

Background: Human myxovirus resistance protein A (MxA) is an antiviral dynamin-related GTPase.
Results: Dimerization of MxA via a GTPase domain interface is required for GTP hydrolysis and antiviral activity.
Conclusion: GTP binding allows GTPase domain dimerization and membrane-associated assembly of MxA, but it is not sufficient to induce a sustained antiviral effect.
Significance: New mechanistic insights into the antiviral action of MxA are provided.
Keywords: G protein, innate immunity, interferon, membrane, viral replication, Mx proteins, antiviral response, catalytic mechanism, dynamin-like protein
Abstract
Myxovirus resistance (Mx) GTPases are induced by interferon and inhibit multiple viruses, including influenza and human immunodeficiency viruses. They have the characteristic domain architecture of dynamin-related proteins with an N-terminal GTPase (G) domain, a bundle signaling element, and a C-terminal stalk responsible for self-assembly and effector functions. Human MxA (also called MX1) is expressed in the cytoplasm and is partly associated with membranes of the smooth endoplasmic reticulum. It shows a protein concentration-dependent increase in GTPase activity, indicating regulation of GTP hydrolysis via G domain dimerization. Here, we characterized a panel of G domain mutants in MxA to clarify the role of GTP binding and the importance of the G domain interface for the catalytic and antiviral function of MxA. Residues in the catalytic center of MxA and the nucleotide itself were essential for G domain dimerization and catalytic activation. In pulldown experiments, MxA recognized Thogoto virus nucleocapsid proteins independently of nucleotide binding. However, both nucleotide binding and hydrolysis were required for the antiviral activity against Thogoto, influenza, and La Crosse viruses. We further demonstrate that GTP binding facilitates formation of stable MxA assemblies associated with endoplasmic reticulum membranes, whereas nucleotide hydrolysis promotes dynamic redistribution of MxA from cellular membranes to viral targets. Our study highlights the role of nucleotide binding and hydrolysis for the intracellular dynamics of MxA during its antiviral action.
Introduction
The myxovirus resistance (Mx)6 proteins are key mediators of the interferon-induced innate immune response in vertebrates (1, 2). In humans, two Mx homologs (MxA and MxB, also called MX1 and MX2, respectively) mediate antiviral activity against a broad range of viruses. MxA shows antiviral activity against several RNA virus families, including orthomyxo-, paramyxo-, bunya-, picorna-, rhabdo-, toga-, and reoviruses (3). MxA also restricts DNA viruses, like African swine fever virus and hepatitis B virus, at the transcriptional level (4, 5). MxB, in contrast, has recently been shown to be a potent inhibitor of HIV and additional lentiviruses (6–9).
Similar to other dynamin superfamily members, MxA binds to negatively charged membranes and forms ring-like oligomers that tubulate liposomes (10–12). In noninfected cells, MxA localizes to membranes of the smooth ER (10, 13). Upon viral infection with La Crosse bunyavirus (LACV), MxA translocates to perinuclear structures containing the viral nucleoprotein (NP) (14). An interaction of Mx proteins with viral NPs or ribonucleoprotein complexes was demonstrated for influenza A virus (FLUAV), Thogoto virus (THOV), and LACV (15–17).
Mx proteins have a three domain architecture comprising an N-terminal G domain, an antiparallel three-helical bundle called the bundle signaling element (BSE), and an antiparallel four-helical bundle called the stalk (Fig. 1, A and B) (18, 19). The globular G domain binds and hydrolyzes GTP and is located at the opposite site of the extended stalk in the elongated MxA monomer. Between the two domains, the BSE is thought to act as a mediator of conformational coupling between the G domain and stalk (20). The stalk mediates ring formation by assembling in a zigzag fashion via three distinct interfaces (18, 19). It also mediates recognition of viral structures and membrane surfaces via the 43 amino acid loop 4 (L4) that extends from the distal end of the stalk (12, 21, 22).
FIGURE 1.

Structure and GTPase domain dimerization of MxA. A, domain architecture; B, structure of human MxA (Protein Data Bank code 3SZR). C, homology model of the MxA GTPase domain dimer (residues 69–340) based on the crystal structure of the human dynamin1 GTPase domain-BSE construct in the GDP-AlF4−-bound state (Protein Data Bank code 2X2E). D, sequence alignment of Mx and dynamin proteins in the G4 loop. Sequences of human MxA (Swiss-Prot accession P20591), human (hs) MxB (P20592), mouse (mm) Mx1 (P09922), mmMx2 (Q9WVP9), chicken (gg) Mx (Q90597), zebrafish (dr) MxA (Q8JH68), human dynamin1 (Q05193), human dynamin2 (P50570), human dynamin3 (Q9UQ16), Drosophila melanogaster (dm) dynamin (P27619), Caenorhabditis elegans (ce) dynamin (Q9U9I9), and Saccharomyces cerevisiae (sc) dynamin-related protein DNM1 (P54861) were aligned and manually adjusted. Residues with a conservation of greater than 70% are color-coded (D and E in red; R, K, and H in blue; N, Q, S, and T in gray; A, L, I, V, F, Y, W, M, and C in green, and P and G in brown). Asp-250 and Asp-253 are marked with a dot. E, details of the catalytic site. The red ball represents the catalytic water, the green ball a Mg2+ ion, and the purple ball a Na+ ion. Asp-250 stabilizes the purine base in cis, and Asp-253 of the neighboring monomer binds to it in trans. F, left, scheme showing the proposed binding mode of the guanine base by Asp-250 in cis and Asp-253 of the opposing molecule in trans. Right, xanthosine base-binding by Asn-250 and envisaged binding mode of Asn-253. Hydrogen bonds are depicted as dashed lines.
The G domain is the most highly conserved region in dynamin superfamily members, with ∼40% sequence identity between dynamin and MxA (23). Dynamin-like GTPases are characterized by a low basal GTPase activity, which is greatly stimulated by GTP-dependent dimerization of their G domains (24). Dimerization in dynamin and the dynamin-1-like protein (DNM1L) is mediated via a highly conserved interface across the nucleotide-binding site, the so-called G interface (25, 26), that was also recently described for MxA (20). We previously proposed that MxA oligomerizes into rings around viral target structures (27) as observed for MxA rings assembling around lipid tubes (12). The G domains dimerize across adjacent rings thereby mediating inter-ring contacts via the G interface (18). Similarly, G domains in dynamin are thought to dimerize across helical turns formed around lipid tubes (28). Formation of this G interface activates GTP hydrolysis by stabilizing the flexible switch regions containing residues crucial for catalysis. GTP hydrolysis then triggers a large scale conformational movement of the adjacent BSE, as shown for dynamin and MxA (20, 28), that may act as a power stroke required for the constriction of the rings or helices. However, the exact role and function of GTP binding and hydrolysis for the antiviral activity of Mx proteins remain unclear.
Based on the crystal structure of the dynamin 1 G domain dimer in the presence of a transition state mimic (25) and previous functional experiments (18), we performed a systematic biochemical characterization of several classical and novel MxA mutants in the G domain to explore the mechanism of nucleotide binding and hydrolysis in MxA. We found that catalytic residues in the G interface and the nucleotide itself are involved in G domain dimerization and GTP hydrolysis. Furthermore, we used these mutants in cell-based assays to dissect the role of nucleotide binding and hydrolysis for the antiviral function of MxA.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
Expression constructs of MxA were described previously (18). Site-directed mutagenesis was carried out using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA).
