

Background: Contribution of SNARE transmembrane domains (TMDs) to membrane fusion is unclear.
Results: Exchange of yeast vacuolar SNARE TMDs against unrelated TMDs yielded normal fusion activities; lipid anchors do not support fusion pore opening.
Conclusion: SNARE TMDs serve as nonspecific membrane anchors in vacuole fusion but they need to be proteinaceous.
Significance: SNARE TMDs participate in fusion pore opening.
Keywords: membrane fusion, membrane trafficking, SNARE proteins, transmembrane domain, yeast, vacuoles
Abstract
Membrane fusion is induced by SNARE complexes that are anchored in both fusion partners. SNAREs zipper up from the N to C terminus bringing the two membranes into close apposition. Their transmembrane domains (TMDs) might be mere anchoring devices, deforming bilayers by mechanical force. Structural studies suggested that TMDs might also perturb lipid structure by undergoing conformational transitions or by zipping up into the bilayer. Here, we tested this latter hypothesis, which predicts that the activity of SNAREs should depend on the primary sequence of their TMDs. We replaced the TMDs of all vacuolar SNAREs (Nyv1, Vam3, and Vti1) by a lipid anchor, by a TMD from a protein unrelated to the membrane fusion machinery, or by artificial leucine-valine sequences. Individual exchange of the native SNARE TMDs against an unrelated transmembrane anchor or an artificial leucine-valine sequence yielded normal fusion activities. Fusion activity was also preserved upon pairwise exchange of the TMDs against unrelated peptides, which eliminates the possibility for specific TMD-TMD interactions. Thus, a specific primary sequence or zippering beyond the SNARE domains is not a prerequisite for fusion. Lipid-anchored Vti1 was fully active, and lipid-anchored Nyv1 permitted the reaction to proceed up to hemifusion, and lipid-anchored Vam3 interfered already before hemifusion. The unequal contribution of proteinaceous TMDs on Vam3 and Nyv1 suggests that Q- and R-SNAREs might make different contributions to the hemifusion intermediate and the opening of the fusion pore. Furthermore, our data support the view that SNARE TMDs serve as nonspecific membrane anchors in vacuole fusion.
Introduction
Membrane fusion reactions at different stations of the exo- and endocytic trafficking pathways share conserved mechanisms and catalysts, such as SNAREs (soluble N-ethylmaleimide-sensitive factor (NSF)2 attachment protein receptors), Rab-GTPases, and their effectors and regulators. Homotypic vacuole fusion allows the study of SNARE-dependent membrane fusion with the intact isolated organelle (1, 2). Fusion activity of isolated vacuoles can be measured by lipid or content mixing. Measurement of content mixing requires vacuoles isolated from two yeast strains that express a pro-enzyme (pro-ALP, strain BJ3505) or the respective maturase (proteinase A, strain DKY6281). Upon fusion, the maturase cleaves and activates pro-ALP, the activity of which is then measured (3). Lipid mixing is assayed via the dequenching of the fluorescence of rhodamine-phosphatidylethanolamine (4), which is incorporated into one fusion partner at self-quenching concentrations. Dilution of the probe by lipid mixing between the fusion partners leads to fluorescence dequenching. A combination of these assays permits us to infer hemifusion intermediates, which can be defined as a state allowing mixing of outer leaflet lipid but not of content (4). Like other membrane fusion reactions, vacuolar fusion is mediated by a number of conserved factors, such as SNARE proteins, the Rab GTPase Ypt7, and the HOPS tethering factors (5–7). SNAREs are classified as Q- or R-SNAREs based on the glutamine (Q) or arginine (R) residue at the center of their SNARE domain (8). Four SNAREs constitute the trans-SNARE complexes that induce homotypic vacuolar fusion, the three Q-SNAREs Vam3, Vti1, and Vam7, and the R-SNARE Nyv1 (9, 10).
SNAREs consist of variable N-terminal domains, a conserved coil-coiled domain (60–70 amino acid residues that contain α-helix-forming heptad repeats), and a C-terminal TMD. A single C-terminal TMD anchors Vam3, Vti1, and Nyv1 in the vacuolar membrane, whereas Vam7 is a soluble protein and is targeted to the membrane by an N-terminal phox homology domain. A fifth protein, Ykt6, probably has regulatory function. It normally does not enter the vacuolar trans-SNARE complex during in vitro fusion reactions (11). The re-activation of cis-SNARE complexes is mediated by the ATPase NSF (Sec18) and α-SNAP (Sec17) (1, 12). It leads to partial separation of SNAREs and to the release of Sec17 from the membrane (13, 14). Via their hydrophilic domains, SNAREs form trans-complexes that hold the membranes of the two fusion partners in close apposition, which is a precondition for subsequent fusion.
Whereas interaction between their cytoplasmic domains is known to be essential for efficient SNARE-driven membrane fusion, the role of the TMDs in the fusion process is less clear. Existing data stem from four major model systems as follows: (i) fusion of pure liposomes by TMD peptides only; (ii) fusion of proteoliposomes containing the entire SNARE proteins; (iii) assay of fusion in physiological membranes containing mutated SNAREs; or (iv) structural studies of SNARE complexes. Liposome experiments and studies in physiological membranes (15–19) suggested that proteinaceous TMDs of the SNAREs cannot be replaced by lipid anchors without affecting fusion activity. Replacement of SNARE TMDs with a phospholipid prevented fusion of the liposomes bearing purified SNAREs. However, if TMDs were substituted with bilayer-spanning polyisoprenoids (undecaprenol, C55), fusion of liposomes was efficient (18). In another study, truncation of half of the SNARE TMDs inhibited inner leaflet mixing, suggesting an essential role of the TMD in fusion pore formation (20). Finally, studies with reconstituted vacuolar SNAREs revealed that lipid-anchored Nyv1 can support full liposome fusion if additional accessory factors (HOPS, Sec18, and Sec17) are added (19). In physiological membranes, the replacement of SNARE TMDs by prenyl anchors was tested using the exocytic SNAREs Snc1 and Sso2 (15) and the vacuolar SNAREs Vam3 and Nyv1 (16, 17). The lipid-anchored versions of these SNAREs were not fully functional, which is in agreement with most of the liposome experiments. In contrast to this, a recent study performed in parallel to our analysis tested the effect of lipid-anchored syntaxin-1 and synaptobrevin2/VAMP2 in cultured neurons, and it found no significant effects on spontaneous neurotransmitter release and moderate reductions of Ca2+-triggered release (21).
