (Table 7). The uncoupling values for K64A are similar to the values obtained for the wild-type enzyme (Table 7). These data indicate that K64A is primarily involved in Lys binding in NbtG as predicted from the three-dimensional structure.
TABLE 7.
Steady-state kinetic parameters for NbtG K64A monitored by Lys hydroxylation
Conditions were 100 mm sodium phosphate, pH 7.5, with 2 mm NAD(P)H. Uncoupling was calculated using the expression (1 − kcat(N-OH)/kcat(O2)) × 100, where kcat(N-OH) is the kcat calculated measuring hydroxylated d/l-Lys, and kcat(O2) is the kcat from the oxygen consumption assay.
| Parameter | NADPH |
NADH |
||
|---|---|---|---|---|
| l-Lys | d-Lys | l-Lys | d-Lys | |
| kcat, s−1 | 0.34 ± 0.01 | 0.130 ± 0.004 | 0.180 ± 0.005 | 0.021 ± 0.001 |
| KLys, mm | 13 ± 1 | 49 ± 4 | 40 ± 3 | 62 ± 5 |
| kcat/KLys, m−1s−1 | 27 ± 3 | 2.7 ± 0.2 | 4.4 ± 0.4 | 0.38 ± 0.03 |
| Uncoupling, % | 78 | 89 | 69 | 94 |
DISCUSSION
NMOs represent a class of flavin monooxygenases involved in the biosynthesis of hydroxamate-containing siderophores whose iron-chelating properties are responsible for the virulence of microbes that secrete them. The l-Lys monooxygenase NbtG from N. farcinica catalyzes the hydroxylation of l-Lys in the pathway leading to the production of the siderophore nocobactin (Scheme 1), similar to what occurs in mycobacteria for mycobactin biosynthesis by the MbsG and MbtG proteins (20). Homologous enzymes are represented by l-Orn monooxygenases such as SidA (13), PvdA (11), and KtzI (12), which specifically hydroxylate l-Orn to generate siderophores. Like other flavin monooxygenases, the NMO catalytic process begins with binding of NADPH, which reduces the flavin cofactor that, in turn, reacts with molecular oxygen generating the FADOOH intermediate capable of donating the distal oxygen to the substrate.
We first studied the NbtG reaction by probing the enzyme cofactor specificity. In contrast to l-Orn monooxygenases, which show a clear preference for NADPH, NbtG can also use NADH as a reducing agent, although at a lower efficiency. The same was observed for MbsG where, nevertheless, the Km values for both cofactors are higher than those measured with NbtG: for the mycobacterial enzyme KNADPH and KNADH are 12.0 and 7.0 mm, respectively, whereas values of 0.20 and 0.36 mm were determined with NbtG (Table 2) (21). In either case the presence of the l-Lys substrate does not significantly alter the affinity for the coenzymes. A comparative analysis of NbtG and SidA structures shows that the interactions in SidA that stabilize the NADPH 2′-OH-phosphate during binding (Arg-279 and Ser-325 side chains) do not form in NbtG (and neither in MbsG) because these residues are not conserved, and therefore, either NADH or NADPH would indifferently bind to these Lys monooxygenases. Substitution of Pro to an Arg at position 238 (the corresponding position for Arg-279 in SidA) converted NbtG into a NADPH-specific monooxygenase but increased the Km value for NADPH. Furthermore, the P238R enzyme was as uncoupled as wild-type NbtG. These results suggest that the mode of binding of NADP(H) in NbtG is different from what is observed in SidA and PvdA.
The kinetics of the NbtG reaction were investigated by measuring the O2 consumption rate under steady-state conditions by using an Oxygraph. A kcat value of 0.58 s−1 was determined for NADPH, and a ∼2-fold increase was obtained in the presence of l-Lys, with this substrate effect being even more pronounced with NADH. We also probed the substrate stereospecificity of NbtG, and we found that the enzyme is also active on d-Lys, although it binds l-Lys with a higher affinity. The results of the K64A variant support a conserved amino acid substrate binding site among members of the NMO group of enzymes.