Protein Expression and Purification
Human MxA (ExPASy accession P20591) and the indicated mutants were expressed as N-terminal His6 fusions from a pET28 plasmid followed by a PreScissionTM cleavage site in the Escherichia coli BL21 (DE3) Rosetta strain (Novagen) (19). Bacterial cultures were grown in TB medium at 37 °C. At an A600 >0.4, cultures were cooled to 18 °C, and protein expression was induced by the addition of 40 μm isopropyl β-d-thiogalactopyranoside. Following centrifugation, bacterial pellets were resuspended in ice-cold 50 mm HEPES (pH 7.5), 800 mm NaCl, 30 mm imidazole, 5 mm MgCl2, 1 μm DNase I, 2.5 mm β-mercaptoethanol (β-ME), 500 μm Pefabloc SC (Roth) and lysed in a microfluidizer (Microfluidics). A soluble cell extract was prepared by ultracentrifugation at 40,000 × g for 45 min at 4 °C. After filtration, it was applied to a Ni2+-nitrilotriacetic acid column (GE Healthcare) equilibrated with 50 mm HEPES (pH 7.5), 400 mm NaCl, 30 mm imidazole, 5 mm MgCl2, 2.5 mm β-ME. The column was extensively washed with 20 mm HEPES (pH 7.5), 800 mm NaCl, 5 mm MgCl2, 45 mm imidazole, 2.5 mm β-ME, 1 mm ATP, 10 mm KCl and afterwards with 20 mm HEPES (pH 7.5), 400 mm NaCl, 5 mm MgCl2, 45 mm imidazole, 2.5 mm β-ME. Following protein elution by 20 mm HEPES (pH 7.5), 400 mm NaCl, 300 mm imidazole, 5 mm MgCl2, 2.5 mm β-ME, the protein was incubated overnight at 4 °C in the presence of 250 μg of GST-tagged PreScission protease to cleave the N-terminal His6 tag. The cleaved protein was concentrated and applied to a Superdex 200 16/60 (GE Healthcare) gel filtration column equilibrated with 20 mm HEPES (pH 7.5), 500 mm NaCl, 2 mm MgCl2, 2 mm DTT. PreScission protease was removed using a GST column. Fractions containing MxA were pooled, concentrated, and frozen in small aliquots.
Nucleotide Binding Studies
Nucleotide dissociation constants were determined at 8 °C on a VP-isothermal titration calorimetry (VP-ITC) system (MicroCalTM, GE Healthcare). 1 mm nucleotide in ITC Buffer (50 mm HEPES (pH 7.5), 150 mm NaCl, 5 mm MgCl2, 5 mm KCl) was titrated in 8-μl steps into a reaction chamber containing 50 μm MxAM527D (or the indicated M527D mutants) in the same buffer. For the K83A mutant, an iTC200 (Microcal) was used with 200 μm protein and 4 mm guanosine 5′-O-[γ-thio]triphosphate (GTPγS). The resulting heat change upon injection was integrated over a time range of 240 s, and the obtained values were fitted to a standard single-site binding model using Origin®.
Nucleotide Hydrolysis Assay
GTPase activities of human MxA mutants were determined at 37 °C in 50 mm HEPES (pH 7.5), 150 mm NaCl, 5 mm MgCl2, 5 mm KCl. Saturating concentrations of GTP or xanthosine 5′-triphosphate (XTP) (1 mm) were used for each reaction. Reactions were initiated by the addition of protein to the final reaction solution. For the heteromeric stimulation reactions, the concentration of MxAM527D was kept constant at 2.5 μm, and increasing concentrations of the indicated MxA mutants were added. At different time points, reaction aliquots were 20-fold diluted in GTPase buffer (50 mm HEPES (pH 7.5), 150 mm NaCl, 5 mm MgCl2, 5 mm KCl) and quickly transferred into liquid nitrogen. Separation of different nucleotides was achieved on a reversed phase HPLC system using a Hypersil ODS-2 C18 column. Nucleotide peaks were detected by measuring adsorption at 254 nm and compared with standard nucleotide samples. GTP and hydrolysis product GDP in the samples were quantified by integration of the corresponding absorption peaks. Rates derived from a linear fit to the initial rate of the reaction (<40% GTP hydrolyzed) were plotted against the protein concentrations, and the kobs values were calculated. For data analysis, the program GraFit5 (Erithacus Software) was used.
Analytical Gel Filtration
The MxA mutants were analyzed using an FPLC Akta purifier (GE Healthcare) equipped with a Superdex 200 10/300 column in the absence or presence of the indicated nucleotides. The running buffer contained 20 mm HEPES (pH 7.5), 150 mm NaCl, 2 mm MgCl2 but no nucleotides. The mutants were preincubated for 15 min with 2 mm GDP/xanthosine 5′-diphosphate (XDP), 2 mm AlCl3, and 20 mm NaF at 4 °C in gel filtration buffer. 50 μl of a 2 mg/ml protein solution was subsequently applied to the column. A flow rate of 0.5 ml/min was used. Chromatograms were recorded at a wavelength of 280 nm.
Homology Modeling
For homology modeling, the fully automated protein structure homology-modeling server, accessible via the ExPASy web server, was used (29). Subsequently, the calculated monomeric model was superimposed to the GDP-AlF4−-bound GTPase dimer of human dynamin 1 (25) using PyMOL (30).
Cells and Viruses
HEK-293T, HeLa, and Vero cells were cultivated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum, 2 mm l-glutamine, penicillin (50 units/ml), and streptomycin (50 μg/ml). THOV strain SiAr126 (31) and LACV (32) were used for the infection experiments.
Minireplicon Assay
To reconstitute the polymerase activity of FLUAV, strain A/Vietnam/1203/04 (H5N1) (33), 293T cells were seeded into 12-well plates and transfected with 10 ng of pCAGGS expression plasmids for the viral polymerase subunits PB2, PB1, and PA as well as 100 ng of NP-encoding plasmids. As a minigenome, 50 ng of plasmids encoding firefly luciferase in negative sense orientation flanked by 5′- and 3′-UTRs from viral segment 8 (pPolI-FFLuc-RT for FLUAV) were co-transfected. 10 ng of pRL-SV40, from which Renilla luciferase is constitutively expressed, was added to normalize transfection efficiency. It was shown that expression of the firefly luciferase reporter gene correlates with the activity of the reconstituted polymerase complex (34).
For reconstitution of the THOV strain SiAr126 minireplicon system (22), 10 ng of pCAGGS expression plasmids for the viral polymerase subunits PB2, PB1, and PA as well as 50 ng of NP-encoding plasmids, 50 ng of plasmids encoding firefly luciferase in negative-sense orientation flanked by 5′- and 3′-UTRs from viral segment 5 (pHH21-vNP-FFLuc for THOV) and 10 ng of pRL-SV40 were co-transfected. To examine Mx-mediated inhibition of virus polymerase activities, 300 ng (FLUAV minireplicon) and 100 ng (THOV minireplicon) of pCAGGS plasmids coding for N-terminally FLAG-tagged Mx proteins were co-transfected, respectively. At 24 h post-transfection, cells were lysed, and firefly and Renilla luciferase activities in the lysates were measured using the dual-luciferase reporter assay (Promega). After normalization of firefly to Renilla luciferase activity, the empty vector control was set to 100%. Each experiment contained technical duplicates, and all experiments were performed three times. Statistical analysis was done using the GraphPad Prism 6 software. For expression control, Western blots with specific antibodies against FLAG or HA tag (Sigma), FLUAV NP (Serotec), as well as β-actin (Sigma) were performed.
Co-immunoprecipitation and Immunoblot Analysis
293T cells were transfected with pCAGGS expression plasmids coding for MxA (1 μg, FLAG-tagged wild type (WT) and mutants) using the Nanofectin transfection reagent (PPA Laboratories). 24 h post-transfection, cells were infected with THOV at a multiplicity of infection of 10. 24 h post-infection, co-immunoprecipitation analysis was performed. The cells were lysed in 50 mm Tris (pH 8.0), 150 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40 and incubated with anti-FLAG-M2 affinity gel (Sigma) for 2 h at 4 °C. The washed precipitates as well as whole cell lysates to control protein expression were subjected to standard Western blot analysis using antibodies against FLAG tag (Sigma), THOV NP (35), as well as β-actin (Sigma).
Immunofluorescence Analysis
To examine the intracellular localization of MxA G domain mutants, HeLa cells were transfected with FLAG-tagged MxA expression plasmids (50 ng per 24-well) and fixed 24 h post transfection with 3% paraformaldehyde. MxA was detected with the monoclonal mouse antibody M143 (36) and an Alexa Fluor 488-conjugated donkey secondary antibody (Invitrogen). Fluorescence staining of syntaxin 17 was performed using a polyclonal goat anti-syntaxin 17 (13) antibody and an Alexa Fluor 555-conjugated donkey secondary antibody. Immunofluorescence analyses were performed with a Zeiss Axio Observer.Z1 microscope. For co-localization studies, a Leica TCSSP2 confocal laser scanning microscope was used.
