

Background: Serum amyloid A is expressed in the liver in acute-phase inflammation.
Results: Saa1 regulates the expression of chemokines and activation of T cells in T cell-mediated hepatitis.
Conclusion: Overexpression of Saa1 aggravates hepatitis via the induction of chemokine expression by the TLR2 signaling pathway.
Significance: Saa1 might be a novel inflammatory factor as a chemokine modulator in hepatitis.
Keywords: chemokine, cytokine, inflammation, liver injury, transgenic mice, Saa1, T cell activation, Toll like receptor, concanavalin A
Abstract
Serum amyloid A is a proinflammatory molecule that induces leukocyte infiltration and promotes neutrophil adhesion to endothelial cells under inflammatory conditions. The aim of this study was to examine whether Saa1 aggravates T cell-mediated hepatitis by inducing chemokines in a liver-specific, Saa1-overexpressing, transgenic (TG) mouse model. We generated TG mice in which Saa1 was overexpressed specifically in liver tissue. The chemokines monocyte chemotactic protein 1 (MCP1), MIP1α, MIP1β, interferon γ-induced protein 10 (IP-10), and eotaxin were induced in Saa1 TG mice. After concanavalin A treatment, Saa1 expression was higher in Saa1 TG mice than in WT mice. More severe liver injury, increased hepatocyte apoptosis, and higher levels of hepatic enzymes were observed in Saa1 TG mice than in WT mice. Liver infiltration of CD4+ T cells and macrophages increased after inducing hepatitis. Activation of T cells was higher in Saa1 TG mice than in WT mice, and the populations of Th17 cells and regulatory T cells were altered by overexpressing Saa1 in TG mice. Secretion of various cytokines, such as interferon γ, tumor necrosis factor α, and interleukin 6, increased in Saa1 TG mice. Injecting a Toll-like receptor 2 (TLR2) antagonist in vivo inhibited chemokine expression and IκBα phosphorylation and showed that the induction of chemokines by Saa1 was dependent on TLR2. Hepatic Saa1 accelerated T cell-mediated hepatitis by inducing chemokine production and activating T cells by TLR2. Therefore, Saa1 might be a novel inflammatory factor that acts as a chemokine modulator in hepatitis.
Introduction
Hepatitis can be caused by various factors, including drugs, alcohol, chemicals, viruses, genetic factors, and the immune response. The disease represents a major public health problem. In most cases, including autoimmune hepatitis, viral hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis, hepatitis is caused by activated T cells that infiltrate and destroy the liver parenchyma, leading to liver injury (1).
Concanavalin A (ConA)3 is a lectin originally purified from Canavalia brasiliensis. It has specific sugar binding sites that can bind to α-d-mannoside, methyl α-d-mannopyranoside, α-d-glucose, and methyl-α-d-glucose (2). ConA-induced hepatitis is a well known mouse model of T cell-mediated hepatitis. After intravenous injection, ConA binds to mannose receptors on the surface of sinusoidal endothelial cells, leading to breakdown of the sinusoidal endothelial cell membrane, bleb formation, and disappearance of the cytoplasm (3). ConA induces systemic immune activation and acute liver damage. CD4+ T helper cells detect MHC class II and T cell receptors on Kupffer cells modified by ConA, resulting in their activation (4). These activated T cells play an important role in ConA-induced hepatitis. Differentiated T cells infiltrate the damaged liver tissue and secrete various cytokines, such as interferon IFN-γ, TNF-α, and IL-6. T cells, natural killer cells, and macrophages have the capacity to induce hepatocyte apoptosis and necrosis by interacting with one another.
Serum amyloid A (SAA) is an acute-phase response protein. Three SAA isoforms have been reported in mice. Saa1 and Saa2 isoforms are mainly expressed by hepatocytes, whereas the Saa3 isoform has been found to be induced in various tissues during inflammation (5, 6). Under acute inflammatory conditions, such as infection, tissue damage, inflammation, and cancer, Saa1 and Saa2 are similarly induced, and serum SAA levels increase as much as 1000-fold (6). Induction of the genes encoding SAA1 and SAA2 during inflammation is triggered by elevated secretion of proinflammatory cytokines, such as IL-6 and TNF-α, into the circulation and in hepatic cells (7, 8). SAA functions as a proinflammatory mediator at the site of inflammation by inducing chemotaxis in neutrophils, monocytes, and T cells, promoting leukocyte infiltration and neutrophil adhesion to endothelial cells (9, 10) as well as stimulating neutrophils and monocytes to release cytokines (11, 12), chemokines (13), and matrix metalloproteinase (14). These findings suggest a major role for SAA in the establishment and maintenance of inflammation.
Chemokines are small proteins with chemotactic properties. Almost 50 chemokines have been identified (15). These proteins provide migratory signals to immune cells (16). Chemokine expression is up-regulated in areas of tissue injury, and their increased expression leads to infiltration of immune cells such as lymphocytes, neutrophils, and monocytes. Chemokines and chemokine receptors play critical roles in liver diseases such as hepatitis (17–19). Chemokines are secreted from immune cells and primary cells, including hepatocytes, Kupffer cells, and endothelial cells (20).