Whether the proteinaceous TMDs have sequence-specific functions in fusion is not clear. A radical approach to the problem was taken by studying the fusion of liposomes by synthetic TMD peptides lacking the hydrophilic SNARE domains, which have been found essential for their fusogenic activity in all other experimental setups. This approach is based on the assumption that the presence of the TMD helices destabilizes the membranes to accelerate fusion. These experiments showed a strong influence of the primary sequence of the transmembrane peptides on fusion kinetics of the liposomes (22–25). Fusogenic activity decreased with increasing stability of the α-helical solution structure, suggesting that structural plasticity of transmembrane segments may be important for SNARE function.
Dimerization of R-SNAREs through their TMDs was observed in intact cells, but it was shown not to have an important functional role (26, 27). Additionally, mutations in the transmembrane helix of the SNARE Vam3 were shown to reduce the ability of the full-length protein to induce content mixing to different extents. Based on additional liposome experiments, the authors suggested that homodimerization of the transmembrane domain of Vam3 controls the transition of a hemifusion intermediate to complete lipid mixing (28). Analysis of the crystal structure of the entire synaptic SNARE complex revealed that the coiled-coil forming helices continued to interact up into the transmembrane regions. This led to the suggestion that trans-SNARE complexes might drive fusion by continuous zippering from the SNARE domains up into the TMD regions, thereby forcing the lipids to mix (29, 30). According to this model, TMDs of SNAREs undergo specific interactions with each other.
In sum, the data regarding the role of SNARE TMDs in membrane fusion are partially contradictory, and corresponding studies in physiological membranes have not been comprehensive. We decided to use the specific advantages of the fusion of intact yeast vacuoles, which allow us to discriminate lipid and content mixing and to investigate for all vacuolar SNARE proteins whether their TMDs are required, whether helical continuity between TMD and SNARE domain is essential, and whether different SNARE TMDs must interact or zip up during fusion. Vacuolar SNAREs were systematically modified by replacing their TMDs with nonrelated sequences, inserting helix-breaking motifs, increasing the distance between SNARE domains and TMDs, or by substituting the TMD by lipid anchors. The established assays for lipid and content mixing of vacuoles and for SNARE activation and pairing were used to study the effects of SNARE TMDs on vacuole fusion.
MATERIALS AND METHODS
Reagents
G418 sulfate was purchased from Calbiochem; ClonNAT was from Werner Bioagents; DMSO was from Sigma; rhodamine-phosphatidylethanolamine was from Invitrogen; monoclonal mouse anti-HA was from Covance; monoclonal mouse antibody to alkaline phosphatase (ALP, Pho8) was from Molecular Probes; and 5-fluoro-orotic acid was from BioVectra. Polyclonal antibodies to Vam3, Nyv1, Vam7, Vti1, Ypt7, and proteinase A (Pep4) were produced by injecting the soluble cytosolic domains subcutaneously into rabbits (31). Antibodies were affinity-purified from the serum using the antigens covalently coupled to NHS-Sepharose. Fluorescently labeled secondary antibodies were from LiCor (goat anti-rabbit IRDye 800 or goat anti-mouse IRDye 800). Gdi1, Vam7, and Sec18 were recombinantly expressed and purified from Escherichia coli and frozen in aliquots (32, 33).
Strains and Genetic Modifications
All strains used for fusion reactions are listed in Table 1. BJ3505 and DKY6281 are standard fusion strains (3).
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Refs. |
|---|---|---|
| BJ3505 | MATa pep4::HIS3 prb1-Δ1.6R lys2-208 trp1-Δ101 ura3-52 gal2 can | 67 |
| DKY6281 | MATa pho8::TRP1 leu2-3 leu2-112 lys2-801 suc2-Δ9 trp1-Δ901 ura3-52 | 3 |
| BJ3505 vam3Δ | BJ3505; vam3::TRP | 35 |
| DKY6281 vam3Δ | DKY6281; vam3::HIS | 35 |
| BJ Vam3-WT-HA | BJ3505; vam3::TRP; pRS416-Vam3-WT-HA3 | This study |
| DKY Vam3-WT-HA | DKY6281; vam3::HIS; pRS416-Vam3-WT-HA3 | This study |
| BJ Vam3-LA | BJ3505; vam3:TRP; pCM189-Vam3-LA | This study |
| DKY Vam3-LA | DKY6281; vam3::HIS; pCM189-Vam3-LA | This study |
| BJ Vam3-ALP-HA | BJ3505; vam3::TRP; pRS416-Vam3-ALP-HA3 | This study |
| DKY Vam3-ALP-HA | DKY6281; vam3::HIS; pRS416-Vam3-ALP-HA3 | This study |
| BJ Vam3-LV-HA | BJ3505; vam3::TRP; pRS416-Vam3-LV-HA3 | This study |