We then investigated the overall reaction leading to substrate hydroxylation by isotope effect analysis and by quantifying the final product using the Csaky assay. Flavin reduction occurs through two phases, one being ∼10-fold faster than the other and both dependent on NADPH concentration. Although hydride transfer has been shown to be the rate-limiting step in SidA, isotope effect experiments showed that in NbtG this step is only partially rate-limiting. The large solvent kinetic isotope effect suggests that a proton exchangeable step is the slow step in the reaction. Similar to MbsG, we found that the kcat of d/l-Lys hydroxylation is significantly lower than that of O2 consumption even in the presence of the substrate, and production of superoxide and hydrogen peroxide was also detected using both NADPH and NADH (Table 4). This means that NbtG undergoes a number of unproductive catalytic cycles with an uncoupling level of 70% for l-Lys hydroxylation from O2 consumption using NADPH as the reducing agent and 52% in the case of NADH (Table 3). Even more pronounced uncoupling effects were detected with d-Lys as the substrate.
NbtG represents the first crystal structure of a l-Lys monooxygenase (Fig. 6A). Structural comparison with l-Orn monooxygenases revealed that although the NADPH domain is individually very similar to those of SidA and PvdA, in NbtG it is differently oriented by about 30° with respect to the FAD domain. Importantly, such a domain rotation alters the NADPH binding site, which becomes physically inaccessible to the dinucleotide cofactor. Although domain rotations have been found among members of this family of flavoenzymes (41) (including NMOs, Baeyer-Villiger enzymes, and flavin-containing monooxygenases), such a large change so drastically affecting the active center has not been observed thus far. The characteristic mechanistic feature of this family of enzymes is stabilization of the FADOOH intermediate by NADP+, which remains bound throughout the catalytic reaction ensuring a high degree of coupling. This effect is absent in NbtG as even high NADP+ concentrations do not promote FADOOH stabilization. Consistently, the enzyme exhibits a rather loose binding of NADP+ as demonstrated by the measured KD value of 2 mm, which indicates a much weaker affinity compared with that observed in the family members (typical values in the 2–100 μm range) (11, 12, 15–17, 39, 42, 43). Collectively, these findings indicate that in NbtG the catalytic uncoupling of O2 consumption from substrate hydroxylation is related to the pronounced stability of an NADP-domain conformation, which occludes the NADP(H) binding site. After reduction of the flavin by NADPH, the enzyme might quickly release NADP+ and adopt the occluded conformation observed in the crystal structure. Along with this, flavin reoxidation will occur without any stable formation of the FADOOH, whose stability requires bound NADP+ (16, 44). Alternatively, the FADOOH might form but will quickly decay upon release of NADP+. In either case, flavin reoxidation occurs without stable formation of the FADOOH, which is fully consistent with the low NADP+ affinity, lack of a detectable intermediate in flavin reoxidation, and the substantial uncoupling featured by the enzyme (Fig. 4 and Table 3).
These findings raise the intriguing question as to the biological significance of these functional and structural features. The possibility exists that NbtG and the homologous mycobacterial MbsG are uncoupled because they dually function as both Lys hydroxylases and NADPH oxidases, with this second activity possibly being involved in ROS generation and signaling. Alternatively, these properties might be instrumental in enzyme regulation by yet-unknown factors that control the oxidase versus the hydroxylase activities, for instance through stabilization of the functionally distinct enzyme conformations.
Acknowledgments
We thank the European Synchrotron Radiation Facility (ESRF) and the Swiss Light Source (SLS) for providing beam time and assistance and the BioStruct-X program (project 5275) for funding synchrotron trips. We thank the anonymous reviewers for suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grants P41RR012408 (National Center for Research Resources) and P41GM103473 (NIGMS). This work was also supported by the Offices of Biological and Environmental Research and of Basic Energy Sciences of the United States Department of Energy and by National Science Foundation Grant MCB-1021384 (to P. S.).
The atomic coordinates and structure factors (code 4d7e) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NMO
- N-hydroxylating monooxygenase
- FMOC-Cl
- fluorenylmethoxycarbonyl chloride.