We investigated the relationship between Saa1 and ConA-induced hepatitis using Saa1-overexpressing transgenic (TG) mice. Induction of chemokines by Saa1 increased hepatocyte necrosis and apoptosis, liver injury, proinflammatory cytokine levels, and T cell activation. Therefore, Saa1 might be a novel proinflammatory chemokine modulator in hepatitis.
EXPERIMENTAL PROCEDURES
Generation of Transgenic Mice
The full-length open reading frame of mouse Saa1, containing SalI and NotI sites at either end, was cloned into the pCl-neo vector under the control of the albumin promoter and enhancer. The expression cassette, which was subcloned into the vector by digestion with restriction enzymes and subsequent ligation, was used to produce transgenic mice following the methods described previously in detail by Hogan et al. (21). The offspring produced by mating these transgenic founder mice were genotyped by PCR analysis using DNA extracted from tail lysates as the template. Animals were raised and maintained under conventional conditions in a room with a 12-h light/dark cycle, a controlled temperature of 25 °C, and 50% humidity. Animals were given free access to food and water. All animal experiments were carried out in accordance with the guidelines for animal experimentation and with permission from the Animal Use and Care Committee of Kyungpook National University.
Animal Experiments
For ConA-induced hepatitis, age-matched, 7- to 10-week-old C57BL/6J mice and Saa1-overexpressing TG mice were injected intravenously with 10 mg/kg ConA (Sigma-Aldrich, St. Louis, MO). Control mice were injected with the same volume of PBS. For inhibition of TLR2 in vivo, mice were injected intraperitoneally with 3 mg/kg CU-CPT22, a TLR2 inhibitor (Calbiochem, Billerica, MA) (22).
Assessment of Hepatotoxicity
At 0, 6, and 24 h after ConA injection, blood was collected from the eye. Serum was harvested after centrifugation at 10,000 × g for 10 min at 4 °C. The concentrations of ALT and AST in serum were measured using a Hitachi 704 autoanalyzer (Hitachi, Tokyo, Japan).
Histological Analysis
Mice were sacrificed after 24 h of ConA treatment. Livers were fixed with 4% (w/v) paraformaldehyde solution for 2 days and embedded in paraffin. Tissue sections (5-μm-thick) were stained with H&E or TUNEL staining using an in situ cell apoptosis detection kit (Trevigen, Gaithersburg, MD). For semiquantitative analysis, the number of TUNEL-positive cells was counted in three randomly selected fields of view in a non-necrotic area at a magnification of ×100 and ×200. The average number of cells counted in each group was presented.
Western Blot Analysis
Twenty-four hours after ConA injection, Saa1 protein levels were measured in liver and spleen extracts. Livers and spleens were isolated from mice after sacrifice and immediately placed in PRO-PREP protein extraction solution (iNtRON, Seoul, Korea). The samples were homogenized on ice by using a homogenizer and then centrifuged at 10,000 × g for 20 min at 4 °C. Supernatants containing 50 μg of protein were resolved by 10% SDS-PAGE and then transferred to nitrocellulose membranes. The membranes were incubated with goat-anti mouse Saa1 (R&D Systems, Minneapolis, MN), IκBα, and phosphorylated IκBα (Ser-32/36) (Cell Signaling Technology, Danvers, MA), and mouse monoclonal β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies, followed by HRP-conjugated anti-goat IgG and anti-mouse IgG antibodies (Santa Cruz Biotechnology).
FACS Analysis
Single cell suspensions were prepared from the livers and spleens, from which blood was eliminated by perfusion of the heart with saline solution, and then treated with red blood cell lysis solution (0.15 m NH4Cl and 0.1 mm Na2EDTA) for 5 min at 4 °C to eliminate erythrocytes. To isolate mononuclear cells, single cell suspensions were mixed with Percoll (Sigma-Aldrich), and the mixtures were centrifuged. CD4+ T cells were positively isolated to more than 95% purity by using anti-CD4 monoclonal antibody-coupled magnetic cell sorting microbeads (Miltenyi Biotech, San Diego, CA). Cells were stained with fluoro-conjugated anti-mouse CD4, CD69, CD25, IL-17, FoxP3, and CXCR5 antibodies and then analyzed on a FACScan flow cytometer (BD Biosciences) using CellQuest software (BD Biosciences).
ELISA
At 0, 6, and 24 h after ConA injection, cytokine levels were measured in the serum, which was collected as described above for the assessment of hepatotoxicity. ELISA kits (R&D Systems) for mouse TNF-α, IFN-γ, IL-6, and IL-10 were used according to the instructions of the manufacturer.
Quantitative Real-time PCR
Quantitative real-time PCR was performed using a StepOnePlus PCR system (Applied Biosystems, Foster City, CA). Total RNA was isolated from the liver, lung, kidney, intestine, white adipose tissue, and ankle. Tissue was harvested, frozen in liquid nitrogen, and ground into a fine power. TRIzol reagent (Ambion, Austin, TX) was used according to the instructions of the manufacturer. Total RNA was reverse-transcribed at 60 °C for 1 h by using TOPscriptTM RT DryMIX (Enzynomics, Seoul, Korea). The β-actin gene was used as an internal control for quantification. mRNA levels were expressed as the -fold change in expression relative to expression in untreated control mice.