| DKY Vam3-LV-HA | DKY6281; vam3::HIS; pRS416-Vam3-LV-HA3 | This study |
| BJ Vam3-LV-GP-HA | BJ3505; vam3::TRP; pRS416-Vam3-LV-GP- HA3 | This study |
| DKY Vam3-LV-GP-HA | DKY6281; vam3::HIS; pRS416-Vam3-LV-GP- HA3 | This study |
| BJ Vam3-JM-HA | BJ3505; vam3::TRP; pRS416-Vam3-JM-HA3 | This study |
| DKY Vam3-JM-HA | DKY6281; vam3::HIS; pRS416-Vam3-JM-HA3 | This study |
| BJ Vam3-PP-HA | BJ3505; vam3::TRP; pRS416-Vam3-PP-HA3 | This study |
| DKY Vam3-PP-HA | DKY6281; vam3::HIS; pRS416-Vam3-PP-HA3 | This study |
| BJ Vam3-plus3-HA | BJ3505; vam3::TRP; p4RS16-Vam3-plus3-HA3 | This study |
| DKY Vam3-plus3-HA | DKY6281; vam3::HIS; pRS416-Vam3-plus3-HA3 | This study |
| BJ Vam3-plus6-HA | BJ3505; vam3::TRP; pRS416-Vam3-plus6-HA3 | This study |
| DKY Vam3-plus6-HA | DKY6281; vam3::HIS; pRS416-Vam3-plus6-HA3 | This study |
| BJ Vti1-WT-HA | BJ3505; vti1::kanMX; pRS416-Vti1-WT-HA3 | This study |
| DKY Vti1-WT-HA | DKY6281; vti1::kanMX; pRS416-Vti1-WT-HA3 | This study |
| BJ Vti1-LA | BJ3505; vti1::kanMX; pRS414-Vti1-LA | This study |
| DKY Vti1-LA | DKY6281; vti1::kanMX; pRS413-Vti1-LA | This study |
| BJ Vti1-WT-HA | BJ3505; vti1::kanMX; pRS414-Vti1-WT- HA3 | This study |
| DKY Vti1-WT-HA | DKY6281; vti1::kanMX; pRS413-Vti1-WT-HA3 | This study |
| BJ Vti1-ALP-HA | BJ3505; vti1::kanMX; pRS414-Vti1-ALP- HA3 | This study |
| DKY Vti1-ALP-HA | DKY6281; vti1::kanMX; pRS413-Vti1-ALP-HA3 | This study |
| BJ Vti1-LV-HA | BJ3505; vti1::kanMX; pRS414-Vti1-LV- HA3 | This study |
| DKY Vti1-LV-HA | DKY6281; vti1::kanMX; pRS413-Vti1-LV- HA3 | This study |
| BJ3505 nvy1Δ | BJ3505; nvy1::TRP1 | 35 |
| DKY6281 nvy1Δ | DKY6281; nyv1::HIS3 | 35 |
| BJ Nyv1-WT | BJ3505; nvy1::TRP1; pRS406-Nyv1-WT | This study |
| DKY Nyv1-WT | DKY6281; nyv1::HIS3; pRS406-Nyv1-WT | This study |
| BJ Nyv1-LA | BJ3505; nvy1::TRP1; pCM189-Nyv1-LA | This study |
| DKY Nyv1-LA | DKY6281; nyv1::HIS3; pCM189-Nyv1-LA | This study |
| BJ Nyv1-ALP | BJ3505; nvy1::TRP1; pRS416-Nyv1-ALP | This study |
| DKY Nyv1-ALP | DKY6281; nyv1::HIS3; pRS416-Nyv1-ALP | This study |
| BJ Nyv1-LV | BJ3505; nvy1::TRP1; pRS406-Nyv1-LV | This study |
| DKY Nyv1-LV | DKY6281; nyv1::HIS3; pRS406-Nyv1-LV | This study |
| BJ Nyv1-LV-GP | BJ3505; nvy1::TRP1; pRS416-Nyv1-LV-GP | This study |
| DKY Nyv1-LV-GP | DKY6281; nyv1::HIS3; pRS416-Nyv1-LV-GP | This study |
| BJ3505 Vti1-WT-HA/Vam3-WT-HA | BJ3505; vti1::kanMX; vam3::NAT, pRS414-endo-Vti1-WT- HA3; pRS416-Vam3-WT-HA3 | This study |
| DKY6281 Vti1-WT-HA/Vam3-WT-HA | DKY6281; vti1::kanMX; vam3::NAT; pRS413-endo-Vti1-WT-HA3; pRS416-Vam3-WT-HA3 | This study |
| BJ3505 Vti1-ALP-HA/Vam3-ALP-HA | BJ3505; vti1::kanMX; vam3::NAT; pRS414-endo-Vti1-ALP-HA3; pRS416-Vam3-ALP-HA3 | This study |
| DKY6281 Vti1-ALP-HA/Vam3-ALP-HA | DKY6281; vti1::kanMX; vam3::NAT; pRS413-endo-Vti1-ALP-HA3; pRS416-Vam3-ALP-HA3 | This study |
| BJ3505 Vti1-ALP-HA/Vam3-LV-HA | BJ3505; vti1::kanMX; vam3::NAT; pRS414-endo-Vti1-ALP-HA3; pRS416-Vam3-LV-HA3 | This study |
| DKY6281 Vti1-ALP-HA/Vam3-LV-HA | DKY6281; vti1::kanMX; vam3::NAT; pRS413-endo-Vti1-ALP-HA3; pRS416-Vam3-LV-HA3 | This study |
VAM3 Mutants
All VAM3 constructs were generated using site-directed oligonucleotide mutagenesis (34) on a single-stranded template of pRS416-VAM3 containing the endogenous VAM3 promoter and a sequence encoding for a triple HA tag on the 3′ end of the VAM3 open reading frame. All mutations were verified by sequencing. Plasmids carrying VAM3 variants were transformed into BJ3505 vam3Δ and DKY6281 vam3Δ strains (35). Clones growing on uracil-deficient plates were analyzed for Vam3 expression by immunoblotting.
Lipid-anchored VAM3 Mutants
The VAM3-LA construct was generated by PCR on a template containing the VAM3 open reading frame. The PCR fragment was cut using BamHI/NotI, purified, and ligated into a pCR-XL-TOPO vector (Invitrogen). Mutations were verified by sequencing. Subsequently, TOPO plasmids were cut using BamHI/NotI, and the excised fragment was ligated into the pCM189 vector. Transformants expressing vacuolar VAM3 levels similar to the WT were selected after screening for Vam3 expression by Western blot.
NYV1 Mutants
The NYV1-LA construct was generated by PCR using the primer pair Nyv1-BamHI forward and NYV1-NotI reverse on a template containing the NYV1 open reading frame without the intron sequence. The PCR fragment was cut using BamHI/NotI, purified, and ligated into a pCR-XL-TOPO vector (Invitrogen). Mutations were verified by sequencing. Subsequently, TOPO plasmids were cut using BamHI/NotI, and the excised fragment was ligated into the pCM189 vector.