To detect LACV nucleoprotein-MxA aggregate formation, Vero cells were transfected with FLAG-tagged MxA expression plasmids (50 ng per 24-well) for 24 h and then infected with LACV at a multiplicity of infection of 10 for an additional 20 h. Subsequently, the cells were prepared and stained for MxA proteins and viral antigens by indirect immunofluorescence, as described previously (14). MxA was detected as described above and LACV nucleoprotein (N) protein with a polyclonal rabbit antibody. Alexa Fluor 555- and Alexa Fluor 488-conjugated donkey secondary antibodies (Invitrogen) and an ApoTome fluorescence microscope (Zeiss) were used for the detection of the proteins.
RESULTS
Analysis of the GTPase Mechanism of MxA
To gain insights into the GTPase mechanism of MxA, we modeled the G domain dimer of MxA based on the structure of the GDP-AlF4−-bound dynamin (Fig. 1C) (25). Most of the residues in the catalytic center are highly conserved between MxA and dynamin, for example the G4 loop mediating specificity for guanine binding (Fig. 1D), suggesting a conserved catalytic mechanism of these proteins. The modeled dimer interface was recently confirmed by crystal structures of a stalkless MxA construct determined in the absence of nucleotide, in the presence of GDP or of the nonhydrolyzable GTP analogue GMP-PCP (20).
For this study, we focused on several positions in or close to the catalytic center of the G domain (Fig. 1E) as follows: Lys-83 in the highly conserved phosphate-binding loop (37); Thr-103 in the switch I region that is crucial for stabilizing the transition-state of GTP hydrolysis (38); Asp-250 contacting the purine base in cis; and Asp-253 stabilizing the G interface by contacting the purine base of the opposing G domain in trans (Fig. 1E). To test the contribution of individual positions for GTP binding and hydrolysis in biochemical assays, we used a previously described monomeric stalk mutant of MxA, MxAM527D, and introduced the respective mutations. MxAM527D cannot assemble into higher order oligomers (18), shows greater solubility than the WT protein, and can be prepared in high quantities from bacterial lysates without aggregation. Nucleotide binding to this mutant can reliably be measured by ITC, without considering competing reactions such as higher order assembly via the stalk domains.
Initially, we determined the nucleotide binding affinities of MxAM527D. It bound to the slowly hydrolysable GTP analogue GTPγS with an affinity of 15 μm (Fig. 2A and “Experimental Procedures”, Table 1 for a summary of all data), in good agreement with previous measurements using fluorescence analysis (18). In gel filtration experiments, this mutant eluted as a monomer when incubated with GTPγS, GDP or in the absence of nucleotide (Fig. 3A). However, in the presence of GDP-AlF4− that mimics the transition state of GTP hydrolysis, MxAM527D dimerized, suggesting that trapping of the GTPase transition state is accompanied by G domain dimerization (Fig. 3A). In agreement with previous results (18), MxAM527D showed a robust intrinsic GTPase activity that cooperatively increased with increasing protein concentrations (Fig. 4A), again indicating that the formation of a G interface leads to stimulated GTP hydrolysis.
FIGURE 2.

Nucleotide binding analysis. 1 mm solutions of the indicated nucleotide were titrated stepwise into 50 μm solutions of the indicated MxA mutants at 8 °C in an ITC device. Resulting heat changes were integrated, and the obtained values were fitted to a quadratic binding equation. The following KD values were derived from the fittings. A, M527D for GTPγS: KD = 15 ± 1 μm, n = 0.92 ± 0.02. B, M527D/K83A for GTPγS: KD = 39 ± 6 μm, n = 0.42 ± 0.03. C, M527D/T103A for GTPγS: KD = 28 ± 2 μm, n = 0.81 ± 0.02. D, M527D/D250N for XTPγS: KD = 7.8 ± 0.5 μm, n = 0.82 ± 0.01. E, M527D/D253N for GTPγS: KD = 9 ± 1 μm, n = 0.73 ± 0.01 and F, M527D/D250N/D253N for XTPγS: KD = 5.6 ± 0.4 μm, n = 0.87 ± 0.01. GTPγS (□) and XTPγS (○). Because of the reduced heat signal upon nucleotide binding, higher protein and ligand concentrations were used for the T103A and K83A mutants. For K83A, this resulted in increased protein precipitation that may explain the lowered binding number.
TABLE 1.
Summary of the biochemical and antiviral features of the examined mutants
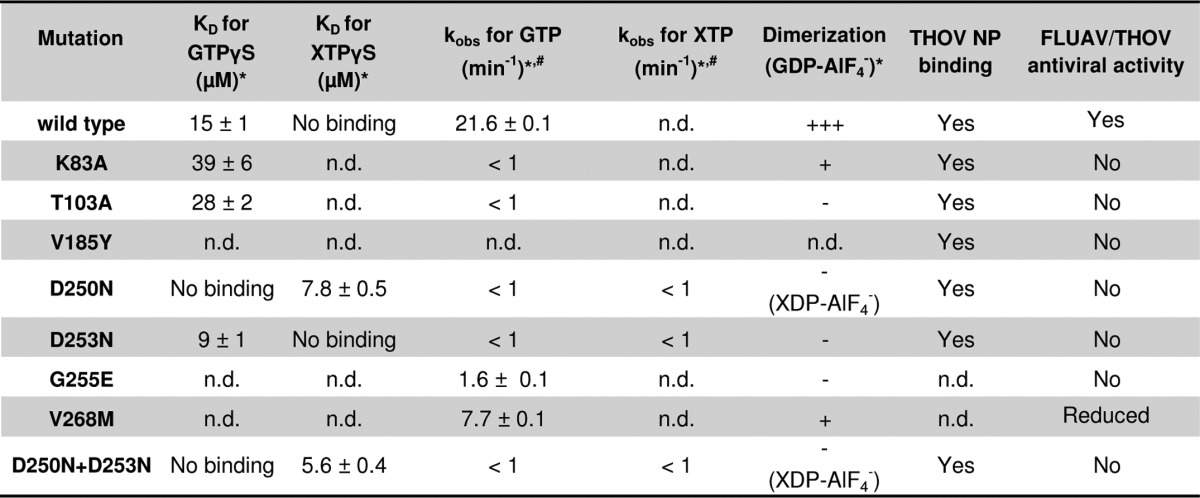
*Experiments were performed with the monomeric M527D mutant.
#Data are at a 20 μm protein concentration.
n.d. indicates not determined.
FIGURE 3.

Analytical gel filtration analysis. Upon 15 min of incubation with 2 mm of the indicated nucleotide solutions, 50 μl of the indicated MxA mutants at a concentration of 2 mg/ml were applied to an S200 gel filtration column. A, M527D with GTPγS, GDP-AlF4−, GDP, AlF4− alone or in the absence of nucleotides. B, M527D/K83A. C, M527D/T103A. D, M527D/D250N. E, M527D/D253N. F, M527D/D250N/D253N in the absence and presence of the indicated nucleotides.
FIGURE 4.

Analysis of GTPase activities.
A, protein concentration-dependent GTPase/XTPase activities of M527D and representative mutants. All reactions were carried out in the presence of 1 mm nucleotide at 37 °C, and GTP/XTP hydrolysis was monitored by HPLC analysis. The mean kobs was calculated from two independent experiments for each concentration, with the error bar showing the range of the two data points. M527D (GTP) (▿), M527D/K83A (GTP) (♢), M527D/T103A (GTP) ( ), M527D/D250N (XTP) (Δ), M527D/D253N (GTP) (□), and M527D/D250N/D253N (XTP) (○). B, GTPase activity of M527D can be stimulated by monomeric G domain mutants of MxA. 2.5 μm M527D was incubated with the indicated concentrations of the corresponding mutant (see x axis). The mean kobs calculated from two independent experiments is indicated, with the error bar showing the range of the two data points. M527D+M527D/T103A (○), M527D+M527D/D250N (Δ), M527D+M527D/D253N (□). C, mixed GTPase assays, similar as in B, using 37.5 μm of each of the indicated M527D mutants. The mean kobs was calculated from two independent experiments, with the error bar showing the range of the two data points. When two monomeric G domain mutants were incubated together, their GTPase reactions were mostly additive.
