

Background: Mycobacteria utilize cAMP for regulating transcription and protein acetylation.
Results: We identify and structurally characterize a universal stress protein as an abundant and specific cAMP-binding protein.
Conclusion: The USP domain is a novel cAMP-binding module.
Significance: Certain members of the universal stress protein family serve to sequester cAMP, acting as sinks to regulate the availability of cAMP for downstream effectors.
Keywords: Crystal Structure, Cyclic AMP (cAMP), Isothermal Titration Calorimetry (ITC), Mycobacteria, Surface Plasmon Resonance (SPR), Universal Stress Protein
Abstract
Mycobacteria are endowed with rich and diverse machinery for the synthesis, utilization, and degradation of cAMP. The actions of cyclic nucleotides are generally mediated by binding of cAMP to conserved and well characterized cyclic nucleotide binding domains or structurally distinct cGMP-specific and -regulated cyclic nucleotide phosphodiesterase, adenylyl cyclase, and E. coli transcription factor FhlA (GAF) domain-containing proteins. Proteins with cyclic nucleotide binding and GAF domains can be identified in the genome of mycobacterial species, and some of them have been characterized. Here, we show that a significant fraction of intracellular cAMP is bound to protein in mycobacterial species, and by using affinity chromatography techniques, we identify specific universal stress proteins (USP) as abundantly expressed cAMP-binding proteins in slow growing as well as fast growing mycobacteria. We have characterized the biochemical and thermodynamic parameters for binding of cAMP, and we show that these USPs bind cAMP with a higher affinity than ATP, an established ligand for other USPs. We determined the structure of the USP MSMEG_3811 bound to cAMP, and we confirmed through structure-guided mutagenesis, the residues important for cAMP binding. This family of USPs is conserved in all mycobacteria, and we suggest that they serve as “sinks” for cAMP, making this second messenger available for downstream effectors as and when ATP levels are altered in the cell.
Introduction
Cyclic nucleotides act as second messengers in diverse signaling cascades throughout all kingdoms of life (1). Not only do cAMP and cGMP modulate critical signaling pathways within individual organisms (2), but their presence across the different kingdoms of life has allowed pathogens to exploit these pathways during their infective as well as disease-causing life cycles (2–7). The actions of cAMP and cGMP are mediated by downstream effector proteins (8). These proteins usually harbor an evolutionarily conserved cyclic nucleotide binding (CNB)6 domain (9) fused to an effector domain that can either have catalytic activity (10), regulate the transport of ions and small molecules (11), or bind DNA (12). Structural studies have provided details of the mode of cyclic nucleotide binding to the CNB domain, as well as the mechanism of allosteric regulation of the fused effector domain following binding of the cyclic nucleotide to the CNB domain (8). Apart from the CNB domain, GAF (cGMP-specific and -regulated cyclic nucleotide phosphodiesterase, adenylyl cyclase, and E. coli transcription factor FhlA) domains have also been shown to bind cAMP or cGMP (13, 14). GAF domains are structurally distinct from the CNB domain, and the mechanisms of allosteric regulation of the downstream catalytic domains following cyclic nucleotide binding are also understood (14, 15).
Mycobacterium tuberculosis is the causative agent of the severe pulmonary disease, tuberculosis. With the availability of the genome sequences of various species of mycobacteria, it was recognized that these bacteria harbor a number of genes that encode adenylyl cyclases (16–18). Indeed, intracellular levels of cAMP can reach hundreds of micromolars (19, 20). Cyclic AMP is also effectively secreted by mycobacteria (19, 21, 22), perhaps modulating host macrophage function during disease establishment and the formation of the caseous granuloma-containing dormant mycobacteria. In concert with the high levels of cAMP, the genome of M. tuberculosis H37Rv encodes 10 proteins that harbor the CNB domain, and two proteins containing the GAF domain (5). Both M. tuberculosis and the fast growing Mycobacterium smegmatis express proteins that are similar to the bacterial transcription factor, CRP, or cAMP receptor protein (4, 23, 24). Mycobacterial CRPs contain a CNB domain, but cAMP binding is not essential for binding DNA, in contrast to the well studied Escherichia coli CRP (23–25). This perhaps is an adaptation to the normally high levels of cAMP in these bacteria. In a previous study, we also characterized unique proteins where a cAMP binding domain is fused to a Gcn5-related N-acetyltransferase-like domain, and we showed that mycobacteria regulate protein acylation in a cAMP-dependent manner (26, 27).
Although bioinformatic and therefore predictive approaches to identify putative cyclic nucleotide-binding proteins have proved valuable so far, in this study we chose to use an unbiased biochemical approach to identify novel cAMP-binding proteins, beyond those homologous to established ones. Using cAMP affinity chromatography, we have isolated and characterized a universal stress protein (USP) Rv1636, which binds cAMP specifically and with high affinity and ATP with lower affinity. We show that orthologs of this protein are conserved in both fast and slow growing mycobacteria, and we have structurally characterized the cAMP-bound form of this USP. The absence of an additional domain associated with the USP domain suggests that these proteins may function as sinks for cAMP, thereby regulating downstream actions of the cyclic nucleotide in the cell.
EXPERIMENTAL PROCEDURES
Mycobacterial Strains and Culture Conditions
M. smegmatis mc2 155 cells were grown in Middlebrook 7H9 medium (BD Biosciences) supplemented with 0.2% glycerol and 0.05% Tween 80 at 37 °C with shaking at 200 rpm. Mycobacterium bovis BCG cultures were grown in the same medium containing oleic acid/albumin/dextrose/catalase (OADC, Difco) supplement at a final concentration of 10% (v/v) in static cultures.
Measurement of Free and Bound cAMP
Both M. smegmatis and M. bovis BCG cultures were harvested at log (A600 ∼1) and early stationary (A600 ∼3) phases. For M. smegmatis, 9 ml of culture was used in the log phase and 3 ml in the stationary phase. For M. bovis BCG, double the volumes were taken at each stage of growth. The cells were pelleted, and the supernatants were collected for measurement of extracellular cAMP. The cell pellets were washed with cold phosphate-buffered saline and resuspended in 400 μl of lysis buffer containing 50 mm Tris-Cl (pH 8.2), 100 mm NaCl, 10 mm 2-mercaptoethanol (2-ME), 10% glycerol, and 1 mm phenylmethylsulfonyl fluoride (PMSF). Cells were lysed by bead beating, centrifuged at 1000 × g, followed by harvesting of the supernatant and further centrifugation at 17,000 × g at 4 °C. Protein (400 μg) in the supernatant (cytosolic fraction) was subjected to centrifugation through a 3-kDa cutoff membrane filter (Amicon Ultra-0.5 3-kDa Ultracell, Millipore) at 4 °C. Cyclic AMP was measured in the eluate (which would represent “free” cAMP) and the original cytosolic fraction (representing “total” cAMP). Subtraction of the free cAMP from the total would provide an estimate of the fraction of cAMP that was “bound” to protein.
Culture supernatant was also subjected to centrifugation through the 3-kDa cutoff membrane filter, and the eluate was used to measure the free cAMP. Neat culture supernatant was used to measure the total extracellular cAMP concentrations. Cyclic AMP was measured by radioimmunoassay following acidification of the samples, as described previously (19).
Identification of cAMP-binding Proteins by Affinity Chromatography
Lyophilized cytosol of M. tuberculosis CDC1551 was obtained from BEI Resources, NIAID, National Institutes of Health, and reconstituted in buffer containing 50 mm Tris-Cl (pH 8.2), 100 mm NaCl, 10 mm 2-ME, 10% glycerol, 1 mm benzamidine, and 2 mm PMSF. For preparing M. smegmatis mc2 155 cytosol, cells were lysed by bead beating (19), and the lysate was centrifuged at 100,000 × g. The supernatant was used for subsequent interaction with N6-(6-aminohexyl)adenosine 3′,5′-cyclic-monophosphate immobilized on agarose beads (6-AH-cAMP-agarose; BioLog). Interaction was carried out at 4 °C for 2 h. The beads were washed three times with lysis buffer, and bound proteins were eluted with buffer containing 50 mm Tris-Cl (pH 8.2), 100 mm NaCl, 10 mm 2-ME, 10% glycerol, and 1 mm cAMP. The eluted fractions were subjected to 12% SDS-PAGE followed by silver staining.