The NYV1-ALP and NYV1-LV constructs were generated on pRS406-NYV1 plasmids with the endogenous NYV1 promoter by site-directed mutagenesis. The mutated pRS406-NYV1-ALP and pRS406-NYV1-LV plasmids were digested with StuI, and 1 μg of linearized plasmid was transformed into the nyv1Δ deletion strains (35) to stably integrate the plasmid into the yeast genome. Transformants having vacuolar Nyv1 levels similar to WT were selected by Western blotting. To use plasmids encoding for nyv1 mutants in the vam3Δ nyv1Δ double knock out, we replaced the URA marker on the pRS416 plasmids by a LEU2 marker using PCR subcloning techniques, thereby creating the pRS415 series. First, the LEU2 sequence was amplified by PCR using a pRS415 template plasmid. Second, the purified PCR fragment was used as a mega-primer for a second PCR with the pRS416 plasmid as template DNA. Following PCR, methylated template DNA was digested with DpnI.
VTI1 Mutants
BJ3505 and DKY6281 yeast strains were transformed with a pRS416-VTI1-WT plasmid containing the WT open reading frame of VTI1, the VTI1 promotor, and a triple HA tag on the 3′ end. The genomic VTI1 was then deleted by PCR-mediated gene disruption using the KANMX cassette amplified from plasmid pYM14 (Euroscarf). Because of the rescue plasmid, the strains were still viable. Clones growing on −URA/+G418 plates were verified for the knock out of VTI1 by colony PCR. All constructs carrying VTI1 mutations were generated in pRS416. The URA marker was replaced on the plasmid by either HIS (generating pRS413) or by TRP (generating pRS414) using PCR subcloning techniques. First, the HIS or TRP sequence was amplified by PCR using a pRS413 or pRS414 template plasmid. Second, the purified PCR fragment was used as a mega-primer for a second PCR with the pRS416 plasmid as template DNA. Following PCR, methylated template DNA was digested with DpnI. Using the plasmid shuffling technique (36), mutagenized VTI1 plasmids were transformed into BJ3505 vti1Δ or DKY6281 vti1Δ cells containing the rescue plasmid pRS416-VTI1-WT. Cells were selected for the newly introduced plasmids and in a second selection with 5′-fluoro-orotic acid for the loss of the pRS416-VTI1-WT plasmid.
Vacuole Isolation
Overnight cultures were inoculated in baffled 2-liter Erlenmeyer flasks with 1 liter of YPD medium and were incubated overnight at 30 °C. At and A600 of 1–1.6, cells were centrifuged (1 min, 4000 × g, room temperature, JA-10 rotor), resuspended in 50 ml of 0.1 m Tris/HCl, pH 8.9, with 10 mm DTT, and incubated for 5 min at 30 °C. Cells were again centrifuged (1 min, 4000 × g, room temperature, JA-10 rotor), resuspended in 15 ml of spheroblasting buffer (50 mm potassium phosphate, pH 7.5, 0.6 m sorbitol in YPD with 0.2% glucose, and 3600 units/ml lyticase (37)), and transferred into 30-ml Corex tubes. Cells were incubated for 25 min at 30 °C, re-isolated (2 min, 1500 × g, 4 °C, JA20 rotor), and resuspended in 2.5 ml of 15% Ficoll 400 (Eurobio) in PS buffer (10 mm PIPES/KOH, pH 6.8, 0.2 m sorbitol) by gentle stirring with a glass rod. DEAE-dextran (250 μl for BJ3505 and 200 μl for DKY6281) was added from a frozen stock (0.4 g/liter) in 15% Ficoll in PS buffer. Spheroplasts were incubated for 2 min at 0 °C and 80 s at 30 °C, chilled again, transferred to an SW41 tube, and overlaid with 2.5 ml of Ficoll, 3.5 ml of 4% Ficoll, and 1.5 ml of 0% Ficoll (all in PS buffer). After centrifugation (90 min, 150,000 × g, 4 °C), the vacuoles were harvested from the 0/4% interface.
Lipid and Content Mixing Assay
Lipid mixing and content mixing were assayed as described (4). In brief, 30 μg of unlabeled BJ3505 vacuoles and 6 μg of rhodamine-phosphatidylethanolamine-labeled DKY6281 vacuoles were mixed in 190 μl of 1 mg/ml cytosol, 0.3 mm MnCl2, 75 mm KCl in PS buffer. The amount of vacuoles taken for the reaction was determined by vacuolar protein mass, using bovine serum albumin as a standard. Inhibitors were pre-warmed to 27 °C before adding them to the tubes. Fusion reactions were started by adding 9.5 μl of 20× ATP-regeneration system, yielding 0.125 mg/ml creatine kinase, 20 mm creatine phosphate, 0.5 mm ATP, 0.5 mm MgCl2. 100 μl were used to assay lipid mixing in a fluorescent plate reader at 27 °C for 32 min. 80 μl were incubated separately for 65 min prior to measuring content mixing. To this end, pre-warmed alkaline phosphatase developing buffer was added; the samples were incubated for 6 min at 27 °C, and the activity was measured via the absorption of the produced p-nitrophenol at 400 nm. Recombinant Vam7 (rVam7) was used at 1.2 μm, recombinant Sec18 at 60 nm, Gdi1 at 0.1 mg/ml, and anti-Vam3 at 20 μg/ml. Cytosol had been prepared from the K91-1A strain as described previously (4).
FM4-64 Staining
Cells were inoculated from a pre-culture in stationary phase and grown overnight to logarithmic phase (A600 between 0.2 and 0.8) in YPD. After dilution to A600 = 0.2 in 2 ml of culture, FM4-64 in (10 mm in DMSO) was added to a final concentration of 25 μm. Cells were stained for 1 h, followed by three washing steps (2 min, 3000 × g) with YPD and a subsequent chase of 1 h in YPD.