), M527D/D250N (XTP) (Δ), M527D/D253N (GTP) (□), and M527D/D250N/D253N (XTP) (○). B, GTPase activity of M527D can be stimulated by monomeric G domain mutants of MxA. 2.5 μm M527D was incubated with the indicated concentrations of the corresponding mutant (see x axis). The mean kobs calculated from two independent experiments is indicated, with the error bar showing the range of the two data points. M527D+M527D/T103A (○), M527D+M527D/D250N (Δ), M527D+M527D/D253N (□). C, mixed GTPase assays, similar as in B, using 37.5 μm of each of the indicated M527D mutants. The mean kobs was calculated from two independent experiments, with the error bar showing the range of the two data points. When two monomeric G domain mutants were incubated together, their GTPase reactions were mostly additive.
Lys-83 in MxA is part of the phosphate-binding loop. In dynamin, the corresponding K44A mutant was originally designed as a GTP-binding deficient mutant, based on homology to the Ras GTPase (39). In dynamin, this mutant is now widely used to block clathrin-mediated endocytosis (40). Interestingly, recent cryo-EM data showed that the dynamin K44A mutant can form a super-constricted helix in the presence of GTP (41). In MxA, K83A was suggested to affect GTP binding (37). In our ITC assays, however, MxAM527D/K83A still bound GTPγS with a dissociation constant of 39 μm, i.e. a slightly reduced affinity compared with MxAM527D (Fig. 2B). Dimerization of this mutant in the presence of GDP-AlF4− was greatly reduced (Fig. 3B), and nucleotide hydrolysis was completely blocked for MxAM527D/K83A (Fig. 4A).
The T103A exchange in MxA was considered to block GTP hydrolysis but not GTP binding (38). The corresponding Thr-65 in dynamin is located in switch I and was shown to stabilize the attacking water molecule for GTP hydrolysis via a main chain interaction (25). Furthermore, the side chain of Thr-65 coordinates the catalytic Mg2+ ion. The T65A mutation was proposed to affect only GTP hydrolysis (42), although others suggested it may reduce nucleotide binding as well (43). In ITC experiments, MxAM527D/T103A bound GTPγS with a slightly reduced affinity (KD = 28 μm) compared with MxAM527D, indicating that the T103A exchange did not grossly affect nucleotide binding (Fig. 2C). However, this mutant did not dimerize in the presence of GDP-AlF4− (Fig. 3C) and showed a complete loss of basal and stimulated GTPase activity (Fig. 4A), suggesting that it cannot stabilize a GTPase transition state.
To create a bona fide GTP-binding deficient mutant, we turned towards an amino acid exchange in the guanine nucleotide specificity (G4) motif. In the small GTPase Ras and the signal recognition particle GTPase (44–46), the mutation of an aspartate to an asparagine in G4 abrogates GTP binding and allows binding of the related XTP. In agreement with these observations, also the corresponding MxA mutant MxAM527D/D250N lost its ability to bind GTPγS but bound XTPγS with a dissociation constant of 7.8 μm (Fig. 1F, 2D), whereas MxAM527D did not bind to XTPγS with appreciable affinity (Fig. 2A). However, addition of XDP-AlF4− did not result in dimerization of MxAM527D/D250N (Fig. 3D), and the mutant did not show stimulated XTPase activity (Fig. 4A) nor GTPase activity (data not shown). Thus, not only nucleotide binding but also the nature of the bound nucleotide itself contributes to dimerization and dimerization-stimulated GTP hydrolysis.
In dynamin, Asp-211 following the G4 motif mediates dimerization by contacting the nucleotide in the opposing molecule via a contact to the guanine base (25). This residue is also conserved in MxA (Asp-253) and contacts the guanine base (Fig. 1E) (20). We reasoned that the lack of XTPase reaction in MxAM527D/D250N mutant might be caused by the inability of Asp-253 to contact the xanthosine base in trans. To explore this hypothesis, we sought to restore this putative contact in MxA by additionally introducing the D253N mutation, which in principle should be able to form two hydrogen bonds with the xanthosine base in trans (Fig. 1F). The MxAM527D/D253N mutant bound GTPγS with comparable affinity to MxAM527D (KD = 9 μm) (Fig. 2E), but it did not dimerize in the presence of GDP-AlF4− (Fig. 3E). It also did not display any GTPase activity (Fig. 4A), indicating an involvement of Asp-253 in dimerization-induced catalysis of MxA. The MxAM527D/D250N/D253N mutant bound with a dissociation constant of 5.6 μm to XTPγS but not to GTPγS (Fig. 2F). However, also for this mutant, we did not observe XDP-AlF4−-induced dimerization (Fig. 3F) and no stimulated XTPase (Fig. 4A) and GTPase (data not shown) activity, indicating that the D253N mutation cannot restore a putative contact to XTP in trans. We speculate that the D250N mutant binds the xanthosine base with a slightly different geometry compared with the genuine guanine-base binding mode. Such a difference may then affect the overall geometry of the catalytic site in a way that is not compatible with G domain dimerization and initiation of nucleotide hydrolysis.
One Intact Catalytic Center Is Sufficient for Nucleotide Hydrolysis
To further characterize the requirements for formation of the G domain interface, we analyzed the ability of the different mutants to stimulate the GTPase activity of the WT G domain of monomeric MxAM527D in a mixed GTPase assay. For these experiments, we employed a constant low concentration of 2.5 μm MxAM527D and added increasing amounts of MxAM527D mutants defective in GTP hydrolysis (T103A), nucleotide binding (D250N), or G interface formation (D253N). Strikingly, the monomeric T103A mutant stimulated the GTPase activity of MxAM527D as efficiently as monomeric MxAM527D (compare Fig. 4, A with B), whereas the G interface mutant D253N was a less potent activator, indicating that one functional GTPase site can be sufficient for the formation of a functional G interface with a GTP-binding competent mutant. The GTP-binding deficient D250N mutant showed only minor GTPase activation of MxAM527D, indicating that the loss of nucleotide binding (D250N) in one molecule interferes with dimerization-induced GTP hydrolysis. Interestingly, GTP hydrolysis by the monomeric T103A, D250N, or D253N mutants could not be restored by any of the other defective mutants (Fig. 4C), indicating that defects in both partner molecules completely abrogate GTPase activity.
Nucleotide Binding and Hydrolysis Are Required for Antiviral Function of MxA
Equipped with a biochemically validated set of G domain mutants, we explored the role of nucleotide binding and hydrolysis for the antiviral activity of MxA. Initially, we assessed inhibition of the polymerase complex of THOV, a FLUAV-like orthomyxovirus, that shows high sensitivity to the antiviral effect of MxA (47, 48). We used a minireplicon reporter assay to study the polymerase activity of THOV (22). Co-expression of the viral polymerase subunits and the viral NP, together with an artificial RNA minigenome encoding the firefly luciferase reporter gene, reconstitutes viral ribonucleoprotein complexes that are active in transcription and replication of the minigenome. Additional expression of WT MxA suppressed viral polymerase activity to 2% (Fig. 5A). However, the introduction of the previously characterized point mutations in the G domain all led to a partial or complete loss of the antiviral function (Fig. 5A and Table 1) indicating that the full cycle of GTP binding, hydrolysis, and G domain dimerization is crucial for the antiviral effect.
FIGURE 5.