REFERENCES
- 1. Meneely K. M., Lamb A. L. (2007) Biochemical characterization of a flavin adenine dinucleotide-dependent monooxygenase, ornithine hydroxylase from Pseudomonas aeruginosa, suggests a novel reaction mechanism. Biochemistry 46, 11930–11937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kang H. Y., Brickman T. J., Beaumont F. C., Armstrong S. K. (1996) Identification and characterization of iron-regulated Bordetella pertussis alcaligin siderophore biosynthesis genes. J. Bacteriol. 178, 4877–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Macheroux P., Plattner H. J., Romaguera A., Diekmann H. (1993) FAD and substrate analogs as probes for lysine N6-hydroxylase from Escherichia coli EN 222. Eur. J. Biochem. 213, 995–1002 [DOI] [PubMed] [Google Scholar]
- 4. Barona-Gómez F., Wong U., Giannakopulos A. E., Derrick P. J., Challis G. L. (2004) Identification of a cluster of genes that directs desferrioxamine biosynthesis in Streptomyces coelicolor M145. J. Am. Chem. Soc. 126, 16282–16283 [DOI] [PubMed] [Google Scholar]
- 5. Lynch D., O'Brien J., Welch T., Clarke P., Cuív P. O., Crosa J. H., O'Connell M. (2001) Genetic organization of the region encoding regulation, biosynthesis, and transport of rhizobactin 1021, a siderophore produced by Sinorhizobium meliloti. J. Bacteriol. 183, 2576–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olucha J., Lamb A. L. (2011) Mechanistic and structural studies of the N-hydroxylating flavoprotein monooxygenases. Bioorg. Chem. 39, 171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hissen A. H., Wan A. N., Warwas M. L., Pinto L. J., Moore M. M. (2005) The Aspergillus fumigatus siderophore biosynthetic gene sidA, encoding l-ornithine N5-oxygenase, is required for virulence. Infect. Immun. 73, 5493–5503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takase H., Nitanai H., Hoshino K., Otani T. (2000) Impact of siderophore production on Pseudomonas aeruginosa infections in immunosuppressed mice. Infect. Immun. 68, 1834–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Voss J. J., Rutter K., Schroeder B. G., Su H., Zhu Y., Barry C. E., 3rd. (2000) The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U.S.A. 97, 1252–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Franceschini S., Fedkenheuer M., Vogelaar N. J., Robinson H. H., Sobrado P., Mattevi A. (2012) Structural insight into the mechanism of oxygen activation and substrate selectivity of flavin-dependent N-hydroxylating monooxygenases. Biochemistry 51, 7043–7045 [DOI] [PubMed] [Google Scholar]
- 11. Olucha J., Meneely K. M., Chilton A. S., Lamb A. L. (2011) Two structures of an N-hydroxylating flavoprotein monooxgenase: the ornithine hydroxylase from Pseudomonas aeruginosa. J. Biol. Chem. 286, 31789–31798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Setser J. W., Heemstra J. R., Jr., Walsh C. T., Drennan C. L. (2014) Crystallographic evidence of drastic conformational changes in the active site of a flavin-dependent N-hydroxylase. Biochemistry 53, 6063–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chocklett S. W., Sobrado P. (2010) Aspergillus fumigatus SidA is a highly specific ornithine hydroxylase with bound flavin cofactor. Biochemistry 49, 6777–6783 [DOI] [PubMed] [Google Scholar]
- 14. Meneely K. M., Barr E. W., Bollinger J. M., Jr., Lamb A. L. (2009) Kinetic mechanism of ornithine hydroxylase (PvdA) from Pseudomonas aeruginosa: substrate triggering of O2 addition but not flavin reduction. Biochemistry 48, 4371–4376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mayfield J. A., Frederick R. E., Streit B. R., Wencewicz T. A., Ballou D. P., DuBois J. L. (2010) Comprehensive spectroscopic, steady state, and transient kinetic studies of a representative siderophore-associated flavin monooxygenase. J. Biol. Chem. 285, 30375–30388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Romero E., Fedkenheuer M., Chocklett S. W., Qi J., Oppenheimer M., Sobrado P. (2012) Dual role of NADP(H) in the reaction of a flavin dependent N-hydroxylating monooxygenase. Biochim. Biophys. Acta 1824, 850–857 [DOI] [PubMed] [Google Scholar]
- 17. Alfieri A., Malito E., Orru R., Fraaije M. W., Mattevi A. (2008) Revealing the moonlighting role of NADP in the structure of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 105, 6572–6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malito E., Alfieri A., Fraaije M. W., Mattevi A. (2004) Crystal structure of a Baeyer-Villiger monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 101, 13157–13162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plattner H. J., Pfefferle P., Romaguera A., Waschütza S., Diekmann H. (1989) Isolation and some properties of lysine N6-hydroxylase from Escherichia coli strain EN222. Biol. Met. 2, 1–5 [DOI] [PubMed] [Google Scholar]