Statistical Analysis
The results were expressed as mean ± S.E. of at least three independent experiments. The significance of the differences between groups was calculated using two-tailed Student's t test. Differences with a p value of less than 0.05 were considered statistically significant.
RESULTS
Generation of Saa1-overexpressing TG Mice by Using a Liver-specific Promoter
In a previous study, a proinflammatory stimulus was found to induce Saa1 expression in the liver (6). Therefore, we created TG mice using an albumin promoter and enhancer to drive liver tissue-specific overexpression of Saa1 (Fig. 1A). We screened TG mice using genomic polymerase chain reaction with one primer pair (albumin-F and Saa1-R) (Fig. 1B). Livers from 6-week-old WT and TG mice were isolated to determine Saa1 protein expression by Western blot analysis. Saa1 expression was higher in the livers of TG mice than in any other tissue (Fig. 1C). These results demonstrate the successful generation of TG mice with liver-specific overexpression of Saa1.
FIGURE 1.

Saa1-overexpressing transgenic mice were generated using a liver-specific promoter and expressed various chemokines. A, we used an albumin enhancer and promoter to induce liver-specific expression of Saa1. B, to identify potential transgenic mice, total genomic DNA, extracted from tail biopsy specimens from 6-week-old pups, was screened with PCR using the primer pair albumin-F and Saa1-R. P, positive control (vector used for template); N, negative control (no vector). C, Saa1 protein was detected in the livers of WT and Saa1 TG mice by Western blot analysis. β-actin was used as a loading control. D, the mRNA expression of the chemokines MCP1 (CCL2), MIP1α (CCL3), MIP1β (CCL4), Rantes (CCL5), IP-10 (CXCL10), and eotaxin (CCL11) in the livers of WT and Saa1 TG mice was detected by real-time PCR. β-actin was used as a control for normalization. *, p < 0.05; **, p < 0.01 versus WT control. WT, C57BL/6 wild-type mice; TG, Saa1-overexpressing transgenic mice.
Saa1 Induces Expression of Various Chemokines in the Liver
We selected mice with the highest Saa1 expression and used them for all experiments. Saa1 has a critical role in inflammation-related cells such as macrophages, T cells, and neutrophils (9, 11, 13). However, no in vivo evidence exists to support the role of Saa1 as a chemokine inducer in the liver. Therefore, we assessed the expression of various chemokines to determine the role of Saa1 in chemokine induction. The mRNA expression of MCP-1, MIP-1α, MIP1β, IP-10, and eotaxin was higher in the livers of Saa1 TG mice than in those of WT mice (Fig. 1D). These results indicate that Saa1 overexpression induces hepatic chemokine expression.
Expression of Saa1 Is Induced in ConA-induced Hepatitis
Two studies have reported that the levels of SAA were increased in hepatitis (23, 24). To determine whether Saa1 expression was altered in ConA-induced hepatitis, we induced hepatitis by injecting ConA into WT and TG mice. After 24 h, the liver was harvested, and Saa1 expression was determined. The increase in Saa1 expression was greater in TG mice than in WT mice (Fig. 2A). These results demonstrate that ConA-induced hepatitis increases Saa1 expression in the liver of WT mice and induces a greater increase in Saa1 expression in TG mice.
FIGURE 2.

Overexpression of Saa1 promotes liver injury and apoptosis of hepatocytes in T cell-mediated hepatitis. A, Saa1 was detected in the livers of WT and Saa1 TG mice 24 h after injection with ConA. β-actin was used as a loading control. B, liver specimens were stained from WT and Saa1 TG mice collected 24 h after ConA injection for histological analysis. C, liver specimens were stained from WT and Saa1 TG mice collected 24 h after ConA injection using the TUNEL assay. **, p < 0.01 versus WT control.
Saa1 Promotes Liver Injury, Necrosis, and Apoptosis in Hepatitis
We examined the area of liver injury with H&E) staining 24 h after ConA injection. The necrotic area was larger in liver sections from ConA-treated Saa1 Tg mice than in those from ConA-treated WT mice. Infiltration of mononuclear cells was greater in the livers of Saa1 TG mice than in the livers of WT mice (Fig. 2B). We performed a TUNEL assay to detect apoptotic cells in the liver. Apoptosis of hepatocytes was increased in both necrotic and non-necrotic areas of the liver sections obtained from TG mice. The increase in the number of TUNEL-positive cells in non-necrotic areas indicated a higher rate of apoptosis in TG mice than in WT mice (Fig. 2C). These results indicate that increased Saa1 expression induced extensive liver injury and promoted apoptosis in ConA-induced hepatitis.