G domain mutants interact with the viral NP. A, 293T cells were co-transfected with expression plasmids for the THOV minireplicon system, consisting of the viral polymerase subunits (10 ng each), NP (50 ng), and the pPolI-FF-Luc minigenome encoding firefly luciferase (50 ng) as well as expression plasmids for MxA or MxA mutants (100 ng) for 24 h. Firefly luciferase activity determined in the cell lysates was normalized to the activity of Renilla luciferase encoded by the co-transfected pRL-SV40 plasmid (10 ng). The activity in the absence of MxA, empty vector control, was set to 100%. Results are presented as means of technical duplicates of three independent experiments. Protein expression of FLAG-tagged Mx, viral NP, and β-actin was determined by Western blot analysis. B, co-precipitation of viral NP with MxA. 293T cells were transfected with FLAG-tagged MxA constructs and infected with THOV (10 multiplicities of infection). At 24 h post-infection, the cell lysates were subjected to FLAG-specific immunoprecipitation (IP). FLAG-MxA and co-precipitated THOV-NP as well as whole cell lysates (WCL) were analyzed by Western blot. Results of one experiment that is representative for three individual experiments are shown.
A direct and robust interaction of MxA with the NP of THOV, the major constituent of the viral nucleocapsids, has previously been demonstrated (16). It involves loop L4 at the tip of the stalk that directly interacts with the viral NP (22). To test the influence of our G domain mutations on viral target interaction, cells expressing the respective FLAG-tagged MxA mutants were infected with THOV, and viral NP was co-precipitated from the cell lysates using a FLAG-specific antibody. The viral NP was co-precipitated by WT MxA, but not by an MxA mutant with a deletion of loop L4 (ΔL4) that lacks the putative NP interaction site (Fig. 5B, lane 9) (22). Interestingly, in pulldown experiments, the G domain mutants K83A, T103A, D250N, D253N, and D250N/D253N efficiently precipitated THOV NP from the infected cells (Fig. 5B). V185Y was included in the analysis because this exchange was recently shown to disrupt formation of the G interface (20). This mutant also bound efficiently to the NP (Fig. 5B, lane 5), but it showed severely reduced antiviral activity (Fig. 5A). These results indicate that nucleotide binding and hydrolysis of MxA are not required for THOV RNP binding but are essential for executing the subsequent antiviral effect against THOV.
Next, we assessed inhibition of a highly pathogenic H5N1 influenza A virus (49) using a previously described minireplicon reporter assay for FLUAV (33). In these assays, WT MxA inhibited viral replication by 80% (Fig. 6A). However, all tested G domain mutations completely abrogated antiviral activity. To further explore the effect of these mutants, we co-expressed some of them with WT MxA, expecting a stimulatory effect, as detected for the stimulation of the GTPase activity in Fig. 4B. When co-expressed even at low concentrations, the G domain mutants interfered with the antiviral activity of WT MxA in a dominant-negative fashion (Fig. 6B). However, the oligomerization-defective mutant MxAM527D that was shown to be antivirally inactive (18) did not affect the antiviral activity of WT MxA in these co-expression experiments, presumably because it was not incorporated into WT MxA oligomers. This suggests that incorporation of GTP-binding or GTPase-deficient MxA molecules into a hetero-oligomeric complex can hamper the antiviral activity of WT MxA.
FIGURE 6.

Functional GTPase is crucial for antiviral activity. A, 293T cells were co-transfected with expression plasmids for the FLUAV minireplicon system of VN/04, consisting of the viral polymerase subunits (10 ng each), NP (100 ng), and the pPolI-FF-Luc minigenome encoding firefly luciferase under the control of the viral promoter (50 ng) as well as expression plasmids for MxA or MxA mutants (300 ng). After 24 h, firefly luciferase activity was determined in the cell lysates and normalized to the activity of Renilla luciferase encoded by the co-transfected pRL-SV40 plasmid (10 ng). The activity in the absence of MxA, empty vector control, was set to 100%. Protein expression of FLAG-tagged MxA, viral NP, and β-actin were determined by Western blot analysis. B, dominant-negative effect of MxA mutants on WT MxA activity. HA-tagged WT MxA (300 ng) was co-transfected with the components of the VN/04 minireplicon system as described for A and increasing amounts (50, 100, and 200 ng) of the indicated FLAG-tagged MxA mutants. Results are presented as means of technical duplicates of three independent experiments.
Role of Nucleotide Binding and Hydrolysis in Formation of Cytoplasmic MxA Assemblies
To further study the consequences of the lack of GTP binding and hydrolysis, we analyzed the intracellular distribution of the MxA mutants. For this, HeLa cells were transfected with the various MxA constructs, and their localization was analyzed by immunofluorescence using an MxA-specific monoclonal antibody (36). In agreement with previous data, WT MxA showed a diffuse cytosolic staining with some punctate structures (Fig. 7). These structures co-localized with syntaxin 17, a marker of the smooth ER, indicating an association of WT MxA with this membrane compartment (Fig. 8), as reported previously (13). In contrast, the GTPase-deficient mutants, K83A, T103A, V185Y, and D253N, that showed GTP binding but not hydrolysis, aggregated in large cytosolic clusters (Fig. 7). These clusters also co-localized with the smooth ER marker syntaxin 17 (Fig. 8). However, MxAD250N, the only mutant defective in GTP binding, showed a cytosolic distribution without punctate aggregates (Fig. 7), pointing to a role of GTP binding for the formation of intracellular MxA assemblies that are associated with syntaxin 17-positive membranes. This was supported by the observation that introduction of the D250N mutation into GTPase-deficient MxA mutants also led to an even cytoplasmic distribution, as exemplified for MxAT103A, MxAV185Y, and MxAD253N versus the respective double mutants (Figs. 7 and 8).
FIGURE 7.
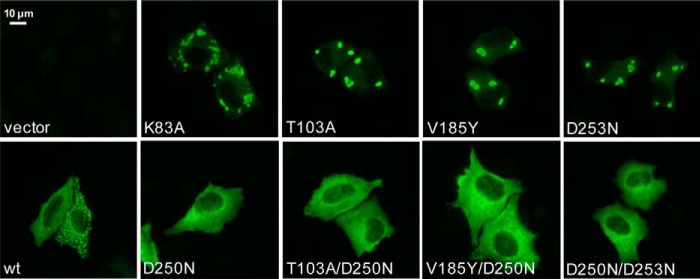
Intracellular distribution of MxA G domain mutants. HeLa cells were transfected with expression plasmids for WT MxA or MxA mutants (50 ng). At 24 h post-transfection, cells were fixed and stained with a specific antibody against MxA. Results are representative for three individual experiments.
FIGURE 8.

Co-localization study of MxA G domain mutants with syntaxin 17. HeLa cells were transfected with expression plasmids for WT MxA or MxA mutants (50 ng). At 24 h post-transfection, cells were fixed and stained with specific antibodies against MxA (green) and syntaxin 17 (red). Immunofluorescence analysis was performed using a confocal laser scanning microscope.
In a further set of experiments, we tested the intracellular redistribution of the MxA mutants induced by LACV infection. LACV replicates in the cytoplasm, and the viral nucleoprotein (N) accumulates in the Golgi area at a late phase of the replication cycle (Fig. 9, vector, white arrow). However, during the process of MxA-mediated restriction of LACV replication, MxA associates with newly synthesized viral N protein and sequesters it into large perinuclear MxA-N assemblies that are a readout for the antiviral effect (14). Accordingly, WT MxA formed the characteristic perinuclear assemblies with the viral N upon infection with LACV (Fig. 9, wt, yellow arrow). In contrast, the GTPase-deficient mutants, MxAT103A and MxAD253N, that spontaneously aggregated into MxA assemblies did not show obvious redistribution upon viral infection and thus did not limit viral replication, as indicated by the localization of N to the Golgi area (Fig. 9, white arrows). Interestingly, the GTP-binding deficient mutants MxAD250N and MxAD250N/D253N showed a diffuse cytoplasmic staining pattern and did not form a complex with viral N (Fig. 9). Taken together, these results suggest that MxA mutants defective in GTP binding, G domain dimerization, and GTP hydrolysis lose antiviral activity against orthomyxoviruses and LACV.
FIGURE 9.