- 20. Robinson R., Sobrado P. (2011) Substrate binding modulates the activity of Mycobacterium smegmatis G, a flavin-dependent monooxygenase involved in the biosynthesis of hydroxamate-containing siderophores. Biochemistry 50, 8489–8496 [DOI] [PubMed] [Google Scholar]
- 21. Robinson R. M., Rodriguez P. J., Sobrado P. (2014) Mechanistic studies on the flavin-dependent N6-lysine monooxygenase MbsG reveal an unusual control for catalysis. Arch. Biochem. Biophys. 550, 58–66 [DOI] [PubMed] [Google Scholar]
- 22. Hoshino Y., Chiba K., Ishino K., Fukai T., Igarashi Y., Yazawa K., Mikami Y., Ishikawa J. (2011) Identification of nocobactin NA biosynthetic gene clusters in Nocardia farcinica. J. Bacteriol. 193, 441–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fox B. G., Blommel P. G. (2009) Autoinduction of protein expression. Curr. Protoc. Protein Sci., 10.1002/0471140864.ps0523s56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doublié S. (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276, 523–530 [PubMed] [Google Scholar]
- 25. Heemstra J. R., Jr., Walsh C. T., Sattely E. S. (2009) Enzymatic tailoring of ornithine in the biosynthesis of the Rhizobium cyclic trihydroxamate siderophore vicibactin. J. Am. Chem. Soc. 131, 15317–15329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Csaky T. (1948) On the estimation of bound hydroxylamine in biological materials. Acta Chem. Scand. 450–454 [Google Scholar]
- 27. Jeong S. S., Gready J. E. (1994) A method of preparation and purification of (4R)-deuterated-reduced nicotinamide adenine dinucleotide phosphate. Anal. Biochem. 221, 273–277 [DOI] [PubMed] [Google Scholar]
- 28. Dhatwalia R., Singh H., Solano L. M., Oppenheimer M., Robinson R. M., Ellerbrock J. F., Sobrado P., Tanner J. J. (2012) Identification of the NAD(P)H binding site of eukaryotic UDP-galactopyranose mutase. J. Am. Chem. Soc. 134, 18132–18138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goldschmidt L., Cooper D. R., Derewenda Z. S., Eisenberg D. (2007) Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 16, 1569–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pape T., Schneider T. R. (2004) HKL2MAP: a graphical user interface for phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–884 [Google Scholar]
- 33. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 35. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 36. McNicholas S., Potterton E., Wilson K. S., Noble M. E. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D 67, 386–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robinson R., Franceschini S., Fedkenheuer M., Rodriguez P. J., Ellerbrock J., Romero E., Echandi M. P., Martin Del Campo J. S., Sobrado P. (2014) Arg-279 is the key regulator of coenzyme selectivity in the flavin-dependent ornithine monooxygenase SidA. Biochim. Biophys. Acta 1844, 778–784 [DOI] [PubMed] [Google Scholar]
- 38. Dudek H. M., Torres Pazmiño D. E., Rodríguez C., de Gonzalo G., Gotor V., Fraaije M. W. (2010) Investigating the coenzyme specificity of phenylacetone monooxygenase from Thermobifida fusca. Appl. Microbiol. Biotechnol. 88, 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Torres Pazmiño D. E., Baas B. J., Janssen D. B., Fraaije M. W. (2008) Kinetic mechanism of phenylacetone monooxygenase from Thermobifida fusca. Biochemistry 47, 4082–4093 [DOI] [PubMed] [Google Scholar]
- 40. Hayward S., Berendsen H. J. (1998) Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins 30, 144–154 [PubMed] [Google Scholar]
- 41. Mirza I. A., Yachnin B. J., Wang S., Grosse S., Bergeron H., Imura A., Iwaki H., Hasegawa Y., Lau P. C., Berghuis A. M. (2009) Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J. Am. Chem. Soc. 131, 8848–8854 [DOI] [PubMed] [Google Scholar]
- 42. Sheng D., Ballou D. P., Massey V. (2001) Mechanistic studies of cyclohexanone monooxygenase: chemical properties of intermediates involved in catalysis. Biochemistry 40, 11156–11167 [DOI] [PubMed] [Google Scholar]
- 43. Beaty N. B., Ballou D. P. (1981) The oxidative half-reaction of liver microsomal FAD-containing monooxygenase. J. Biol. Chem. 256, 4619–4625 [PubMed] [Google Scholar]
- 44. Orru R., Dudek H. M., Martinoli C., Torres Pazmiño D. E., Royant A., Weik M., Fraaije M. W., Mattevi A. (2011) Snapshots of enzymatic baeyer-villiger catalysis: oxygen activation and intermediate stabilization. J. Biol. Chem. 286, 29284–29291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]