Mass Spectrometry
Protein bands were excised from the gel, and subjected to tryptic digestion as described in detail earlier (26). The peptides were analyzed by MALDI-TOF (Ultraflex TOF/TOF, Bruker Daltonics, Germany). Data were obtained using Flex Control software (25 KvA Reflector mode, N2 LASER, 337 nm and 50 Hz). The sum of ion intensities from 300 LASER shots was used for each spectrum. The spectra were analyzed using Flex Analysis version 2.0, and proteins were identified using MASCOT on the M. tuberculosis and M. smegmatis proteomics database.
Sequence and Phylogenetic Analysis
Distant and close homologs of Rv1636 were identified using BLAST analysis. The boundaries of the USP domains were identified using the Pfam database, and multiple sequence alignment was performed using ClustalW (28). Phylogenetic relationships were analyzed by the neighbor-joining method using Mega 6 (29).
Cloning, Expression, and Purification of Rv1636 and MSMEG_3811
PCR was carried out on genomic DNA of M. tuberculosis H37Rv and M. smegmatis mc2 155 using specific primers, sequences of which are available on request. The PCR amplicons were digested with EcoRI and XhoI and cloned into similarly digested pPROEX-HTa to generate pPRO-Rv1636 and pPRO-MSMEG_3811, where both proteins would be expressed with an N-terminal His tag. The clones were confirmed by sequencing (Macrogen, South Korea).
For generating point mutants of Rv1636, a single mutagenic primer-based method was used (30), using pPRO-Rv1636 as template for mutagenesis. All mutants were confirmed by sequencing.
For biochemical studies, plasmids were transformed in E. coli SP850 cyc− strain (31), and proteins were expressed by the addition of 0.5 mm isopropyl β-thiogalactopyranoside at 16 °C. Cells were incubated for 18 h at 16 °C, harvested, and lysed by sonication in buffer containing 50 mm Tris-Cl (pH 8.2), 100 mm NaCl, 10 mm 2-ME, 10% glycerol, 1 mm benzamidine, and 2 mm PMSF. The cytosolic fraction was incubated with Ni-NTA beads (GE Healthcare); beads were washed, and bound protein was eluted with buffer containing 50 mm Tris-Cl (pH 8.2), 100 mm NaCl, 10 mm 2-ΜΕ, 10% glycerol, and 300 mm imidazole. The purified proteins were subjected to gel filtration using a Superose12 10/30 column on an AKTA FPLC system (GE Healthcare) in buffer containing 10 mm HEPES (pH 7.5), 100 mm NaCl, 10 mm 2-ME, and 20% glycerol. The column was calibrated with a commercially available standard (Bio-Rad) having bovine thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobulin (17 kDa), and vitamin B12 (1.35 kDa). Purified proteins were stored in aliquots at −70 °C.
Surface Plasmon Resonance
Surface plasmon resonance (SPR) studies were performed on a BIAcore 3000 platform (GE Healthcare). 8-(6-Aminohexylamino) cAMP (8-AHA-cAMP; BioLog) was dissolved in 100 mm HEPES (pH 8) by careful warming (32). CM5 (carboxymethylated dextran) sensor chip was activated with a mixture of 0.2 m 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and 0.05 m N-hydroxysuccinimide. 8-AHA-cAMP (3 mm) was injected (2 μl/min; 7 min) for immobilization in running buffer containing 100 mm HEPES (pH 8). The surface was deactivated using 1 m ethanolamine-HCl (pH 8.5). A reference channel was activated and deactivated without any 8-AHA-cAMP conjugation. All interactions were studied in buffer containing 10 mm HEPES (pH 7.5), 100 mm NaCl, 10 mm MgCl2, 10 mm 2-ME, and 0.005% P20. Rv1636 was used at a concentration of 1 μm at a flow rate of 5 μl per min. The association and dissociation were both monitored for 10 min at 25 °C. MSMEG_3811 (3 μm) was injected at a flow rate of 30 μl per min, and the association and dissociation were monitored for 100 s at 25 °C. Binding of the proteins to cAMP on the chip during the association phase was also monitored in the presence of different concentrations of free 3′,5′-cAMP, 3′,5′-cGMP, and Mg-ATP. The proportion of Mg-ATP present in the solution was calculated using Mg-ATP calculator version 1.3. Binding data were analyzed using BIAevaluation 3.0 software and plotted using GraphPad Prism 5.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments were carried out on a MicroCal iTC200 system (GE Healthcare) in buffer containing 10 mm HEPES (pH 7.5), 100 mm NaCl, 5 mm 2-ME, and 10% glycerol. Binding studies for monitoring interaction with ATP had 10-fold excess of MgCl2 in the buffer over the ATP used for the injection. Proteins were dialyzed in the buffer mentioned above, and cAMP and ATP were also prepared in the same buffer. All samples and buffers were degassed thoroughly under vacuum. The initial injection volume was 0.4 μl over a duration of 0.8 s. All subsequent injection volumes were 2 μl over 4 s with a spacing of 150 s between two injections. Data from control experiments where only buffer was injected into the protein solution was subtracted from each injection to compensate for the heat of dilution. Data for the initial injection were not considered. The data were analyzed using the built-in single site fitting model of Origin 7.0 software (OriginLab Corp.).
Crystallization
MSMEG_3811 was expressed in E. coli BL21 (DE3). Harvested cells were resuspended in buffer A (50 mm Tris-Cl (pH 8.0), 100 mm NaCl, 10% glycerol) and 2 mm PMSF and protease inhibitor mixture (Roche Applied Science) were added. Cells were lysed by sonication, and debris was removed by centrifugation at 30,000 × g for 30 min. The supernatant was incubated with Ni-NTA beads (Qiagen) for 1–2 h. Ni-NTA beads were washed with 15 column volumes of buffer A and 20 column volumes of 50 mm Tris-Cl (pH 8.0), 300 mm NaCl, 10% glycerol, 20 mm imidazole. His-tagged MSMEG_3811 was eluted in buffer A supplemented with 300 mm imidazole. The His tag was cleaved off with tobacco etch virus protease during dialysis in buffer A overnight. The digested protein was incubated with Ni-NTA beads to remove the His tag and His-tagged tobacco etch virus protease. MSMEG_3811 was concentrated and subjected to gel filtration on an S200 16/60 column (GE Healthcare) in buffer A.
For MSMEG_3811 crystallization, initial screening was performed with 15 mg/ml protein and 5 mm cAMP against the Qiagen JCSG+ and JCSG Core I-IV factorial screens in drops consisting of 0.2 μl of protein stock solution and 0.2 μl of reservoir solution. The sitting-drop experiments were set up at 20 °C using a Phoenix nanodispenser (Art Robbins Instruments) and a total of 35 μl of reservoir solution. The initial condition contained 0.1 m MES (pH 6.0) and 1.6 m ammonium sulfate and was refined to 0.1 m MES (pH 6.0), 1.9 m ammonium sulfate, and 6% (v/v) polypropylene glycol 400 (PPG 400) using the Hampton Research additive screen. Refined crystals were grown at 20 °C in a 24-well format by using the hanging-drop vapor diffusion method with equal volumes of reservoir and protein stock solution (1:1 μl) in the drop and a 1-ml reservoir. The protein stock solution consisted of 25 mg/ml MSMEG_3811 with 5 mm cAMP. Diffraction quality crystals in various shapes appeared after 3 days. Crystals were cryoprotected through soaking in 0.1 m MES (pH 6.0), 2 m ammonium sulfate, 6% (v/v) PPG 400, 20% (v/v) glycerol, and 5 mm cAMP.