Complex formation of MxA with the LACV nucleoprotein. Vero cells were transfected with the indicated MxA expression constructs (50 ng) and infected with LACV. At 18 h post-infection, cells were fixed, and immunofluorescence analysis was performed using specific antibodies against MxA (green) and LACV-N (red). The mock panel shows overlay images of transfected and mock-infected cells. The white arrows indicate accumulation of N protein in the Golgi area that is devoid of an MxA signal. In WT MxA-expressing cells, yellow arrows indicate formation of ER resident MxA-N assemblies.
G255E and V268M Are Natural Genetic Variations in the Putative G Interface
Human MxA is highly conserved with only few allelic variations in the human population. A recent analysis of genomic DNA from healthy individuals identified some rare variants in the MX1 gene. Two of them are located in the G interface and lead to an exchange of glycine to glutamic acid at position 255 and valine to methionine at position 268, respectively (Fig. 10A) (50). We tested whether these mutations affect catalysis and function of MxA. Interestingly, the MxAM527D/V268M and MxAM527D/G255E mutants displayed reduced or a complete loss of GDP-AlF4−-induced dimerization (Fig. 10, B and C). Correspondingly, the mutations led to a reduction and a complete loss of GTPase activation, respectively (Fig. 10D), suggesting that they decrease the capacity to form a proper G interface to different extents. Furthermore, the G255E mutation completely abolished the antiviral activity of MxA in the FLUAV minireplicon assay and led to cytoplasmic aggregates when expressed in HeLa cells. The MxAV268M mutant showed a slightly but significantly reduced antiviral activity (Fig. 10E) and a diffuse cytosolic staining, including small punctate assemblies that were markedly different from the few large aggregates formed by MxAG255E. The pattern was rather reminiscent of that observed for WT MxA (Figs. 7 and 10F).
FIGURE 10.

Effects of MxA polymorphisms in the G interface. A, homology model of the MxA GTPase domain dimer as in Fig. 1C. Interface residues are depicted as balls: Val-185 (purple), Gly-255 (red), and Val-268 (green) with the superscript letter indicating monomer A or B, respectively. B and C, analytical gel filtration analysis of M527D/G255E and M527D/V268M as in Fig. 3. D, protein concentration-dependent GTPase activities of M527D (○), M527D/V268M (□), and M527D/G255E (▵) as in Fig. 4A. E, FLUAV-minireplicon system of VN/04, as described in Fig. 6A. Significance was calculated with Student's t test (n = 3). **, p = 0.0058; ***, p = 0.0002. F, intracellular distribution of G255E and V268M in HeLa cells, as in Fig. 7.
DISCUSSION
A prominent feature of the dynamin superfamily proteins is their concentration-dependent increase of GTPase activity. For dynamin and dynamin-related proteins, such as MxA or the guanylate-binding protein 1, GTPase activation is mediated by nucleotide-dependent dimerization of the G domains via the G interface, which induces rearrangements of catalytic residues in the active site (20, 25, 51). The GTPase activity of dynamin and many dynamin-related proteins has been implicated in the nucleotide-dependent remodeling of cellular membranes. However, the exact function of the GTPase for the antiviral action of MxA has remained unclear. Current knowledge indicates that the antiviral activity of MxA is dependent on GTP hydrolysis (37, 38) and oligomerization via the stalk (18, 19, 52, 53). In this study, we used a panel of G domain mutants to demonstrate that nucleotide binding and formation of the G interface are critical for the intracellular localization and the antiviral function of MxA.
Structural analysis indicates that the G domain dimer of dynamin and MxA is formed by direct contacts between the two switch regions and the trans stabilizing loop (20, 25). Furthermore, Asp-253 from the G4 loop stabilizes this interaction by contacting the guanine base of the opposite G domain in trans. The nucleotide base, in turn, is bound in cis by the highly conserved Asp-250 in the G4 loop that is crucial for nucleotide-binding specificity. Accordingly, the D250N mutation switched the nucleotide binding preference from guanosine to xanthosine triphosphates, as described previously for Ras-like GTPases and the signal recognition particle (46, 54, 55). MxAK83A was previously annotated as a nucleotide-binding deficient mutant (37). Surprisingly, we found that it can still bind to nucleotides, albeit with reduced affinity; instead, it is deficient for GTP hydrolysis and essentially behaves as MxAT103A (38) in all assays that we tested. These two mutants were previously used to assess the effects of GTP binding and hydrolysis for the antiviral effect of MxA against FLUAV and vesicular stomatitis virus (37, 38) and recently of the MxA paralog MxB against HIV-1 (7, 8). In light of our biochemical analysis, some of the interpretations previously drawn from analysis of the K83A mutant may have to be revisited or experiments repeated using, for example, the nucleotide-binding deficient D250N mutant.
In agreement with the data from Rennie et al. (20), we found that monomeric MxAM527D (18) dimerized in solution via the G domain in the presence of the transition state analogue GDP-AlF4−. However, MxAM527D did not dimerize in the presence of GTPγS, GDP, and AlF4− alone or GMP-PCP (data not shown), indicating that formation of the G interface is a low affinity, transient process. Accordingly, MxA variants with defective guanine binding in cis or in trans, like MxAD250N and MxAD253N, lost GDP-AlF4−-induced dimerization via the G interface, as recently described for the V185Y mutation at the center of the G interface (20). We speculate that in native assemblies of WT MxA that oligomerize via the stalk region (18, 19), G interfaces are stabilized by the simultaneous formation of multiple interactions within the large oligomers.
Mixed GTPase assays demonstrated that the G domain mutants, despite being defective in their own hydrolysis, could still effectively stimulate the GTPase activity of an opposing WT G domain. The T103A mutant that is deficient in GTP hydrolysis stimulated the GTPase of an opposing WT G domain to a similar extent as WT MxA. In contrast, loss of GTP binding as exemplified by the D250N mutant greatly reduced GTPase stimulation. These data demonstrate a crucial role of GTP binding for the induction of a G domain dimerization-competent conformation in MxA. Our analysis of the MxAT103A mutant further indicates that the GTPase activity is not coupled between two interacting G domains and that each G domain can independently hydrolyze GTP upon dimerization.
All G domain mutants in the context of the oligomerization-competent WT MxA protein lost their antiviral activity against THOV and FLUAV. In addition, the formation of MxA assemblies with the viral N protein in LACV-infected cells, a measure of an MxA-mediated block of viral replication (14), was not observed for our G domain mutants. Interestingly, all G domain mutants acted in a dominant-negative fashion on the antiviral effect of WT MxA. This supports a model wherein the incorporation of a few GTP-binding or GTPase-deficient mutants into WT MxA oligomers impedes MxA activity that requires formation of MxA oligomers around viral nucleocapsids, coordinated GTP hydrolysis, and structural transformations in the MxA oligomers. Surprisingly, the binding of the viral NP in THOV-infected cells was not affected by G domain mutations in pulldown experiments, indicating that the recognition of the viral target by MxA is independent of the GTPase cycle.
It has been observed in previous studies that nucleotide binding promotes oligomerization of MxA (11, 19). This can be explained by the additional nucleotide-driven association between G domains stabilizing the MxA oligomer. Accordingly, WT MxA that may cycle between GTP- and GDP-bound states showed a typical dispersed cytoplasmic localization with a few small assemblies that co-localized with markers of the smooth ER (13, 52). When analyzing the intracellular localization of the G domain mutants, striking differences to the distribution of WT MxA became apparent. The nucleotide-binding competent but hydrolysis-defective K83A, T103A, and D253A mutants formed few large intracellular aggregates that co-localized with the smooth ER marker syntaxin 17 (56). However, the nucleotide-binding deficient D250N mutant was uniformly dispersed throughout the cytoplasm of the transfected cells. To exclude the possibility that the formation of the large MxA granules is a consequence of the structural disturbance of the mutant proteins rather than a function of the nucleotide binding status of MxA, we combined the G domain mutants that prevent hydrolysis with the D250N mutant that prevents GTP binding. Intriguingly, the double mutants showed the same diffuse distribution as observed previously for the nucleotide-binding deficient mutant, indicating that constant GTP binding stabilizes membrane-associated MxA aggregates. GTP hydrolysis and possibly the release of the nucleotides is thus a prerequisite for dissociating these assemblies. As suggested previously (18, 27), dimeric or tetrameric forms of MxA might be released from these membrane-associated stores to initiate the antiviral action.