Sec17 Release
72 μg of vacuoles from cells in a BJ3505 background were suspended in 500 μl of fusion buffer (Ficoll 0%, 150 mm KCl, 0.5 mm MnCl2), gently vortexed for 3 s, and put on ice. After 10 min, samples were filled up to 1 ml with fusion buffer, and vacuoles were spun down at 4000 × g, 5 min, 4 °C. Vacuoles were taken up in 300 μl of fusion buffer, including 0.1× protease inhibitor cocktail (PIC). 2 × 20 μl of BJ3505 vacuoles were withdrawn and combined with an equal amount of DKY6281 vacuoles to control for their fusion activity. 120 μl of BJ3505 vacuoles and 6 μl of ATP regenerating system with or without ATP were kept in a water bath for 30 min at 27 °C. Then, 280 μl of fusion buffer were added, and vacuoles were spun at 25,000 × g for 5 min at 4 °C. The supernatant was transferred into new Eppendorf tubes and left on ice. Pellets were resuspended in 250 μl of PS buffer and centrifuged (5 min at 25,000 × g in fixed angle rotor or 10 min at 11,700 × g in swing-out rotor) at 4 °C. The pelleted vacuoles were resuspended in 30 μl of 1.5× sample buffer. Samples were heated for 10 min at 65 °C while shaking at 1400 rpm. The recovered supernatants were precipitated by adding 400 μl of MeOH and 200 μl of CHCl3 on ice, vortexed, and spun for 2 min at full speed at room temperature. The upper phase was discarded, and 600 μl of MeOH at room temperature was added. The mixture was vortexed for 2 min at full speed and centrifuged. The pellet was dried and resuspended in 30 μl of sample buffer. Samples were heated at 65 °C for 5 min, vortexed again at full speed for 10 min, and heated again at 65 °C for 5 min. Samples were analyzed by SDS-PAGE and Western blotting.
Statistical Analysis
Values from at least three independent experiments were averaged. Error bars represent the standard deviation of the mean.
RESULTS
Lipid Anchoring Preserves Activity on the Qb-SNARE, but Not on the Qa- and R-SNARE
It has been argued that if force transduction is important, replacement of the TMD by lipid anchors should influence fusion activity (38, 39). Only three of the four vacuolar SNAREs that enter into the trans-SNARE complex carry TMDs, whereas the Qc-SNARE Vam7 binds to the vacuolar membrane via a lipid-binding PX domain (40). To gain insight into the role of the vacuolar SNARE TMDs in fusion, we individually substituted the native TMDs of Vam3, Nyv1, and Vti1 by lipid anchors (LA) (Fig. 1) and tested the effects on vacuole fusion. This substitution was achieved by introducing the prenylation sequence CCIIM at the C-terminal end of the soluble SNARE domains, thereby replacing the native TMD. The CCIIM sequence represents a CAAX box, where C is a cysteine residue that becomes alkylated (AA) (41). The prenyl anchor spans only the outer leaflet and not the inner membrane leaflet.
FIGURE 1.
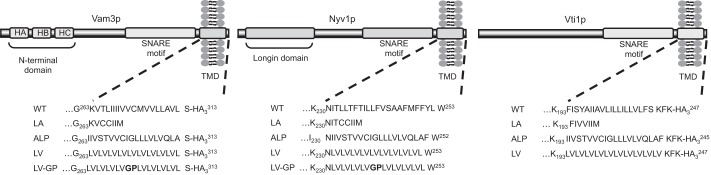
Schematic representation of Vam3, Nyv1, and Vti1 domains and primary sequences of TMD variations used in the study. All VAM3- and VTI1-expressing constructs have a triple HA tag at the C terminus (luminal side). The HA tag does not interfere with VAM3 and VTI1 expression, transport to the vacuole, or vacuole fusion activity. All constructs are under the endogenous promoter of the respective gene.
We prepared isogenic strains in which one of the vacuolar SNAREs had been deleted from the genome. CCIIM or WT alleles of the corresponding SNAREs were expressed from a centromeric plasmid. The same hybrid proteins had been introduced into both fusion partners as the sole source of the respective SNARE. We selected clones that showed expression levels of lipid-anchored SNAREs on the vacuoles that were similar to those of the respective endogenous protein in WT and used those to analyze fusion activity. Of note, the levels of other SNAREs as well as the level of the Rab-GTPase Ypt7 were unaltered, and the reporter proteins pro-ALP and proteinase A were present at normal levels (Fig. 2A). We stained the vacuoles in these strains with the fluorescent vital dye FM4-64 to analyze vacuolar structure in vivo. Vam3-LA showed a fragmented vacuolar morphology in vivo, whereas Nyv1-LA and Vti1-LA did not (Fig. 2B) (16).
FIGURE 2.