Data Collection, Structure Determination, and Refinement
A complete diffraction dataset was collected at 100 K and a wavelength of 0.91841 Å at the Berlin Electron Storage Ring Society for Synchrotron Radiation at synchrotron beamline 14.1 (BESSY BL14.1) operated by Helmholtz-Zentrum Berlin (33). The crystal belonged to space group C2221 and diffracted close to 2 Å resolution. Diffraction data were indexed, integrated, and merged using the x-ray Detector software (XDS (34)), resulting in a 98.8% complete dataset cut at 2.15 Å resolution based on CC1/2 (35). All further programs for structure solution were from the CCP4 suite (36). The program Balbes (37) was used for selecting a suitable search model (PDB code 2Z08)7 and to solve the structure of MSMEG_3811-cAMP through molecular replacement. The Balbes solution contained four monomers in the asymmetric unit even though cell content analysis (39) and electron density analysis in Coot (40) suggested six monomers. The model was therefore adjusted in Coot to contain only residues and side chains that already fitted to the electron density in the partial solution, and molecular replacement was repeated with Molrep (41) searching for three physiological dimers. After successful molecular replacement, refinement was done in Refmac5 (42) resulting in initial crystallographic R and Rfree values of 39.3 and 43.0%, respectively. Coot was used for cycles of manual model building and analysis of model quality followed by TLS (43), and NCS restrained refinement of atom positions and individual B-factors in Refmac5. Figures were generated in PyMOL (44). Coordinates and structure factors for the MSMEG_3811-cAMP complex have been deposited with the PDB under accession number 5ahw.
RESULTS
Significant Fraction of Cytosolic cAMP Is Bound to Protein
Cytosolic as well as extracellular cAMP levels in mycobacteria can reach 100 μm, as measured by radioimmunoassay and ELISA procedures (19, 22). Such measurements incorporate procedures that dissociate cAMP bound to proteins, therefore estimating concentrations of cAMP that reflect the total amount present in the cellular fraction. Cyclic AMP-binding proteins have been identified and characterized from mycobacteria (23, 24, 26), suggesting to us that a significant fraction of the cyclic nucleotide may be bound to proteins within the cell. We therefore attempted to detect this fraction from that which was free or not associated with protein. We performed this by subjecting the cytosolic fraction to filtration through a membrane that retained molecules greater than 3 kDa in size. We argued that free cAMP would readily pass through this filter, whereas cAMP that was bound to protein would be retained. Measuring cAMP in both the filtrate and the cytosolic fraction prior to centrifugation and subtracting the concentration of cAMP in the filtrate from the total cAMP measured in the cytosol could provide an estimate of bound cAMP.
In both exponential and stationary phases of growth of M. smegmatis and M. bovis BCG cultures, we determined that the majority of the extracellular cAMP pool was present as free cAMP (Fig. 1A). However, when we monitored the distribution of intracellular cAMP between bound and free fractions, we observed that ∼50% of the total cytosolic cAMP was bound to protein in M. smegmatis cells (Fig. 1B). Interestingly, almost the entire fraction of cAMP was bound to protein in M. bovis BCG in the log phase, and more than 80% remained bound in the stationary phase of growth. Thus, a significant fraction of cAMP in mycobacteria is protein-associated in the cell.
FIGURE 1.

Determination of the bound fraction of cAMP and identification of an abundant cAMP-binding protein in mycobacteria. A, measurement of the bound (gray) and free (white) cAMP concentrations in the culture supernatant of M. smegmatis and M. bovis BCG cultures at log and stationary phases of growth. The total extracellular concentration of cAMP is the sum of the bound and free fractions. Means ± S.E. are plotted for three biological replicates. B, measurement of the bound (gray) and free (white) amounts of cAMP in the cytosolic fraction of M. smegmatis and M. bovis BCG cultures at log and stationary phases of growth. The sum of bound and free fractions indicates the total levels of intracellular cAMP. Means ± S.E. are plotted for three biological replicates. C, pulldown assays to identify cAMP-binding proteins in mycobacteria. Left panel, proteins bound to 6-AH-cAMP-agarose beads interacting with M. tuberculosis CDC1551 cell lysates were eluted with 1 mm 5′-AMP and -cAMP, respectively. The band corresponding to ∼15 kDa in the cAMP elution lane was identified as MT_1672 (orthologs of Rv1636) by mass spectrometry. Right panel, lane showing MSMEG_3811 (identified by mass spectrometry) that eluted with 1 mm cAMP following interaction of M. smegmatis cell lysate with 6-AH-cAMP-agarose beads. Gels were visualized by silver staining. D, multiple sequence alignment of Rv1636 (M. tuberculosis H37Rv) with its orthologs present in other mycobacterial species as follows: BCG_1674 (M. bovis BCG), MT_1672 (M. tuberculosis CDC1551), MRA_1646 (M. tuberculosis H37Ra), ML1390 (M. leprae TN), MSMEG_3811 (M. smegmatis mc2155), and MAV_3137 (M. avium (strain 104)). The conserved Walker A-like ATP-binding motif present in USPs is highlighted in gray.
Identification of a Highly Expressed cAMP-binding Protein in Mycobacteria
Mycobacterial proteins that can bind cAMP include CRPs and a cAMP-regulated protein acyltransferase, both found in slow growing and fast growing mycobacteria (23, 24, 26). To identify additional proteins that may bind cAMP, we took advantage of affinity chromatography on 6-AH-cAMP agarose beads. We used the cytosolic fraction from M. tuberculosis CDC1551 cells and let it interact with the beads, and following extensive washing of the beads to remove unbound proteins, we eluted bound proteins with 1 mm cAMP. Similar experiments were performed with the cytosol prepared from M. smegmatis. In both cases, a major protein of ∼14 kDa was associated with the beads and specifically eluted with cAMP, but not 5′-AMP (Fig. 1C). Tryptic digestion of the proteins following SDS-gel electrophoresis and by subsequent mass spectrometry analysis identified the two proteins as MT_1672 (identical in amino acid sequence to Rv1636 from M. tuberculosis H37Rv) and MSMEG_3811, with more than 65% sequence coverage. Interestingly, Rv1636 and MSMEG_3811 show more than 80% sequence identity, and their orthologs can be identified in all mycobacteria, including M. leprae, which has undergone a high degree of pseudogenization (Fig. 1D) (45). Surprisingly, neither MSMEG_3811 nor Rv1636 contain a canonical CNB domain, and instead they are annotated as members of the USP superfamily (46, 47).
Rv1636 and MSMEG_3811 Bind cAMP with High Specificity
We cloned, expressed, and purified Rv1636 and MSMEG_3811 with an N-terminal His tag (Fig. 2A), and gel filtration analysis showed that both proteins form a dimer in solution (data not shown). To measure the affinity of cAMP binding to these proteins, we performed SPR experiments, using 8-AHA-cAMP immobilized to a CM5 chip (Fig. 2B). The association (ka) and dissociation (kd) rates for both proteins are shown in Table 1. We incorporated varying concentrations of cAMP in the buffer during the binding reaction and by monitoring the reduction in RU, a similar low micromolar IC50 for both Rv1636 and MSMEG_3811 was observed (Table 1). This result is consistent with the similar Kd values calculated from ka and kd, and we can thus conclude that both Rv1636 and MSMEG_3811 bind cAMP with comparable affinities, reflective of their similarity at the sequence level.
FIGURE 2.

Analysis of kinetic and thermodynamic parameters of binding of Rv1636 and MSMEG_3811 with cAMP. A, Rv1636 and MSMEG_3811 were expressed and purified as an N-terminal His-tagged protein from E. coli SP850 (cyc−) cells. Purified proteins were analyzed by SDS-PAGE and stained with Coomassie dye. Lane M, molecular mass marker. B, SPR sensorgram showing the binding kinetics of Rv1636 (1 μm) and MSMEG_3811 (3 μm) onto 8-AHA-cAMP conjugated to a CM5 sensor chip. Data were analyzed using Langmuir single site-binding model, and data shown are representative of experiments repeated three times. C, either cAMP or cGMP (1 mm) was incubated with Rv1636 (1 μm) following which the mixture was injected over the SPR chip. The RU observed is plotted as a percentage of the RU seen with protein alone. Data shown are the means ± S.D. of experiments repeated twice in duplicate. D, thermodynamic parameters of cAMP-binding were measured using isothermal titration calorimetry, with 50 μm Rv1636 or MSMEG_3811 and 1 mm cAMP as injectant solution. The binding isotherms were plotted using a single site binding model, and binding stoichiometry was determined considering monomeric protein concentrations. Data shown are representative of experiments repeated three times.