The human MX1 gene is highly conserved and only a few single nucleotide polymorphisms are described in the human population (57). However, a recent study analyzing variations in the MxA genes of 267 healthy individuals identified two rare nucleotide changes that result in amino acid exchanges at positions 255 and 268 (50). Based on the structural models of the G domain dimer, these exchanges localize to the G interface and might thus influence its formation or function. Interestingly, the V268M substitution showed diminished GTP hydrolysis but only a slightly reduced antiviral activity. This MxA variation was found homozygotic in a single individual (50). However, the G255E exchange completely abolished GTPase and antiviral activity of MxA in our analysis. The close proximity of Gly-255 to Asp-250 and Asp-253 raises the possibility that the G255E exchange also interferes with the formation of the G interface. The rare occurrence of polymorphic changes in the G domain of MxA, like the G255E exchange that was found only heterozygotic in two individuals (50), underlines the importance of this structural element for the antiviral function of MxA and suggests a central role of MxA for the antiviral host defense in humans. It would be interesting to study whether such polymorphisms are enriched in patients suffering from severe influenza as has been described recently for IFITM3, another IFN-induced antiviral restriction factor against influenza A virus (58).
In summary, our data suggest a dual role for the GTPase activity in MxA function. Upon induction by interferons, newly synthesized MxA is initially deposited close to ER membranes. Following viral infection, GTP hydrolysis is required for the dynamic redistribution of MxA from these depots to the viral ribonucleoproteins or nucleocapsids, the target of MxA action. In the absence of GTPase activity, this release is prevented and MxA, most likely in its GTP-bound state, stays inactive in the membrane-associated deposits. The regular oligomerization of MxA molecules into ring-like structures around the viral nucleocapsids allows multiple contacts with viral NPs, the main component of the nucleocapsids. GTP binding may not be necessary for the initial recognition of viral targets, but it might stabilize the MxA-NP complex. How nucleotide hydrolysis is additionally required for inactivation of the viral target structure or for the recycling of nucleocapsid-bound MxA is a matter of future investigations.
Acknowledgments
We thank Sabine Werner for technical assistance and Otto Haller, Peter Stäheli, and Martin Schwemmle for valuable discussions on the manuscript.
This work was supported by Deutsche Forschungsgemeinschaft Grants Ko 1579/8-2 (to G. K.) and DA 1127/1-1 and SFB958/A12 (to O. D.) and National Natural Science Foundation of China Grant 31200553 (to S. G.).
- Mx
- myxovirus resistance
- β-ME
- β-mercaptoethanol
- ER
- endoplasmic reticulum
- FLUAV
- influenza A virus
- GMP-PCP
- guanosine-5′-[(β,γ)-methyleno]triphosphate
- G domain
- GTPase domain
- GTPγS
- guanosine 5′-O-[γ-thio]triphosphate
- ITC
- isothermal titration calorimetry
- LACV
- La Crosse virus
- N
- nucleoprotein (from bunyaviruses)
- NP
- nucleoprotein (from orthomyxoviruses)
- THOV
- Thogoto virus
- XDP
- xanthosine 5′-diphosphate
- XTPγS
- xanthosine 5′-O-[γ-thio]triphosphate
- BSE
- bundle signaling element.
REFERENCES
- 1. Verhelst J., Hulpiau P., Saelens X. (2013) Mx proteins: antiviral gatekeepers that restrain the uninvited. Microbiol. Mol. Biol. Rev. 77, 551–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haller O., Staeheli P., Schwemmle M., Kochs G. (2015) Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol. 23, 154–163 [DOI] [PubMed] [Google Scholar]
- 3. Haller O., Kochs G. (2011) Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J. Interferon Cytokine Res. 31, 79–87 [DOI] [PubMed] [Google Scholar]
- 4. Netherton C. L., Simpson J., Haller O., Wileman T. E., Takamatsu H. H., Monaghan P., Taylor G. (2009) Inhibition of a large double-stranded DNA virus by MxA protein. J. Virol. 83, 2310–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gordien E., Rosmorduc O., Peltekian C., Garreau F., Bréchot C., Kremsdorf D. (2001) Inhibition of hepatitis B virus replication by the interferon-inducible MxA protein. J. Virol. 75, 2684–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu Z., Pan Q., Ding S., Qian J., Xu F., Zhou J., Cen S., Guo F., Liang C. (2013) The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14, 398–410 [DOI] [PubMed] [Google Scholar]
- 7. Goujon C., Moncorgé O., Bauby H., Doyle T., Ward C. C., Schaller T., Hué S., Barclay W. S., Schulz R., Malim M. H. (2013) Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502, 559–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kane M., Yadav S. S., Bitzegeio J., Kutluay S. B., Zang T., Wilson S. J., Schoggins J. W., Rice C. M., Yamashita M., Hatziioannou T., Bieniasz P. D. (2013) MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502, 563–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Busnadiego I., Kane M., Rihn S. J., Preugschas H. F., Hughes J., Blanco-Melo D., Strouvelle V. P., Zang T. M., Willett B. J., Boutell C., Bieniasz P. D., Wilson S. J. (2014) Host and viral determinants of Mx2 antiretroviral activity. J. Virol. 88, 7738–7752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Accola M. A., Huang B., Al Masri A., McNiven M. A. (2002) The antiviral dynamin family member, MxA, tubulates lipids and localizes to the smooth endoplasmic reticulum. J. Biol. Chem. 277, 21829–21835 [DOI] [PubMed] [Google Scholar]
- 11. Kochs G., Haener M., Aebi U., Haller O. (2002) Self-assembly of human MxA GTPase into highly ordered dynamin-like oligomers. J. Biol. Chem. 277, 14172–14176 [DOI] [PubMed] [Google Scholar]
- 12. von der Malsburg A., Abutbul-Ionita I., Haller O., Kochs G., Danino D. (2011) Stalk domain of the dynamin-like MxA GTPase protein mediates membrane binding and liposome tubulation via the unstructured L4 loop. J. Biol. Chem. 286, 37858–37865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stertz S., Reichelt M., Krijnse-Locker J., Mackenzie J., Simpson J. C., Haller O., Kochs G. (2006) Interferon-induced, antiviral human MxA protein localizes to a distinct subcompartment of the smooth endoplasmic reticulum. J. Interferon Cytokine Res. 26, 650–660 [DOI] [PubMed] [Google Scholar]
- 14. Reichelt M., Stertz S., Krijnse-Locker J., Haller O., Kochs G. (2004) Missorting of LaCrosse virus nucleocapsid protein by the interferon-induced MxA GTPase involves smooth ER membranes. Traffic 5, 772–784 [DOI] [PubMed] [Google Scholar]
- 15. Turan K., Mibayashi M., Sugiyama K., Saito S., Numajiri A., Nagata K. (2004) Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 32, 643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kochs G., Haller O. (1999) Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc. Natl. Acad. Sci. U.S.A. 96, 2082–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kochs G., Janzen C., Hohenberg H., Haller O. (2002) Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proc. Natl. Acad. Sci. U.S.A. 99, 3153–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao S., von der Malsburg A., Paeschke S., Behlke J., Haller O., Kochs G., Daumke O. (2010) Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465, 502–506 [DOI] [PubMed] [Google Scholar]
- 19. Gao S., von der Malsburg A., Dick A., Faelber K., Schröder G. F., Haller O., Kochs G., Daumke O. (2011) Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 35, 514–525 [DOI] [PubMed] [Google Scholar]
- 20. Rennie M. L., McKelvie S. A., Bulloch E. M., Kingston R. L. (2014) Transient dimerization of duman MxA promotes GTP hydrolysis, resulting in a mechanical power stroke. Structure 22, 1433–1445 [DOI] [PubMed] [Google Scholar]