Saa1 Increases Expression of Aspartate Aminotransferase (AST) and Alanine Aminotransferase (ALT) as well as Secretion of Proinflammatory Cytokines during Hepatitis
ALT and AST were detected a little in the liver under normal conditions. However, both of these markers were expressed in injured liver following ConA injection. We measured serum AST and ALT to evaluate liver injury 0, 6, and 24 h after ConA injection. The levels of AST and ALT were higher in Saa1 TG mice than in WT mice (Fig. 3A). Because cytokines orchestrate hepatitis (25), their serum levels in WT and TG mice were examined by ELISA 0, 6, and 24 h after ConA injection. At 6 h after ConA injection, the increase in the secretion of proinflammatory cytokines, including IFN-γ, TNF-α, and IL-6, was greater in Saa1 TG mice than in WT mice. The anti-inflammatory cytokine IL-10 tended to be increased following ConA injection in both WT and TG mice. However, the differences were not significant (Fig. 3B). These results indicate that increased Saa1 expression induces liver enzyme expression and contributes to the increased secretion of proinflammatory cytokines during hepatitis.
FIGURE 3.

Saa1 regulates liver enzyme expression and the secretion of proinflammatory cytokines in ConA-induced hepatitis. A, the levels of AST and ALT were measured from serum 0, 6, and 24 h after ConA injection. B, the inflammatory cytokines IFN-γ, TNF-α, IL-6, and IL-10 in the serum of the WT and Saa1 TG mice were measured by ELISA at 0, 6, and 24 h after ConA injection. ***, p < 0.001 versus WT control.
Saa1 Recruits T Cells and Macrophages during Hepatitis
Because chemokine expression was induced in Saa1 TG mice (Fig. 1D), we performed a FACS analysis to determine the role of chemokines during immune cell infiltration in the liver. Mononuclear cells were isolated from the liver after heart perfusion with saline solution. Infiltration of CD4+ T cells and F4/80+CD11b+ macrophages was greater in the livers of Saa1 TG mice than in those of WT mice (Fig. 4). These results indicate that the induction of chemokines by Saa1 affects the infiltration of immune cells, such as T cells and macrophages, into the liver.
FIGURE 4.

Overexpression of Saa1 increases the percentage of CD4+ T cells and macrophages among mononuclear cells in the liver. T cell (CD4+ and CD8+ cells, A and B), B cell (B220+ cells, C), and macrophage (F4/80+CD11b+ cells, D) populations were counted in mononuclear cells isolated from the perfused livers of WT mice and Saa1 TG mice 24 h after ConA injection. *, p < 0.05; **, p < 0.01 versus WT control.
Saa1 Modulates T Cell Activation by Balancing Th17 and Regulatory T (Treg) Cells
T cell activation was assessed by flow cytometry by using antibodies against CD25 and CD69, which are markers of activated T cells. After ConA injection, the number of CD69+ or CD25+ among CD4+ T cells in the liver was higher in Saa1 TG mice than in WT mice (Fig. 5, A and B). We performed a FACS analysis to detect IL-17 and FoxP3, markers of Th17 and Treg cells, respectively, which contribute to T cell activation. The number of IL-17+ CD4+CD25+ cells was higher in the liver and spleen of Saa1 TG mice than in those of WT mice, but the number of FoxP3+CD4+CD25+ cells was lower in Saa1 TG mice (Fig. 5, C and D). These results indicate that overexpression of Saa1 increases T cell activation by regulating the differentiation of Th17 and Treg cells, consistent with the promotion of inflammation in the liver.
FIGURE 5.
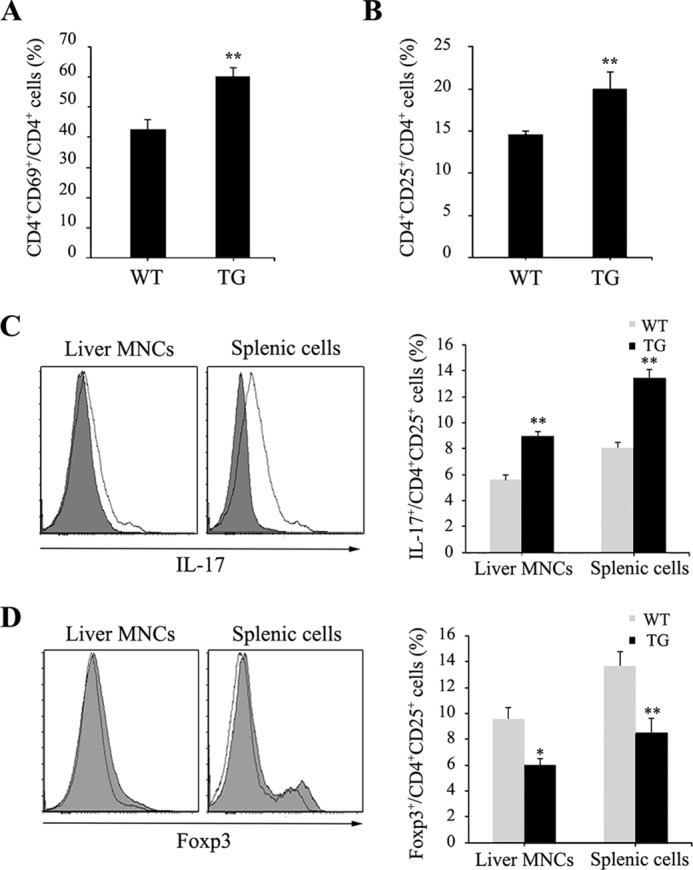
Saa1 modulates T cell activation with regulation of Th17 and Treg cells. A and B, the expression of CD69 and CD25 was assessed by FACS analysis from the livers and spleens of WT and Saa1 TG mice 24 h after ConA injection. C and D, the expression of IL-17 and FoxP3 was assessed by FACS analysis from the livers and spleens of WT and Saa1 TG mice 24 h after ConA injection. MNC, mononuclear cell. *, p < 0.05; **, p < 0.01 versus WT control.