TABLE 1.
Parameters of cAMP binding to Rv1636 and MSMEG_3811 as determined by surface plasmon resonance
Values shown represent the means ± S.D. obtained from experiments repeated at least three times.
| Protein | ka | kd | Kd | IC50 |
|---|---|---|---|---|
| m−1s−1 | s−1 | 10−6 m | 10−6 m | |
| Rv1636 | 1.84 ± 0.7 × 103 | 4.07 ± 1.3 × 10−3 | 1.94 ± 1.0 | 3.34 ± 0.1 |
| MSMEG_3811 | 3.62 ± 1.9 × 103 | 1.98 ± 0.1 × 10−2 | 6.82 ± 1.5 | 6.8 ± 2.3 |
CNB and GAF domains show specificity in their interactions with cyclic nucleotides, binding either cAMP or cGMP (12, 13). Binding of Rv1636 to the sensor chip was inhibited by cAMP but not by cGMP (Fig. 2C), indicating that Rv1636, and presumably its orthologs, are specific cAMP-binding proteins.
This is the first report of the identification of USPs that can bind cAMP. USPs show no sequence similarity to the canonical CNB or GAF domains that are well characterized cyclic nucleotide-binding proteins. We therefore decided to study the nature of the interaction of these proteins with cAMP, using isothermal titration calorimetry that could also provide binding stoichiometry. For both Rv1636 and MSMEG_3811, the tight binding was confirmed (Kd ∼3 μm) and found to be enthalpy-driven (Table 2 and Fig. 2D). One molecule of cAMP bound per monomer of both Rv1636 and MSMEG_3811 (Table 2).
TABLE 2.
Parameters of cAMP binding to Rv1636 and MSMEG_3811 as determined by isothermal titration calorimetry
Values shown represent the means ± S.D. obtained from experiments repeated at least three times.
| Protein | ΔH | ΔS | N | Kd |
|---|---|---|---|---|
| cal/mol | cal/mol/degree | 10−6m | ||
| Rv1636 | −2.11 ± 0.2 × 104 | −45.9 ± 6.5 | 0.9 ± 0.06 | 3.96 ± 0.07 |
| MSMEG_3811 | −2.08 ± 0.6 × 104 | −44.6 ± 2.4 | 1.39 ± 0.08 | 3.32 ± 0.67 |
Rv1636 and MSMEG_3811 Bind ATP
USPs are classified into two groups that differ in their ability to bind ATP (48). A conserved Walker A-like motif, GX2GX9G(S/T), forms an essential region that interacts with the triphosphate of ATP (46). Sequence analysis revealed that this ATP-binding motif is present in Rv1636 and its orthologs (Fig. 1D). We therefore monitored ATP binding to Rv1636 in the presence of MgCl2 by surface plasmon resonance. ATP was able to inhibit binding of Rv1636 to the cAMP-coated sensor chip (Fig. 3A), with an IC50 of 65.6 ± 5.6 μm, 10-fold higher than that of cAMP. This result indicates that ATP can compete with cAMP for its binding site, but it interacts with lower affinity. ITC indeed revealed that Kd for binding of ATP to Rv1636 was ∼ 31 μm, representing a 10-fold lower affinity as compared with cAMP (Fig. 3B). The binding of ATP was also enthalpy driven, and a single binding site for ATP was seen per monomer of protein. Similar binding parameters were observed for MSMEG_3811 (data not shown). Thus, Rv1636 and its orthologs are nucleotide-binding proteins, which show higher affinity for cAMP over ATP.
FIGURE 3.

Parameters of binding of ATP to Rv1636. A, increasing concentrations (micromolars) of free Mg-ATP were incubated with Rv1636 (1 μm) and applied to the SPR chip. Inset, Bmax (binding maximum in RU) versus log[ATP] was plotted to measure the IC50. B, isotherm showing the binding of Mg-ATP (3 mm concentration of injectant solution) to Rv1636 (150 μm) using ITC. The table below represents the changes in enthalpy (ΔH) and entropy (ΔS); the number of binding sites (N) and the dissociation constant (Kd) values were measured. Monomer protein concentrations were used for analysis with a single site binding model. Values represent means ± S.D. from three independent experiments.
Structural Basis of cAMP Binding
USPs form a subfamily of the adenine nucleotide α-hydrolase like superfamily according to the database for the Structural Classification of Proteins. Their generic structure consists of a core of three α/β/α layers and a 5-stranded, parallel β-sheet (47). Crystal structures of several USP family members show that ATP binds between β1 and β4, in a pocket composed of residues from α1, β1, β2, and β4 (47). The triphosphate is bound by a loop featuring a conserved Walker A-like motif (see above) and often coordinated by additional magnesium or manganese ions (47, 49). To understand the unique binding behavior of Rv1636/MSMEG_3811, i.e. their preference for cAMP over ATP, we attempted to crystallize these proteins. For Rv1636, we could only obtain a low resolution structure for its apo-state (data not shown), which was not pursued further due to the availability of a higher resolution structure (PDB 1TQ8). For MSMEG_3811, we could not obtain crystals of the apo-form but obtained well diffracting crystals for its cAMP complex. Initial attempts to solve the structure of the MSMEG_3811-cAMP complex by molecular replacement phasing with the Rv1636 apo-structure failed. Instead, we identified the universal stress protein TTHA0895 from Thermus thermophilus (PDB ID 2Z08) as a search model that enabled successful molecular replacement phasing of our MSMEG_3811-cAMP complex. The MSMEG_3811/cAMP structure was refined at 2.15 Å resolution to R/Rfree values of 17.9 and 23.5% (Fig. 4A; Table 3) and good geometry (98.8% residues in favored regions of the Ramachandran plot, 1.2% in allowed regions, and none in disallowed regions). One cAMP ligand per monomer was well defined by electron density (Fig. 4, B–D). Because of the high similarity of Rv1636 and MSMEG_3811 in their sequence (Fig. 1D) as well as their ligand binding properties (Tables 1 and 2), we used the available Rv1636 apo-structure and our MSMEG_3811-cAMP complex for comparing these two states of this protein family.
FIGURE 4.

Crystal structure of MSMEG_3811 bound to cAMP. A, overall structure of the MSMEG_3811/cAMP homodimer. cAMP is shown in stick presentation and colored according to atom type. Secondary structure elements of monomer 1 are labeled. B, nucleotide binding pocket of the MSMEG_3811-cAMP complex with relevant residues shown in stick presentation. Hydrogen bonds are shown as dashed lines. cAMP is shown as sticks colored according to atom type and overlaid with 2Fo − Fc density contoured at 1 σ. C, cAMP ligand of the MSMEG_3811 complex overlaid with Fo − Fc density contoured at 3 σ (green) and −3 σ (red). cAMP is shown as sticks colored according to atom type. D, scheme of the interactions between cAMP and MSMEG_3811. All atoms are colored according to atom type. Hydrogen bonds are shown as dashed lines labeled with the distance between relevant atoms. MSMEG_3811 residues involved in hydrophobic contacts are shown as half-circles. The figure was generated using LigPlot+ (68). E, topology diagrams of Rv1636 apo (left) and MSMEG_3811/cAMP (right), generated with TopDraw and Pro-Origami (67, 69). Cylinders represent α-helices, and arrows correspond to β-strands. Green-shaded secondary structure elements belong to the corresponding second monomer (Fig. 4A). The gray-shaded α-helix (α2) in Rv1636 apo is not visible in the structure, but it will likely form a helix according to secondary structure prediction and sequence alignment with MSMEG_3811 (Fig. 1D). F, overlay of MSMEG_3811/cAMP (blue) with Rv1636 apo (PDB code 1TQ8; green). 84 Cα atoms overlaid with a root mean square deviation of 0.95 Å. cAMP is shown in stick presentation and colored according to atom type. Hydrogen bonds are shown as dashed lines. Residues of the conserved Walker A-like motif are underlined. G, overlay of MSMEG_3811/cAMP (blue) with the ATP-binding universal stress protein TTHA0895 (yellow) used for molecular replacement. 79 Cα atoms overlaid with a root mean square deviation of 1.022 Å. Ligands are shown as sticks and colored according to atom type. Relevant residues for ATP binding of TTHA0895 are shown in stick presentation and colored according to atom type. The ATP-binding region of TTHA0895 with Gly-109, Gly-111, Ser-120, Gln-121, and Ser-122, as well as two magnesium ions (green), coordinate the β- and γ-phosphate groups of ATP. Residues of the conserved Walker A-like motif are underlined.