- 21. Mitchell P. S., Patzina C., Emerman M., Haller O., Malik H. S., Kochs G. (2012) Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe 12, 598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patzina C., Haller O., Kochs G. (2014) Structural requirements for the antiviral activity of the human MxA protein against Thogoto and influenza A virus. J. Biol. Chem. 289, 6020–6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Staeheli P., Pitossi F., Pavlovic J. (1993) Mx proteins: GTPases with antiviral activity. Trends Cell Biol. 3, 268–272 [DOI] [PubMed] [Google Scholar]
- 24. Gasper R., Meyer S., Gotthardt K., Sirajuddin M., Wittinghofer A. (2009) It takes two to tango: regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 10, 423–429 [DOI] [PubMed] [Google Scholar]
- 25. Chappie J. S., Acharya S., Leonard M., Schmid S. L., Dyda F. (2010) G domain dimerization controls dynamin's assembly-stimulated GTPase activity. Nature 465, 435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wenger J., Klinglmayr E., Fröhlich C., Eibl C., Gimeno A., Hessenberger M., Puehringer S., Daumke O., Goettig P. (2013) Functional mapping of human dynamin-1-like GTPase domain based on x-ray structure analyses. PLoS One 8, e71835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haller O., Gao S., von der Malsburg A., Daumke O., Kochs G. (2010) Dynamin-like MxA GTPase: structural insights into oligomerization and implications for antiviral activity. J. Biol. Chem. 285, 28419–28424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chappie J. S., Mears J. A., Fang S., Leonard M., Schmid S. L., Milligan R. A., Hinshaw J. E., Dyda F. (2011) A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell 147, 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 30. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC, New York [Google Scholar]
- 31. Kochs G., Weber F., Gruber S., Delvendahl A., Leitz C., Haller O. (2000) Thogoto virus matrix protein is encoded by a spliced mRNA. J. Virol. 74, 10785–10789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thompson W. H., Kalfayan B., Anslow R. O. (1965) Isolation of California encephalitis group virus from a fatal human illness. Am. J. Epidemiol. 81, 245–253 [DOI] [PubMed] [Google Scholar]
- 33. Dittmann J., Stertz S., Grimm D., Steel J., García-Sastre A., Haller O., Kochs G. (2008) Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J. Virol. 82, 3624–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lutz A., Dyall J., Olivo P. D., Pekosz A. (2005) Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J. Virol. Methods 126, 13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kochs G., Haller O. (1999) GTP-bound human MxA protein interacts with the nucleocapsids of Thogoto virus (Orthomyxoviridae). J. Biol. Chem. 274, 4370–4376 [DOI] [PubMed] [Google Scholar]
- 36. Flohr F., Schneider-Schaulies S., Haller O., Kochs G. (1999) The central interactive region of human MxA GTPase is involved in GTPase activation and interaction with viral target structures. FEBS Lett. 463, 24–28 [DOI] [PubMed] [Google Scholar]
- 37. Pitossi F., Blank A., Schröder A., Schwarz A., Hüssi P., Schwemmle M., Pavlovic J., Staeheli P. (1993) A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J. Virol. 67, 6726–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ponten A., Sick C., Weeber M., Haller O., Kochs G. (1997) Dominant-negative mutants of human MxA protein: domains in the carboxyl-terminal moiety are important for oligomerization and antiviral activity. J. Virol. 71, 2591–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Herskovits J. S., Burgess C. C., Obar R. A., Vallee R. B. (1993) Effects of mutant rat dynamin on endocytosis. J. Cell Biol. 122, 565–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Damke H., Baba T., Warnock D. E., Schmid S. L. (1994) Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127, 915–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sundborger A. C., Fang S., Heymann J. A., Ray P., Chappie J. S., Hinshaw J. E. (2014) A dynamin mutant defines a superconstricted prefission state. Cell Rep. 8, 734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marks B., Stowell M. H., Vallis Y., Mills I. G., Gibson A., Hopkins C. R., McMahon H. T. (2001) GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 410, 231–235 [DOI] [PubMed] [Google Scholar]
- 43. Song B. D., Leonard M., Schmid S. L. (2004) Dynamin GTPase domain mutants that differentially affect GTP binding, GTP hydrolysis, and clathrin-mediated endocytosis. J. Biol. Chem. 279, 40431–40436 [DOI] [PubMed] [Google Scholar]
- 44. Egea P. F., Shan S. O., Napetschnig J., Savage D. F., Walter P., Stroud R. M. (2004) Substrate twinning activates the signal recognition particle and its receptor. Nature 427, 215–221 [DOI] [PubMed] [Google Scholar]
- 45. Shan S. O., Stroud R. M., Walter P. (2004) Mechanism of association and reciprocal activation of two GTPases. PLoS Biol. 2, e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shan S. O., Walter P. (2005) Molecular crosstalk between the nucleotide specificity determinant of the SRP GTPase and the SRP receptor. Biochemistry 44, 6214–6222 [DOI] [PubMed] [Google Scholar]
- 47. Frese M., Kochs G., Meier-Dieter U., Siebler J., Haller O. (1995) Human MxA protein inhibits tick-borne Thogoto virus but not Dhori virus. J. Virol. 69, 3904–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pavlovic J., Arzet H. A., Hefti H. P., Frese M., Rost D., Ernst B., Kolb E., Staeheli P., Haller O. (1995) Enhanced virus resistance of transgenic mice expressing the human MxA protein. J. Virol. 69, 4506–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maines T. R., Lu X. H., Erb S. M., Edwards L., Guarner J., Greer P. W., Nguyen D. C., Szretter K. J., Chen L. M., Thawatsupha P., Chittaganpitch M., Waicharoen S., Nguyen D. T., Nguyen T., Nguyen H. H., et al. (2005) Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J. Virol. 79, 11788–11800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Duc T. T., Farnir F., Michaux C., Desmecht D., Cornet A. (2012) Detection of new biallelic polymorphisms in the human MxA gene. Mol. Biol. Rep. 39, 8533–8538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ghosh A., Praefcke G. J., Renault L., Wittinghofer A., Herrmann C. (2006) How guanylate-binding proteins achieve assembly-stimulated processive cleavage of GTP to GMP. Nature 440, 101–104 [DOI] [PubMed] [Google Scholar]
- 52. Melén K., Ronni T., Broni B., Krug R. M., von Bonsdorff C. H., Julkunen I. (1992) Interferon-induced Mx proteins form oligomers and contain a putative leucine zipper. J. Biol. Chem. 267, 25898–25907 [PubMed] [Google Scholar]
- 53. Di Paolo C., Hefti H. P., Meli M., Landis H., Pavlovic J. (1999) Intramolecular backfolding of the carboxyl-terminal end of MxA protein is a prerequisite for its oligomerization. J. Biol. Chem. 274, 32071–32078 [DOI] [PubMed] [Google Scholar]
- 54. Schmidt G., Lenzen C., Simon I., Deuter R., Cool R. H., Goody R. S., Wittinghofer A. (1996) Biochemical and biological consequences of changing the specificity of p21ras from guanosine to xanthosine nucleotides. Oncogene 12, 87–96 [PubMed] [Google Scholar]
- 55. Cool R. H., Schmidt G., Lenzen C. U., Prinz H., Vogt D., Wittinghofer A. (1999) The Ras mutant D119N is both dominant negative and activated. Mol. Cell. Biol. 19, 6297–6305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steegmaier M., Oorschot V., Klumperman J., Scheller R. H. (2000) Syntaxin 17 is abundant in steroidogenic cells and implicated in smooth endoplasmic reticulum membrane dynamics. Mol. Biol. Cell 11, 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tazi-Ahnini R., di Giovine F. S., McDonagh A. J., Messenger A. G., Amadou C., Cox A., Duff G. W., Cork M. J. (2000) Structure and polymorphism of the human gene for the interferon-induced p78 protein (MX1): evidence of association with alopecia areata in the Down syndrome region. Hum. Genet. 106, 639–645 [DOI] [PubMed] [Google Scholar]
- 58. Everitt A. R., Clare S., Pertel T., John S. P., Wash R. S., Smith S. E., Chin C. R., Feeley E. M., Sims J. S., Adams D. J., Wise H. M., Kane L., Goulding D., Digard P., Anttila V., et al. (2012) IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484, 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]