Induction of Chemokines by Saa1 Occurs in a TLR2-dependent Manner
An interaction between TLR2 and Saa1 has been reported (26). We used a TLR2 antagonist in Saa1 TG mice to determine whether the induction of chemokines by Saa1 in hepatocytes involved TLR2. 16 h after injection of the TLR2 antagonist, MIP1α and eotaxin mRNA levels were decreased in TLR2 antagonist-treated Saa1 TG mice (Fig. 6A). The NFκB pathway, which is downstream of TLR2 signaling, is activated by the phosphorylation and subsequent degradation of IκBα. 2 h after the injection of a TLR2 antagonist, the phosphorylation of IκBα was decreased in TLR2 antagonist-treated Saa1 TG mice (Fig. 6B). These results demonstrate that the induction of chemokines by Saa1 is mediated by the TLR2-dependent NFκB pathway.
FIGURE 6.
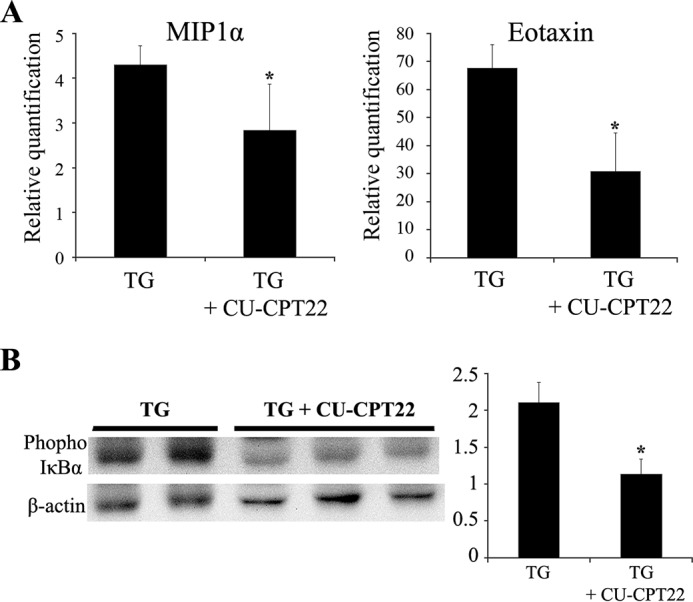
Induction of chemokines by Saa1 is regulated by TLR2-dependent NF-κB signaling. A, the expression of MIP1α (CCL3) and eotaxin (CCL11) mRNA was detected in the perfused liver of Saa1 TG mice treated with or without 3 mg/kg CU-CPT22 (a TLR2 antagonist) for 16 h. B, phosphorylation of IκBα in the perfused liver of Saa1 TG mice treated with or without 3 mg/kg CU-CPT22 for 2 h was detected by Western blot analysis. *, p < 0.05 versus TG control.
DISCUSSION
The ConA injection method to induce T cell-mediated hepatitis is a well known mouse disease model. However, little is known about the mechanism by which Saa1 contributes to the progression of T cell-mediated hepatitis. Our results demonstrate that, following ConA injection, Saa1 TG mice had larger areas of liver injury and higher levels of ALT and AST, which are indicators of liver injury, compared with the same measurements made in WT mice. In addition, Saa1 overexpression increased the infiltration of T cells and macrophages, promoted T cell activation, modulated the balance between Th17 and Treg cells, and induced the secretion of proinflammatory cytokines such as TNF-α, IFN-γ, and IL-6. Therefore, we suggest that Saa1 has a possible role in aggravating T cell-mediated hepatitis.
A recent study found that virus-infected patients had increased serum SAA levels (23). In the case study of a woman with alcoholic liver cirrhosis, an existing liver nodule was diagnosed as SAA-positive by immunohistochemistry (27). These studies suggest that hepatitis might lead to increased SAA expression and that SAA can exacerbate hepatitis. However, other studies have reported that SAA has antiviral activity against hepatitis C virus infection in cultured cells, which it exerts by binding hepatitis C virions and specifically blocking hepatitis C virus entry (28, 29). Therefore, further study is needed to determine the correlation between SAA1 expression and various forms of hepatitis, such as viral hepatitis, autoimmune hepatitis, and primary biliary cirrhosis, and to establish the role of Saa1 in hepatitis.