TABLE 3.
Diffraction data and refinement statistics for MSMEG_3811/cAMP

The monomer structure of MSMEG_3811/cAMP has the typical open, twisted, 5-stranded parallel β-sheet with topology 32145, sandwiched by six α-helices of different lengths (Fig. 4A). Like other USPs of this family, e.g. MJ0577 from Methanococcus jannaschii (PDB code 1MJH), MSMEG_3811 forms a type 1 homodimer (Fig. 4A) as defined by Tkaczuk et al. (47), consistent with its behavior in size exclusion chromatography. The dimer interface is formed mainly by the C terminus of each monomer through a conserved, strongly hydrophobic VL(J/V)V motif in β5 (Fig. 1D) (47). In contrast to Rv1636 apo, the loop with the Walker A-like ATP-binding motif in MSMEG_3811 is closer to the nucleotide-binding pocket due to the parallel orientation of β4 and β5, whereas β5 of Rv1636 apo is swapped between monomers and inserted between β4- and β5 of the partner monomer (Fig. 4, E and F). Variations in the topology of this region between USP members have been reported before (see “Discussion”). Cyclic AMP is bound in the same nucleotide binding pocket as ATP in other USPs, with its adenine base making conserved interactions with the amino and carbonyl groups of Ala-40 (47), and the hydroxyl group of the ribose forming hydrogen bonds to Gly-10 and Gly-114 (Fig. 4, B and D).
Based on the details provided by the crystal structure, the Gly-10 residue in Rv1636 interacts with the 2′-OH group of the ribose moiety (Fig. 4, B and D). Because the interaction is through the main chain of the peptide backbone, we introduced a bulkier side chain at this position by mutating the Gly residue to a Thr. In addition, Rv1636-Gly-113 (Gly-114 in MSMEG_3811) is part of the ATP-binding loop and interacts with the oxygen that is part of the ester bond between the phosphate and the 3′-C of the ribose. We mutated this Gly to Ala to reduce the flexibility of the loop. The mutant proteins were expressed and purified, and their ability to bind cAMP was monitored using SPR. Both Rv1636 G10T and G113A were significantly compromised in their binding ability, resulting in 24.4 ± 8.5 and 40.0 ± 12.2%, respectively, of the ligand binding signal observed for wild type Rv1636. Moreover, no binding to ATP was detected by isothermal titration calorimetry for either of the mutant proteins (data not shown). Therefore, our mutational analysis supports the conclusion from our competition experiment that cAMP and ATP bind to the same pocket in the protein, albeit with different affinities.
To identify structural features hindering ATP binding to MSMEG_3811 and thus causing its cAMP preference, we overlaid MSMEG_3811/cAMP with the TTHA0895-ATP complex (Fig. 4G). Although the binding mode of the adenine and ribose moieties is comparable (Fig. 4, B and G), the cyclic phosphodiester moiety of cAMP forms interactions with Ser-14 and Ser-16 in α1, and with Val-116 due to the close proximity of the binding loop comprising the Walker A-like motif (Fig. 4, B and D). The additional residues of this loop are oriented away from the nucleotide-binding site; however, due to the short but bulky α5-helix in its middle in MSMEG_3811 (Fig. 4G). In contrast, the corresponding loop of TTHA0895, which does not comprise this helical insertion, is conformationally less restricted and forms a binding pocket for the β- and γ-phosphate groups (Fig. 4G). In the MSMEG_3811-binding site, the terminal ATP phosphates would cause only small clashes, and the protein might in fact adjust and avoid them, but the lack of a significant interaction surface for these phosphates likely contributes to the weak interaction with ATP. Also, the triphosphate of the ATP-binding USP is complexed through two magnesium ions and further interactions with Ser-120, Gln-121, and Ser-122 in the TTHA0895-α4-helix (Fig. 4G). Ser-120 is part of the conserved Walker A-like motif and also present in MSMEG_3811/Rv1636. The Gln is replaced by Val in MSMEG_3811/Rv1636 and also in the ATP-binding USP MJ0577. However, Ser-122 is replaced by a Pro in MSMEG_3811/Rv1636, causing a tight and rigid bend of the protein main chain into α6. This rigid arrangement likely prevents the small conformational adjustment needed to allow the slightly rotated ribose position of ATP in TTHA0895 (Fig. 4G). Interestingly, the Pro in this position in MSMEG_3811/Rv1636 is absent in all the other USPs with ligands other than cAMP and thus might be part of a signature module for cAMP-binding USPs.
Potential Binding of Other Cyclic Nucleotides
To understand how the nucleotide-binding site of MSMEG_3811 discriminates between cAMP and cGMP, we inserted a hypothetical cGMP ligand by overlaying it with cAMP (Fig. 5A). Although the binding mode of the phosphodiester and the ribose moiety would be comparable, the amino group of the cGMP guanine moiety would have a distance of 2.45 Å to Ala-38 leading to a slight clash, and the carbonyl groups of the guanine moiety and of Ala-40 would be only 3.02 Å apart (Fig. 5A). As a result of the latter, two potential proton acceptors would be in close contact instead of a favorable hydrogen bond as observed for adenine, likely contributing to the observed specificity of Rv1636/MSMEG_3811 for cAMP.
FIGURE 5.
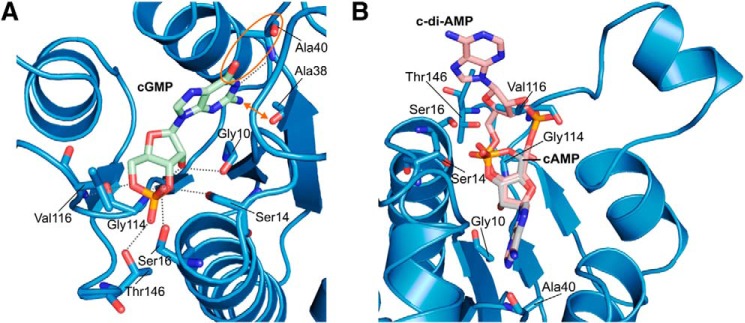
Potential binding of other cyclic nucleotides to MSMEG_3811. A, overlay of MSMEG_3811/cAMP with the hypothetical ligand cGMP, with relevant residues shown in stick presentation and colored according to atom type. Hypothetical hydrogen bond interactions are shown as dashed lines. The guanine moiety of cGMP would experience some slight clashes with Ala-38 indicated by an outward arrow. The unfavorable interaction between the carbonyl groups of the guanine moiety and of Ala-40 is highlighted by an orange circle. B, placing the cyclic nucleotide c-di-AMP (PDB code 2BA) into the MSMEG_3811/cAMP complex (blue) would lead to a strong clash of one phosphate group with Val-116. Ligand cAMP and hypothetical ligand c-di-AMP are shown as sticks and colored according to atom type. Relevant residues of MSMEG_3811 are labeled and shown in stick presentation.
In mycobacteria, bis-(3′,5′) cyclic dimeric nucleotides also play an important role as second messengers (50, 51), and we therefore analyzed the potential binding of c-di-AMP or c-di-GMP to Rv1636/MSMEG_3811. Because of the observed adenine nucleotide preference, we focused on c-di-AMP. Placing a hypothetical c-di-AMP ligand into the MSMEG_3811-cAMP complex by overlaying it with cAMP would lead to a strong clash of one phosphate group with Val-116 (Fig. 5B). This Val is one of the first residues of the loop featuring the conserved Walker A-like binding motif of USPs (Fig. 1D). The observed clash is probably due to the close proximity of this loop to the nucleotide binding pocket because of the parallel arrangement of β4 and β5 in MSMEG_3811/cAMP (Fig. 5B). However, based on this structural analysis we assume that c-di-AMP does not bind to Rv1636/MSMEG_3811.