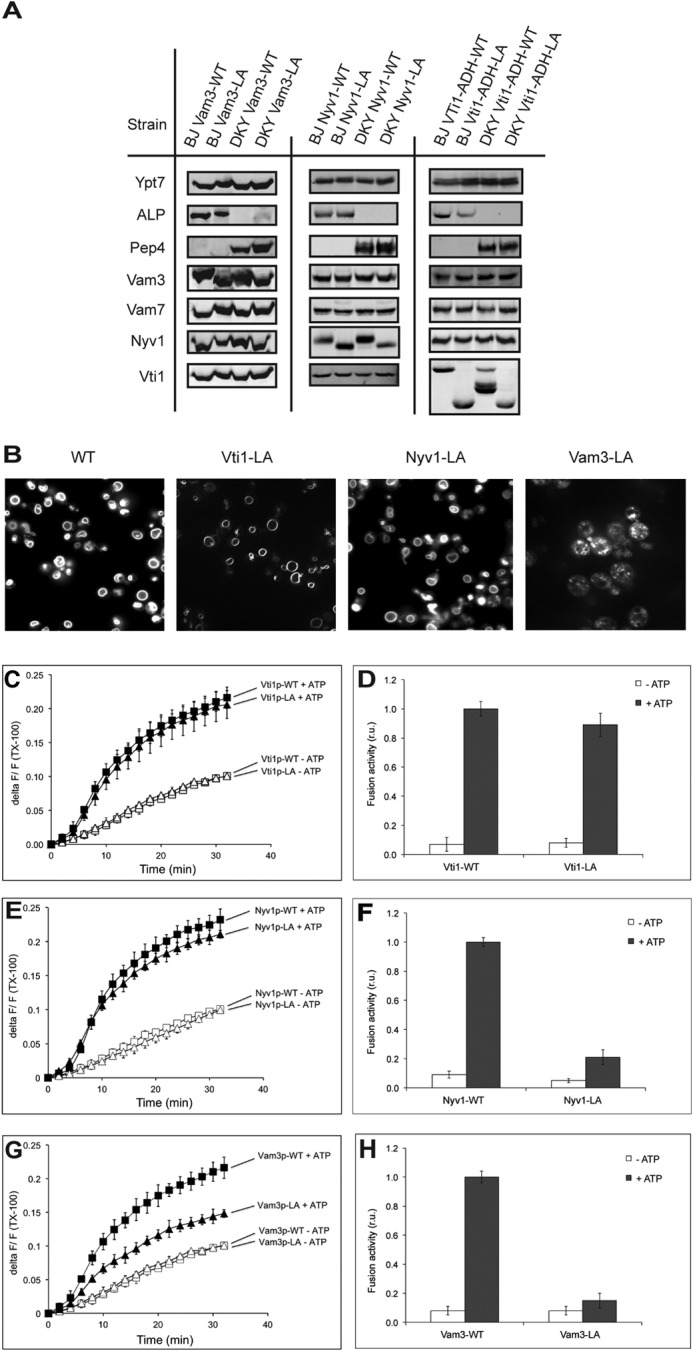
Fusion reactions with vacuoles isolated from Vam3-LA, Nyv1-LA, and Vti1-LA mutants. A, vacuoles were purified from the indicated strains and analyzed by SDS-PAGE and Western blotting for various components of the fusion machinery and for the indicator proteins pro-ALP and proteinase A. B, vacuole morphology of Vam3-LA, Nyv1-LA, and Vti1-LA mutants compared with WT. The cells were grown in YPD and stained with FM4-64, and the structure of vacuoles was analyzed by confocal fluorescence microscopy. C–H, vacuoles were isolated from the indicated strains and used for in vitro fusion reactions in the presence or absence of ATP. Lipid (C, E, and G) and content (D, F, and H) mixing were assayed in parallel. Means ± S.D. are shown for three independent experiments.
We isolated vacuoles from these strains and determined their vacuole fusion activity. In the case of Vti1-LA, neither lipid mixing nor content mixing was affected, suggesting that the proteinaceous anchor of Vti1 is dispensable for fusion (Fig. 2, C and D). In the case of Nyv1-LA and Vam3-LA, content mixing was reduced by 70–80% in comparison with the WT proteins (Fig. 2, F and H, respectively). Part of this defect in fusion of vacuoles with lipid-anchored Vam3 and Nyv1 might arise from improper SNARE activation reactions, as indicated by the impaired ATP-dependent release of Sec17 from the membranes (Fig. 3A) (16, 17). Therefore, we tested the effect of an excess of recombinant Sec18/NSF, which stimulates SNARE priming (42), and of purified Vam7, which stimulates the assembly of existing nonpaired SNAREs into trans-SNARE complexes and allows fusion independently of the priming step (10, 33). Fusion of Nyv1-LA vacuoles could be partially rescued by the addition of recombinant Vam7 and Sec18/NSF, which was not the case for vacuoles with Vam3-LA (Fig. 3B). Lipid mixing of vacuoles bearing Nyv1-LA and Vam3-LA was also affected in different ways. Nyv1-LA vacuoles showed lipid mixing comparable with WT, whereas vacuoles with Vam3-LA were strongly deficient for lipid mixing (Fig. 2, E and G). This suggests that the contributions of the TMDs of Vam3 and Nyv1 are not identical. The TMD of Nyv1 is dispensable for lipid mixing but participates in the transition from hemifusion to fusion. The TMD of Vam3, by contrast, is already necessary for the earlier steps.
FIGURE 3.
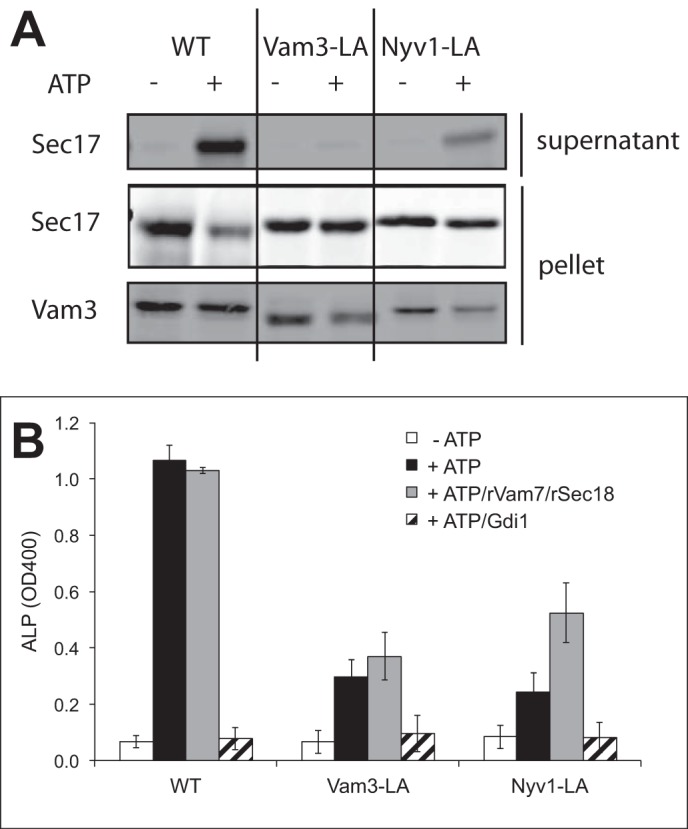
SNARE activation in Vam3-LA and Nyv1-LA mutants. A, vacuoles with lipid-anchored Vam3 and Nyv1 were incubated in fusion reactions in the presence or absence of ATP. The release of Sec17 was assayed by pelleting the vacuoles and blotting for the released protein. B, content mixing was analyzed with vacuoles bearing Vam3-LA and Nyv1-LA or the respective WT proteins. Fusion reactions had been incubated in the presence or absence of rVam7 (1.2 μm) and rSec18 (60 nm). Means ± S.D. are shown for three independent experiments.