Chemokine expression in injured areas of the liver promotes immune cell infiltration (17–19). Our results also show that Saa1 overexpression induced MCP-1, MIP-1α, MIP-1β, eotaxin, and IP-10 mRNA expression in the livers of TG mice. MIP-1α acts as a proinflammatory factor in T cell-mediated hepatitis by recruiting CD4+ T cells (19), and IP-10 is expressed by hepatocytes during hepatitis (30). Chemokines bind CC and CXC chemokine receptors to induce signal transduction. The function of CCR5, a MIP-1α and MIP-1β receptor, has been reported in hepatitis (18, 31). The function of CXCR3, an IP-10 receptor, has also been reported in hepatitis (32–34). Further studies are needed to examine the relationship between chemokine receptors and chemokine induction by SAA1.
Here we showed that overexpression of Saa1 exacerbated liver injury by increasing T cell activation. The numbers of CD4+CD25+ and CD4+CD69+ T cells, which express markers of T cell activation, were increased in Saa1 TG mice compared with those observed in WT mice. The number of IL-17+ cells was increased in the population of activated T cells. However, the number of FoxP3+ cells was decreased. The balance between Th17 and Treg cells mediates T cell activation (35, 36), and this mechanism plays an important role in modulating T cell activation in hepatitis. The shift in the Th17/Treg balance could contribute to the T cell activation observed in Saa1 TG mice. Regulation of T cell activation by chemokines via CC chemokine receptors on the T cell surface could also contribute to increased T cell activation (37). Following ConA injection, Saa1 was found to be expressed not only in the liver but also in the spleen. Therefore, Saa1 could have a role in regulating inflammation in both of these tissues. Hence, we suggest that the overexpression of Saa1 might regulate T cell activation by altering chemokine expression or the balance between Th17 and Treg cells.
TLRs play a major role in the innate immune system. They are expressed on immune and non-immune cells. TLR2 has a critical role in mouse hepatitis (38). In this study, we showed that TLR2-dependent chemokine secretion is regulated by Saa1 and that the role of Saa1 in T cell-mediated hepatitis is TLR2-dependent. Further study of the interaction between SAA1 and TLR2 is needed to clarify the role of cross-talk between immune and non-immune cells.
Our in vivo data show that treatment with a TLR2 antagonist decreased the induction of chemokines but did not return chemokine expression to the basal level. Previous studies have shown that Saa1 binds to TLR4, CD36, and FPRL1 (39–41). Therefore, the effects of Saa1 overexpression in mice might be mediated by the binding of Saa1 not only to TLR2 but also to other receptors, such as TLR4, CD36, and FPRL1.
In this study, Saa1 was expressed in the livers and spleens of WT mice with ConA-induced hepatitis. The substantial induction of Saa1 during hepatitis was similar to that described in a previous study (5). We generated Saa1-overexpressing TG mice in which Saa1 was expressed specifically in the liver, as shown in Fig. 1. Our Saa1 TG mouse model may be useful for studying the role of Saa1 in many diseases.
For the first time, we determined that Saa1 plays a critical role in ConA-induced hepatitis by inducing chemokines and regulating T cell activation in a TLR2-dependent manner. TG mice with liver-specific overexpression of Saa1 may be a useful animal model to study metabolic diseases and other inflammatory diseases. In the future, Saa1 might be a therapeutic target for hepatitis.
This research was supported by Next Generation BioGreen21 Program Grant PJ 009573 and National Research Foundation of Korea grant 2008-0062618 funded by the Korean government (MSIP).
- ConA
- concanavalin A
- SAA
- serum amyloid A
- TG
- transgenic
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- Treg cell
- regulatory T cell
- TLR
- Toll-like receptor.
REFERENCES
- 1. Eggink H. F., Houthoff H. J., Huitema S., Gips C. H., Poppema S. (1982) Cellular and humoral immune reactions in chronic active liver disease: I: lymphocyte subsets in liver biopsies of patients with untreated idiopathic autoimmune hepatitis, chronic active hepatitis B and primary biliary cirrhosis. Clin. Exp. Immunol. 50, 17–24 [PMC free article] [PubMed] [Google Scholar]
- 2. Kanellopoulos P. N., Pavlou K., Perrakis A., Agianian B., Vorgias C. E., Mavrommatis C., Soufi M., Tucker P. A., Hamodrakas S. J. (1996) The crystal structure of the complexes of concanavalin A with 4′-nitrophenyl-α-d-mannopyranoside and 4′-nitrophenyl-α-d-glucopyranoside. J. Struct. Biol. 116, 345–355 [DOI] [PubMed] [Google Scholar]