DISCUSSION
In this study, we have attempted to empirically detect cAMP-binding proteins in mycobacteria, and we identified Rv1636/MSMEG_3811 as low molecular weight and highly abundant cAMP/ATP-binding proteins. The existence of a noncanonical cAMP-binding module identified in these USPs adds a further dimension to our understanding of the diverse functions of this family of proteins. Of note, cAMP-binding USPs as exemplified by Rv1636 and MSMEG_3811 do not contain additional domains in the full-length protein, as is normally observed in CNB and GAF domain containing proteins.
The overall architecture of Rv1636/MSMEG_3811 is a homodimer, assembled via a conserved, hydrophobic VL(J/V)V dimerization motif in β5. This arrangement groups them with the type 1 USP proteins as defined by Tkaczuk et al. (47). Although the dimer interface is formed via this C-terminal motif in both proteins, MSMEG_3811/cAMP retains two independently folded monomers, whereas in apo-Rv1636 the β5-strand is swapped, leading to additional intermolecular interaction between β5 and the partner monomer's β4 (Fig. 4F). This interaction is mediated by an LLVVG motif in β4, but the motif is also present in MSMEG_3811, where it mediates the corresponding intramolecular interaction. Slight variations in the USP β-sheet organization in the interaction region have been observed before (47). However, the extreme case of secondary structure exchanges between monomers (“domain swapping”) appears to be of physiological relevance only in few proteins and is more often attributed to the nonphysiological crystallization conditions such as low pH or the high salt concentration in our condition (52). We therefore assume that the observed difference between Rv1636 and MSMEG_3811 is not related to ligand binding.
The affinity of Rv1636/MSMEG_3811 for cAMP is higher than that of the CRPs and the cAMP-regulated protein acyltransferase from M. tuberculosis, KATmt, or Rv0998 (23, 26). A recent study has shown Rv1636 to be the 20th most abundant protein in the M. tuberculosis proteome (53), in agreement with our ability to readily purify it through affinity chromatography. Because a major fraction of the intracellular cAMP is bound to proteins in mycobacteria (Fig. 1B), we can speculate that a significant fraction of this cAMP is bound to Rv1636/MSMEG_3811. Thus, the ability of these proteins to act as “sinks” for cAMP will limit the fraction of free cAMP available for binding to other cAMP-binding proteins, which in turn could be an effective way of regulating signaling events mediated by cAMP, and perhaps secretion of cAMP from the bacterial cell.
USPs are classified into two groups based on their ability to bind ATP. The UspFG-type binds ATP and UspAs and UspA-like proteins do not (48). All known ATP-binding USPs possess a conserved ATP-binding motif (46), but the ability to bind ATP has to date been characterized only from the co-crystal structures. The conserved ATP-binding motif is present in Rv1636/MSMEG_3811, and we report the first biochemical and thermodynamic analysis of ATP binding by a USP. A few USPs lack a fully conserved ATP-binding motif but continue to bind ATP. These include Klebsiella pneumoniae USP KPN01444, Himanthalia elongata USP HELO4277, and the C-terminal USP domain of T. thermophilus TTHA0350 (47). None of the ATP-binding USPs have been shown to have ATPase activity, and we were also unable to detect ATPase activity in Rv1636 or MSMEG_3811 (data not shown). Therefore, the functional significance of the ATP-binding by USPs still remains unknown.
Aravind et al. (54) had suggested that UspA-like proteins evolved from the UspFG-type ATP-binding proteins. It is probable that cAMP-binding USPs, such as Rv1636/MSMEG_3811, may have independently evolved from UspFG-type proteins, retaining their ATP binding properties while achieving a higher affinity for cAMP. It is not immediately obvious why Rv1636/MSMEG_3811 assume a different conformation in the Walker A-type motif region, with a short helix α5, but this structural difference certainly contributes to the unique specificity for cAMP by providing a binding site fitting the cyclic phosphodiester and not providing favorable interactions for ATP phosphates. Formation of α5 might be the structural reason for this difference, because accommodation of this helix leaves little flexibility to the remainder of the loop. Rv1636/MSMEG_3811 have identical sequences in this region, in particular in the IAGRLL motif that forms the helix, and its helix formation propensity, possibly together with the Pro described above, appears to be a key feature discriminating this cAMP-specific USP subfamily from other ATP-binding members.
The genome of M. tuberculosis encodes for 10 USPs, and Rv1636 is the only protein that contains a single USP domain (46). All other USPs have either two tandem USP domains or are associated with other domains of unknown function. The single USP present in Mycobacterium leprae, ML1390, is the ortholog of Rv1636 sharing 89% identity (55). Shown in Fig. 6 is a phylogenetic analysis of all USP domains from M. tuberculosis, M. bovis BCG, M. smegmatis, and M. leprae. One USP from M. smegmatis, MSMEG_3308, clusters closely with Rv1636/BCG_1674 and MSMEG_3811/ML1390, but an insertion of 14 amino acids is seen in MSMEG_3308 after Rv1636-Glu-45/MSMEG_3811-Glu-46. This region is not structured in the Rv1636 apo- or MSMEG_3811/cAMP crystal structures. The conserved IAGRLL motif in Rv1636/MSMEG_3811 that forms the α5 helix has the slightly altered sequence VAGRLL in MSMEG_3308. In the mass spectrometric analysis of proteins purified by cAMP affinity chromatography using M. smegmatis cytosol, we did not detect peptide fragments corresponding to MSMEG_3308, although its size (17 kDa) is very similar to MSMEG_3811. Ser-14 and Ser-16 of MSMEG_3811 (conserved through this subfamily) coordinate the cAMP phosphate moiety (Fig. 4D). However, the Val of MSMEG_3308 between those Ser residues might create a different environment from that generated by Asp in MSMEG_3811/Rv1636/ML1390/BCG_1674, thus preventing cAMP binding to MSMEG_3308.
FIGURE 6.

Phylogenetic tree depicting the relationships between USPs in mycobacteria. USP domain-containing proteins were identified in the genomes of M. tuberculosis (Rv), M. bovis BCG (BCG), M. smegmatis (MSMEG), and M. leprae (ML). Alignment and tree building was performed by MEGA 6. Highlighted as an arc are orthologs of Rv1636, described in this study. Tandem USP domains in proteins are named as USP1 and USP2, respectively.
Rv1636 and MSMEG_3811 bind cAMP and also ATP, albeit with ∼10-fold lower affinity. Because cellular ATP concentrations normally exceed cAMP levels, the intracellular concentrations of these two adenine nucleotides will strongly influence which nucleotide is bound to these USPs. Interestingly, the formation of cAMP can be dependent on the concentration of ATP in the cell (56), and Rv1636/MSMEG_3811 may therefore act as protein regulators of downstream effectors of cAMP-binding proteins, coupling this action with the energy status of the cell. Expression levels of these USPs may also determine the extent of free cAMP present in the cell. MSMEG_3811 is induced upon heat stress, along with a number of other heat-shock proteins (57). Levels of MSMEG_3811 appear to be higher during the stationary phase, and the authors (57) suggest that expression of this protein may serve as a generalized stress response. It would be interesting to monitor, for example, levels of free cAMP and, as an example, the consequent acetylation of substrates of the cAMP-regulated protein acetyltransferases (58) under these conditions. Several proteomic studies have suggested Rv1636 to be present in the cell membrane and cell wall fractions (59–62), perhaps again regulating the activities of cAMP-binding proteins in various subcellular locations in the mycobacterial cell. Interestingly, Rv1636 has been found in the culture supernatant as a secreted protein (63, 64) and also expressed during infection in mice (65), again raising the possibility of depleting free cAMP levels either secreted from the bacteria or present in the bacterial cells within the host.