Nonrelated and Artificial TMDs Allow SNARE-driven Fusion of Yeast Vacuoles
Structural data led to postulates that SNARE zippering, initiated in the coiled-coil domains, could continue into the TMDs (30). We tested this model by replacing the native TMDs with an artificial leucine-valine (LV) repeat motif of the same length, or with a TMD from another protein. We chose the TMD of the vacuolar alkaline phosphatase Pho8p, because Pho8p is a monomeric vacuolar protein not implicated in fusion (Fig. 1). Its TMD should hence be best adapted to the thickness and lipid environment of the vacuolar membrane, a factor that can influence the sorting of TMDs (43). The resulting hybrid SNARE proteins were expressed in the background of respective deletion mutants, making the hybrid proteins the sole source of the respective SNARE in the cells. The selected clones expressed the introduced hybrid genes, the other SNAREs, the Rab-GTPase Ypt7, as well as the reporter proteins pro-ALP and proteinase A at normal levels, i.e. at levels similar to those found on isolated vacuoles of WT cells expressing the unaltered, endogenous version of the respective SNARE (data not shown). This suggests that the mutations in TMDs of SNAREs did not induce significant protein sorting defects.
We tested the fusion activity of vacuoles in which one of the vacuolar SNAREs had been replaced by a TMD of ALP or by an artificial TMD consisting of LV repeats (Fig. 4, A–F). The results for Vam3, Nyv1, and Vti1 TMD mutants were similar and showed that neither lipid mixing nor content mixing were compromised. This suggests that the primary sequence of the SNARE TMDs is not critical for the fusion of vacuoles.
FIGURE 4.

Fusion activity of SNAREs with alternative proteinaceous membrane anchors. Vacuoles were prepared from cells carrying Vti1, Vam3, or Nyv1 anchored by the TMD of ALP or a synthetic poly-LV sequence. Lipid (A, C, and E) and content (B, D, and F) mixing were assayed in parallel in the presence or absence of ATP. Gdi1 had been added to reactions where indicated to inhibit docking and fusion. Means ± S.D. are shown for three independent experiments.
Helix-breaking Residues within TMDs Are Tolerated
We next tested whether helical continuity within TMDs of these SNAREs might influence fusion. If the TMDs continue coiled-coil formation into the bilayer, as had been suggested based on a crystal structure of an intact SNARE complex (30), fusion should be sensitive to helix-breaking residues within the TMD. Proline residues can serve this purpose because they distort the helical structure and diminish the α-helical content (44, 45). In the TMDs with the LV repeats, we replaced the fifth LV pair by a glycine-proline (GP) motif to destabilize the α-helix (Fig. 1). This motif increases the β-sheet/α-helical ratio (25). Vam3 and Nyv1 derivatives carrying these GP motifs showed normal lipid and content mixing (Fig. 5, A–D).
FIGURE 5.

Fusion of vacuoles carrying Vam3-LV-GP and Nyv1-LV-GP TMDs. Lipid (A and C) and content (B and D) mixing were assayed in parallel in the presence or absence of ATP. Gdi1 had been added to inhibit docking and fusion where indicated. Means ± S.D. are shown for three independent experiments.
Simultaneous Exchange of Two SNARE TMDs Supports Fusion Activity
In the previous approach, we had modified the TMD in only one out of the three membrane-integral vacuolar SNAREs. This leaves the possibility of specific binary interactions between the remaining two native TMDs. To eliminate this constraint, we constructed strains in which two of the three native SNARE TMDs had been replaced by an LV repeat motif or the TMD of vacuolar ALP. This leaves only one of the three vacuolar SNAREs with its native TMD, thus eliminating the possibility of specific TMD-TMD interactions. We constructed strains co-expressing Vam3-LV/Vti1-ALP and Vam3-ALP/Vti1-ALP. The mutated alleles for these SNAREs were expressed in cells deleted for the respective WT alleles, i.e. the mutated alleles were the sole source of the respective SNARE. The amounts of these SNAREs found on purified vacuoles were comparable with the WT (Fig. 6A). We also attempted to generate cells co-expressing Vam3-LV/Nyv1-LV and Vam3-ALP/Nyv1-LV. Although the proteins were expressed in whole cells at significant levels, virtually nothing was detectable in purified vacuoles, suggesting that sorting of these SNAREs was perturbed by the double TMD substitution (data not shown). By contrast, Nyv1-LV, Vam3-ALP, and Vam3-LV were sorted correctly in cells expressing only one SNARE with modified TMD (see above). This suggests that the TMDs of SNARE complexes might be relevant for sorting them to vacuoles or for stabilizing them on this organelle. Such a sorting function might be related to the role of TMDs in partitioning SNAREs into membrane domains of specific lipid composition (46). We did not pursue this aspect because it goes beyond the scope of this study.
FIGURE 6.