- 3. Knolle P. A., Gerken G., Loser E., Dienes H. P., Gantner F., Tiegs G., Meyer zum Buschenfelde K. H., Lohse A. W. (1996) Role of sinusoidal endothelial cells of the liver in concanavalin A-induced hepatic injury in mice. Hepatology 24, 824–829 [DOI] [PubMed] [Google Scholar]
- 4. Tsui T. Y., Obed A., Siu Y. T., Yet S. F., Prantl L., Schlitt H. J., Fan S. T. (2007) Carbon monoxide inhalation rescues mice from fulminant hepatitis through improving hepatic energy metabolism. Shock 27, 165–171 [DOI] [PubMed] [Google Scholar]
- 5. Ray A., Schatten H., Ray B. K. (1999) Activation of Sp1 and its functional co-operation with serum amyloid A-activating sequence binding factor in synoviocyte cells trigger synergistic action of interleukin-1 and interleukin-6 in serum amyloid A gene expression. J. Biol. Chem. 274, 4300–4308 [DOI] [PubMed] [Google Scholar]
- 6. Meek R. L., Benditt E. P. (1986) Amyloid A gene family expression in different mouse tissues. J. Exp. Med. 164, 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ito A., Takii T., Matsumura T., Onozaki K. (1999) Augmentation of type I IL-1 receptor expression and IL-1 signaling by IL-6 and glucocorticoid in murine hepatocytes. J. Immunol. 162, 4260–4265 [PubMed] [Google Scholar]
- 8. Ganapathi M. K., Rzewnicki D., Samols D., Jiang S. L., Kushner I. (1991) Effect of combinations of cytokines and hormones on synthesis of serum amyloid A and C-reactive protein in Hep 3B cells. J. Immunol. 147, 1261–1265 [PubMed] [Google Scholar]
- 9. Xu L., Badolato R., Murphy W. J., Longo D. L., Anver M., Hale S., Oppenheim J. J., Wang J. M. (1995) A novel biologic function of serum amyloid A: induction of T lymphocyte migration and adhesion. J. Immunol. 155, 1184–1190 [PubMed] [Google Scholar]
- 10. Badolato R., Wang J. M., Murphy W. J., Lloyd A. R., Michiel D. F., Bausserman L. L., Kelvin D. J., Oppenheim J. J. (1994) Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J. Exp. Med. 180, 203–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He R., Sang H., Ye R. D. (2003) Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 101, 1572–1581 [DOI] [PubMed] [Google Scholar]
- 12. Furlaneto C. J., Campa A. (2000) A novel function of serum amyloid A: a potent stimulus for the release of tumor necrosis factor-α, interleukin-1β, and interleukin-8 by human blood neutrophil. Biochem. Biophys. Res. Commun. 268, 405–408 [DOI] [PubMed] [Google Scholar]
- 13. Leow K. Y., Goh W. W., Heng C. K. (2012) Effect of serum amyloid A1 treatment on global gene expression in THP-1-derived macrophages. Inflamm. Res. 61, 391–398 [DOI] [PubMed] [Google Scholar]
- 14. Lee H. Y., Kim M. K., Park K. S., Bae Y. H., Yun J., Park J. I., Kwak J. Y., Bae Y. S. (2005) Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem. Biophys. Res. Commun. 330, 989–998 [DOI] [PubMed] [Google Scholar]
- 15. Bromley S. K., Mempel T. R., Luster A. D. (2008) Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat. Immunol. 9, 970–980 [DOI] [PubMed] [Google Scholar]
- 16. Weber S. N., Wasmuth H. E. (2010) Liver fibrosis: from animal models to mapping of human risk variants. Best Pract. Res. Clin. Gastroenterol. 24, 635–646 [DOI] [PubMed] [Google Scholar]
- 17. Simpson K. J., Henderson N. C., Bone-Larson C. L., Lukacs N. W., Hogaboam C. M., Kunkel S. L. (2003) Chemokines in the pathogenesis of liver disease: so many players with poorly defined roles. Clin. Sci. 104, 47–63 [DOI] [PubMed] [Google Scholar]
- 18. Moreno C., Gustot T., Nicaise C., Quertinmont E., Nagy N., Parmentier M., Le Moine O., Devière J., Louis H. (2005) CCR5 deficiency exacerbates T-cell-mediated hepatitis in mice. Hepatology 42, 854–862 [DOI] [PubMed] [Google Scholar]
- 19. Ajuebor M. N., Hogaboam C. M., Le T., Proudfoot A. E., Swain M. G. (2004) CCL3/MIP-1α is pro-inflammatory in murine T cell-mediated hepatitis by recruiting CCR1-expressing CD4+ T cells to the liver. Eur. J. Immunol. 34, 2907–2918 [DOI] [PubMed] [Google Scholar]
- 20. Saiman Y., Friedman S. L. (2012) The role of chemokines in acute liver injury. Front. Physiol. 3, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hogan B., Beddington R., Constantin F., Lacy E. (1994) Manipulating the mouse embryo. [Google Scholar]
- 22. Cheng K., Wang X., Zhang S., Yin H. (2012) Discovery of small-molecule inhibitors of the TLR1/TLR2 complex. Angew. Chem. 51, 12246–12249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lannergård A., Larsson A., Kragsbjerg P., Friman G. (2003) Correlations between serum amyloid A protein and C-reactive protein in infectious diseases. Scand. J. Clin. Lab. Invest. 63, 267–272 [PubMed] [Google Scholar]