To the best of our knowledge, the evidence for cellular sinks for second messengers has not been alluded to until now. We have earlier suggested that GAF domains can act as sinks for cGMP, in the absence of an associated effector domain (66). The USPs that we have characterized, being devoid of an associated effector domain, perhaps represent the first example of a “bank” for cAMP, where the currency is released depending on the needs of the organism. Thus, the free cAMP in the cell available for regulating cAMP-regulated effectors (such as CRP or the protein acetyltransferase, KATmt) will be determined by the levels of Rv1636 in the cell. The USPs characterized here possess an affinity for cAMP that is higher than that of mycobacterial CRPs (23, 24). Thus, cAMP-dependent, CRP-regulated transcription may operate only when levels of Rv1636/MSMEG_3811 fall sufficiently in the cell, thereby increasing free cellular cAMP to levels that will saturate CRP. In a similar manner, cAMP-regulated protein acetylation may also depend on levels of Rv1636 in the cell.
Because Rv1636/MSMEG_3811 can bind both cAMP and ATP, we also suggest an additional role for these sinks. Because ATP is the substrate for adenylyl cyclases, the concentration of free ATP may indirectly regulate cAMP levels in the cell. As cAMP levels increase in the cell, Rv1636/MSMEG_3811 may preferentially be bound to cAMP, thereby releasing ATP into the cell. Thus, cAMP could be an indirect regulator of energy (i.e. ATP)-dependent pathways in mycobacteria. Perhaps bacteria have evolved these USPs to serve this function, adding another twist to the unique ways that mycobacteria utilize cAMP as a second messenger (5).
In conclusion, we report that a group of enigmatic universal stress proteins bind cAMP with high specificity and affinity, and we suggest that although this finding has important implications in thinking about cAMP signaling in mycobacteria, it is conceivable that similar USPs in a variety of bacteria are in fact utilized for regulating the actions of this ancient second messenger within and between cells.
Acknowledgments
We thank Dr. Michael Weyand for supporting structure refinement and deposition. We thank Helmholtz-Zentrum Berlin for the allocation of neutron/synchrotron radiation beamtime and for travel support.
The atomic coordinates and structure factors (code 5ahw) have been deposited in the Protein Data Bank (http://wwpdb.org/).
H. Iino, S. Yokoyama, and S. Kuramitsu, unpublished data.
- CNB
- cyclic nucleotide-binding
- 2-ME
- β-mercaptoethanol
- CRP
- cAMP receptor protein
- GAF
- cGMP-specific and -regulated cyclic nucleotide phosphodiesterase, adenylyl cyclase, and E. coli transcription factor FhlA
- ITC
- isothermal titration calorimetry
- SPR
- surface plasmon resonance
- USP
- universal stress protein
- Ni-NTA
- nickel-nitrilotriacetic acid
- PDB
- Protein Data Bank
- 6-AH-cAMP
- N6-(6-aminohexyl)adenosine 3′,5′-cyclic-monophosphate
- 8-AHA-cAMP
- 8-(6-aminohexylamino)-cAMP
- RU
- response unit.
REFERENCES
- 1. Antoni F. A. (2000) Molecular diversity of cyclic AMP signalling. Front. Neuroendocrinol. 21, 103–132 [DOI] [PubMed] [Google Scholar]
- 2. McDonough K. A., Rodriguez A. (2012) The myriad roles of cyclic AMP in microbial pathogens: from signal to sword. Nat. Rev. Microbiol. 10, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Laxman S., Beavo J. A. (2007) Cyclic nucleotide signaling mechanisms in trypanosomes: possible targets for therapeutic agents. Mol. Interv. 7, 203–215 [DOI] [PubMed] [Google Scholar]
- 4. Bai G., Knapp G. S., McDonough K. A. (2011) Cyclic AMP signalling in mycobacteria: redirecting the conversation with a common currency. Cell. Microbiol. 13, 349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shenoy A. R., Visweswariah S. S. (2006) New messages from old messengers: cAMP and mycobacteria. Trends Microbiol. 14, 543–550 [DOI] [PubMed] [Google Scholar]
- 6. Sánchez J., Holmgren J. (2008) Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell. Mol. Life Sci. 65, 1347–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weiglmeier P. R., Rösch P., Berkner H. (2010) Cure and curse: E. coli heat-stable enterotoxin and its receptor guanylyl cyclase C. Toxins 2, 2213–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rehmann H., Wittinghofer A., Bos J. L. (2007) Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 8, 63–73 [DOI] [PubMed] [Google Scholar]
- 9. Kannan N., Wu J., Anand G. S., Yooseph S., Neuwald A. F., Venter J. C., Taylor S. S. (2007) Evolution of allostery in the cyclic nucleotide binding module. Genome Biol. 8, R264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taylor S. S., Kim C., Cheng C. Y., Brown S. H., Wu J., Kannan N. (2008) Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design. Biochim. Biophys. Acta 1784, 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Borland G., Smith B. O., Yarwood S. J. (2009) EPAC proteins transduce diverse cellular actions of cAMP. Br. J. Pharmacol. 158, 70–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harman J. G. (2001) Allosteric regulation of the cAMP receptor protein. Biochim. Biophys. Acta 1547, 1–17 [DOI] [PubMed] [Google Scholar]
- 13. Zoraghi R., Corbin J. D., Francis S. H. (2004) Properties and functions of GAF domains in cyclic nucleotide phosphodiesterases and other proteins. Mol. Pharmacol. 65, 267–278 [DOI] [PubMed] [Google Scholar]
- 14. Martinez S. E., Beavo J. A., Hol W. G. (2002) GAF domains: two-billion-year-old molecular switches that bind cyclic nucleotides. Mol. Interv. 2, 317–323 [DOI] [PubMed] [Google Scholar]
- 15. Heikaus C. C., Pandit J., Klevit R. E. (2009) Cyclic nucleotide binding GAF domains from phosphodiesterases: structural and mechanistic insights. Structure 17, 1551–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCue L. A., McDonough K. A., Lawrence C. E. (2000) Functional classification of cNMP-binding proteins and nucleotide cyclases with implications for novel regulatory pathways in Mycobacterium tuberculosis. Genome Res. 10, 204–219 [DOI] [PubMed] [Google Scholar]
- 17. Shenoy A. R., Sivakumar K., Krupa A., Srinivasan N., Visweswariah S. S. (2004) A survey of nucleotide cyclases in actinobacteria: unique domain organization and expansion of the class III cyclase family in Mycobacterium tuberculosis. Comp. Funct. Genomics 5, 17–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shenoy A. R., Visweswariah S. S. (2006) mycobacterial adenylyl cyclases: biochemical diversity and structural plasticity. FEBS Lett. 580, 3344–3352 [DOI] [PubMed] [Google Scholar]
- 19. Dass B. K., Sharma R., Shenoy A. R., Mattoo R., Visweswariah S. S. (2008) Cyclic AMP in mycobacteria: characterization and functional role of the Rv1647 ortholog in Mycobacterium smegmatis. J. Bacteriol. 190, 3824–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Padh H., Venkitasubramanian T. A. (1976) Cyclic adenosine 3′,5′-monophosphate in mycobacteria. Indian J. Biochem. Biophys. 13, 413–414 [PubMed] [Google Scholar]
- 21. Lowrie D. B., Jackett P. S., Ratcliffe N. A. (1975) Mycobacterium microti may protect itself from intracellular destruction by releasing cyclic AMP into phagosomes. Nature 254, 600–602 [DOI] [PubMed] [Google Scholar]
- 22. Bai G., Schaak D. D., McDonough K. A. (2009) cAMP levels within Mycobacterium tuberculosis and Mycobacterium bovis BCG increase upon infection of macrophages. FEMS Immunol. Med. Microbiol. 55, 68–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stapleton M., Haq I., Hunt D. M., Arnvig K. B., Artymiuk P. J., Buxton R. S., Green J. (2010) Mycobacterium tuberculosis cAMP receptor protein (Rv3676) differs from the Escherichia coli paradigm in its cAMP binding and DNA binding properties and transcription activation properties. J. Biol. Chem. 285, 7016–7027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharma R., Zaveri A., Gopalakrishnapai J., Srinath T., Thiruneelakantan S., Varshney U., Visweswariah S. S. (2014) Paralogous cAMP receptor proteins in Mycobacterium smegmatis show biochemical and functional divergence. Biochemistry 53, 7765–7776 [DOI] [PubMed] [Google Scholar]
- 25. Kolb A., Busby S., Buc H., Garges S., Adhya S. (1993) Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62, 749–795 [DOI] [PubMed] [Google Scholar]
- 26. Nambi S., Basu N., Visweswariah S. S. (2010) cAMP-regulated protein lysine acetylases in mycobacteria. J. Biol. Chem. 285, 24313–24323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Podobnik M., Siddiqui N., Rebolj K., Nambi S., Merzel F., Visweswariah S. S. (2014) Allostery and conformational dynamics in cAMP-binding acyltransferases. J. Biol. Chem. 289, 16588–16600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shenoy A. R., Visweswariah S. S. (2003) Site-directed mutagenesis using a single mutagenic oligonucleotide and DpnI digestion of template DNA. Anal. Biochem. 319, 335–336 [DOI] [PubMed] [Google Scholar]
- 31. Shah S., Peterkofsky A. (1991) Characterization and generation of Escherichia coli adenylate cyclase deletion mutants. J. Bacteriol. 173, 3238–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bertinetti D., Schweinsberg S., Hanke S. E., Schwede F., Bertinetti O., Drewianka S., Genieser H. G., Herberg F. W. (2009) Chemical tools selectively target components of the PKA system. BMC Chem. Biol. 9, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mueller U., Darowski N., Fuchs M. R., Förster R., Hellmig M., Paithankar K. S., Pühringer S., Steffien M., Zocher G., Weiss M. S. (2012) Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 19, 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Long F., Vagin A. A., Young P., Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deleted in proof.
- 39. Kantardjieff K. A., Rupp B. (2003) Matthews coefficient probabilities: improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 12, 1865–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 42. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schomaker V., Trueblood K. N. (1968) On the rigid-body motion of molecules in crystals. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mat. 64, 63–76 [Google Scholar]
- 44. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.5.0.3, Schrödinger, LLC, New York [Google Scholar]
- 45. Fsihi H., Cole S. T. (1995) The Mycobacterium leprae genome: systematic sequence analysis identifies key catabolic enzymes, ATP-dependent transport systems and a novel polA locus associated with genomic variability. Mol. Microbiol. 16, 909–919 [DOI] [PubMed] [Google Scholar]
- 46. O'Toole R., Williams H. D. (2003) Universal stress proteins and Mycobacterium tuberculosis. Res. Microbiol. 154, 387–392 [DOI] [PubMed] [Google Scholar]
- 47. Tkaczuk K. L., A Shumilin I., Chruszcz M., Evdokimova E., Savchenko A., Minor W. (2013) Structural and functional insight into the universal stress protein family. Evol. Appl. 6, 434–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kvint K., Nachin L., Diez A., Nyström T. (2003) The bacterial universal stress protein: function and regulation. Curr. Opin. Microbiol. 6, 140–145 [DOI] [PubMed] [Google Scholar]
- 49. Zarembinski T. I., Hung L. W., Mueller-Dieckmann H. J., Kim K. K., Yokota H., Kim R., Kim S. H. (1998) Structure-based assignment of the biochemical function of a hypothetical protein: a test case of structural genomics. Proc. Natl. Acad. Sci. U.S.A. 95, 15189–15193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Corrigan R. M., Campeotto I., Jeganathan T., Roelofs K. G., Lee V. T., Gründling A. (2013) Systematic identification of conserved bacterial c-di-AMP receptor proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 9084–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hengge R. (2009) Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 7, 263–273 [DOI] [PubMed] [Google Scholar]
- 52. Gronenborn A. M. (2009) Protein acrobatics in pairs–dimerization via domain swapping. Curr. Opin. Struct. Biol. 19, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schubert O. T., Mouritsen J., Ludwig C., Röst H. L., Rosenberger G., Arthur P. K., Claassen M., Campbell D. S., Sun Z., Farrah T., Gengenbacher M., Maiolica A., Kaufmann S. H., Moritz R. L., Aebersold R. (2013) The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe 13, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aravind L., Anantharaman V., Koonin E. V. (2002) Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide binding domains: implications for protein evolution in the RNA. Proteins 48, 1–14 [DOI] [PubMed] [Google Scholar]
- 55. Hingley-Wilson S. M., Lougheed K. E., Ferguson K., Leiva S., Williams H. D. (2010) Individual Mycobacterium tuberculosis universal stress protein homologs are dispensable in vitro. Tuberculosis 90, 236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steegborn C. (2014) Structure, mechanism, and regulation of soluble adenylyl cyclases -similarities and differences to transmembrane adenylyl cyclases. Biochim. Biophys. Acta 1842, 2535–2547 [DOI] [PubMed] [Google Scholar]
- 57. Ntolosi B. A., Betts J., Zappe H., Powles R., Steyn L. M. (2001) Growth phase-associated changes in protein expression in Mycobacterium smegmatis identify a new low molecular weight heat shock protein. Tuberculosis 81, 279–289 [DOI] [PubMed] [Google Scholar]
- 58. Nambi S., Gupta K., Bhattacharyya M., Ramakrishnan P., Ravikumar V., Siddiqui N., Thomas A. T., Visweswariah S. S. (2013) Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J. Biol. Chem. 288, 14114–14124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. de Souza G. A., Leversen N. A., Målen H., Wiker H. G. (2011) Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J. Proteomics 75, 502–510 [DOI] [PubMed] [Google Scholar]
- 60. Gu S., Chen J., Dobos K. M., Bradbury E. M., Belisle J. T., Chen X. (2003) Comprehensive proteomic profiling of the membrane constituents of a Mycobacterium tuberculosis strain. Mol. Cell. Proteomics 2, 1284–1296 [DOI] [PubMed] [Google Scholar]
- 61. Målen H., Pathak S., Søfteland T., de Souza G. A., Wiker H. G. (2010) Definition of novel cell envelope associated proteins in Triton X-114 extracts of Mycobacterium tuberculosis H37Rv. BMC Microbiol. 10, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rosenkrands I., Weldingh K., Jacobsen S., Hansen C. V., Florio W., Gianetri I., Andersen P. (2000) Mapping and identification of Mycobacterium tuberculosis proteins by two-dimensional gel electrophoresis, microsequencing and immunodetection. Electrophoresis 21, 935–948 [DOI] [PubMed] [Google Scholar]
- 63. Bahk Y. Y., Kim S. A., Kim J. S., Euh H. J., Bai G. H., Cho S. N., Kim Y. S. (2004) Antigens secreted from Mycobacterium tuberculosis: identification by proteomics approach and test for diagnostic marker. Proteomics 4, 3299–3307 [DOI] [PubMed] [Google Scholar]
- 64. Mattow J., Schaible U. E., Schmidt F., Hagens K., Siejak F., Brestrich G., Haeselbarth G., Müller E. C., Jungblut P. R., Kaufmann S. H. (2003) Comparative proteome analysis of culture supernatant proteins from virulent Mycobacterium tuberculosis H37Rv and attenuated M. bovis BCG Copenhagen. Electrophoresis 24, 3405–3420 [DOI] [PubMed] [Google Scholar]
- 65. Talaat A. M., Lyons R., Howard S. T., Johnston S. A. (2004) The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc. Natl. Acad. Sci. U.S.A. 101, 4602–4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Biswas K. H., Sopory S., Visweswariah S. S. (2008) The GAF domain of the cGMP-binding, cGMP-specific phosphodiesterase (PDE5) is a sensor and a sink for cGMP. Biochemistry 47, 3534–3543 [DOI] [PubMed] [Google Scholar]
- 67. Bond C. S. (2003) TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics 19, 311–312 [DOI] [PubMed] [Google Scholar]
- 68. Laskowski R. A., Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 69. Stivala A., Wybrow M., Wirth A., Whisstock J. C., Stuckey P. J. (2011) Automatic generation of protein structure cartoons with Pro-origami. Bioinformatics 27, 3315–3316 [DOI] [PubMed] [Google Scholar]