- 24. Saha A., Theis J. D., Vrana J. A., Dubey N. K., Batra V. V., Sethi S. (2011) AA amyloidosis associated with hepatitis B. Nephrol. Dial. Transplant. 26, 2407–2412 [DOI] [PubMed] [Google Scholar]
- 25. Tilg H., Kaser A., Moschen A. R. (2006) How to modulate inflammatory cytokines in liver diseases. Liver Int. 26, 1029–1039 [DOI] [PubMed] [Google Scholar]
- 26. Cheng N., He R., Tian J., Ye P. P., Ye R. D. (2008) Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol. 181, 22–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim S. R., Kondo F., Otono Y., Imoto S., Ando K., Hirakawa M., Fukuda K., Sasaki M., Kim S. K., Komaki T., Tsuchida S., Kobayashi S., Matsuoka T., Kudo M. (2014) Serum amyloid A and C-reactive protein positive nodule in alcoholic liver cirrhosis, hard to make definite diagnosis. Hepatol. Res. 44, 584–590 [DOI] [PubMed] [Google Scholar]
- 28. Cai Z., Cai L., Jiang J., Chang K. S., van der Westhuyzen D. R., Luo G. (2007) Human serum amyloid A protein inhibits hepatitis C virus entry into cells. J. Virol. 81, 6128–6133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lavie M., Voisset C., Vu-Dac N., Zurawski V., Duverlie G., Wychowski C., Dubuisson J. (2006) Serum amyloid A has antiviral activity against hepatitis C virus by inhibiting virus entry in a cell culture system. Hepatology 44, 1626–1634 [DOI] [PubMed] [Google Scholar]
- 30. Harvey C. E., Post J. J., Palladinetti P., Freeman A. J., Ffrench R. A., Kumar R. K., Marinos G., Lloyd A. R. (2003) Expression of the chemokine IP-10 (CXCL10) by hepatocytes in chronic hepatitis C virus infection correlates with histological severity and lobular inflammation. J. Leukocyte Biol. 74, 360–369 [DOI] [PubMed] [Google Scholar]
- 31. Ajuebor M. N., Wondimu Z., Hogaboam C. M., Le T., Proudfoot A. E., Swain M. G. (2007) CCR5 deficiency drives enhanced natural killer cell trafficking to and activation within the liver in murine T cell-mediated hepatitis. Am. J. Pathol. 170, 1975–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Erhardt A., Wegscheid C., Claass B., Carambia A., Herkel J., Mittrücker H. W., Panzer U., Tiegs G. (2011) CXCR3 deficiency exacerbates liver disease and abrogates tolerance in a mouse model of immune-mediated hepatitis. J. Immunol. 186, 5284–5293 [DOI] [PubMed] [Google Scholar]
- 33. Zeremski M., Hooker G., Shu M. A., Winkelstein E., Brown Q., Des Jarlais D. C., Tobler L. H., Rehermann B., Busch M. P., Edlin B. R., Talal A. H. (2011) Induction of CXCR3- and CCR5-associated chemokines during acute hepatitis C virus infection. J. Hepatol. 55, 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeremski M., Petrovic L. M., Chiriboga L., Brown Q. B., Yee H. T., Kinkhabwala M., Jacobson I. M., Dimova R., Markatou M., Talal A. H. (2008) Intrahepatic levels of CXCR3-associated chemokines correlate with liver inflammation and fibrosis in chronic hepatitis C. Hepatology 48, 1440–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hanidziar D., Koulmanda M. (2010) Inflammation and the balance of Treg and Th17 cells in transplant rejection and tolerance. Curr. Opin. Organ Transplant. 15, 411–415 [DOI] [PubMed] [Google Scholar]
- 36. Su Z. J., Yu X. P., Guo R. Y., Ming D. S., Huang L. Y., Su M. L., Deng Y., Lin Z. Z. (2013) Changes in the balance between Treg and Th17 cells in patients with chronic hepatitis B. Diagn. Microbiol. Infect. Dis. 76, 437–444 [DOI] [PubMed] [Google Scholar]
- 37. Luther S. A., Cyster J. G. (2001) Chemokines as regulators of T cell differentiation. Nat. Immunol. 2, 102–107 [DOI] [PubMed] [Google Scholar]
- 38. Zhou M., Zhu X., Ye S., Zhou B. (2014) Blocking TLR2 in vivo attenuates experimental hepatitis induced by concanavalin A in mice. Int. Immunopharmacol. 21, 241–246 [DOI] [PubMed] [Google Scholar]
- 39. Baranova I. N., Vishnyakova T. G., Bocharov A. V., Kurlander R., Chen Z., Kimelman M. L., Remaley A. T., Csako G., Thomas F., Eggerman T. L., Patterson A. P. (2005) Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J. Biol. Chem. 280, 8031–8040 [DOI] [PubMed] [Google Scholar]
- 40. Sandri S., Rodriguez D., Gomes E., Monteiro H. P., Russo M., Campa A. (2008) Is serum amyloid A an endogenous TLR4 agonist? J. Leukocyte Biol. 83, 1174–1180 [DOI] [PubMed] [Google Scholar]
- 41. Shim J. W., Jo S. H., Kim S. D., Lee H. Y., Yun J., Bae Y. S. (2009) Lysophosphatidylglycerol inhibits formyl peptide receptorlike-1-stimulated chemotactic migration and IL-1β production from human phagocytes. Exp. Mol. Med. 41, 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]