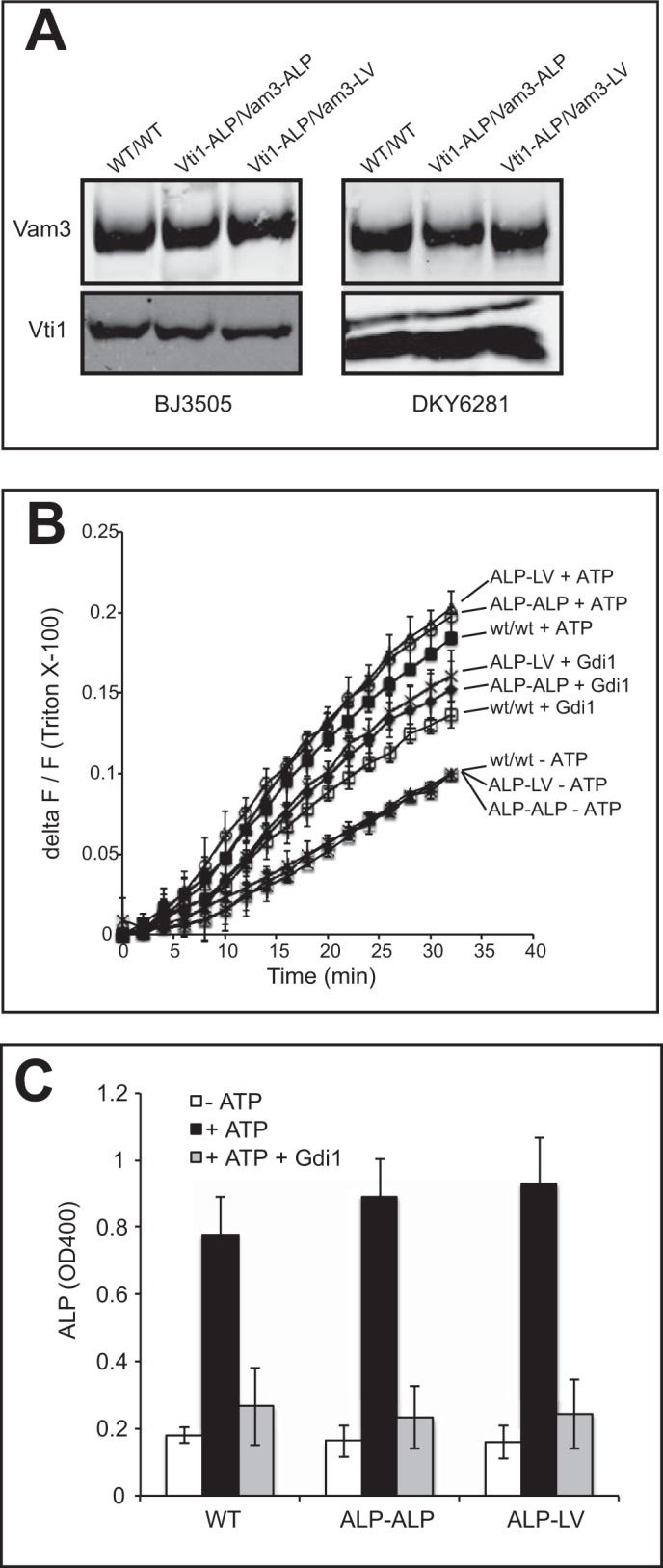
Fusion activity of vacuoles carrying two TMD substitutions. Vacuoles were isolated from cells carrying combinations of Vam3 and Vti1 with the indicated substitutions of their TMDs. A, aliquots were TCA-precipitated and analyzed by SDS-PAGE and Western blotting against Vam3 and Vti1. Mixing of lipid (B) and content (C) was assayed for these vacuoles in parallel in the presence or absence of ATP and Gdi1. Means ± S.D. are shown for three independent experiments.
The vacuoles carrying Vam3-LV/Vti1-ALP and Vam3-ALP/Vti1-ALP were subjected to assays for lipid and content mixing (Fig. 6, B and C). These combinations of mutant TMDs supported content and lipid mixing similarly as the corresponding WT TMDs. The fact that vacuoles with substitutions of two of the three TMDs of their SNAREs remain fully fusion-competent renders it highly unlikely that SNARE TMDs engage in specific interactions among each other, with other fusion-relevant proteins, to promote vacuole fusion. The TMDs of Vam3 and Nyv1 do contribute to fusion, however, because they could not be replaced by lipid anchors without losing fusion activity (see above). Taken together, these results favor the view that the TMDs serve as force-transducing anchors that contribute to fusion by exerting mechanical strain. They do not seem to utilize sequence-specific features to destabilize the bilayers.
Distance and Helical Continuity between the SNARE Domain and the TMD Are Critical
The role of SNAREs as force-transducing molecules should not be compatible with modifications leading to an increase in the distance between SNARE domains and TMDs or to a disruption of the helical continuity between them (47). To test this hypothesis, we analyzed fusion of vacuoles bearing Vam3 variants with a single or double glycine-serine (GGS) motif insertion in their juxta-membrane region (Fig. 7A). The insertion is expected to increase the distance between the TMD and the SNARE domain. Fusion activity was reduced to ∼30% of the WT value by a single GGS insertion and to 20% by a double GGS insertion (Fig. 7B). This agrees with previous observations using similar manipulations (47, 48). Because the GGS insertions lack side chains, they are also predicted to be flexible and to prefer a random coil conformation. To test whether the observed loss of fusion might be due to increased flexibility of the juxta-membrane region rather than the increase in distance, we constructed a VAM3 version in which nine amino acids of the juxta-membrane sequence were replaced by GGS repeats, but the length of the sequence was not changed. This version yielded less than 15% fusion activity. As an alternative approach, we substituted the juxta-membrane sequence with a motif containing two proline residues, which should also break helical continuity between the SNARE domain and the TMD. This manipulation decreased fusion activity to less than 15%. The observed effects are consistent with a force-transducing role of the SNARE-TMD and support a role for mechanical coupling via helical continuity between the SNARE domain and the TMD.
FIGURE 7.

Effect of juxta-membrane substitutions in Vam3 on fusion activity. A, schematic representation of Vam3 and primary sequences of the juxta-membrane region variants used. All constructs had been expressed from the endogenous VAM3 promoter. B, fusion reactions with vacuoles isolated from Vam3 juxta-membrane mutants. Content mixing was assayed in the presence or absence of ATP. Means ± S.D. are shown for three independent experiments.
DISCUSSION
SNAREs dock membranes in tight apposition and perturb bilayer structure to induce fusion. Experimental evidence, mostly based on liposome studies, has supported various but not mutually exclusive hypotheses on the function of SNARE TMDs as follows: transmission of mechanical strain on the membrane, localization of other membrane-destabilizing proteins to the fusion site (e.g. synaptotagmin), or genuine membrane destabilizing activity of TMDs (15, 18, 23, 24, 49). Additionally, SNARE TMDs were proposed to undergo α-helix to β-sheet transitions, thereby favoring reorientation of associated lipids (23, 50). A crystal structure of the neuronal SNARE complex inspired the attractive proposal that SNARE TMDs might perpetuate the zippering process of the coiled-coil domains into the hydrophobic regions of the bilayer, thereby forcing the associated lipids into the fused state (30). It was also proposed that homodimerization of Vam3 TMDs may control the transition from hemifusion to full fusion (28).
Few studies on SNARE TMDs used physiological membranes. Replacement of SNARE TMDs by the isoprenoid lipid geranylgeranyl (chain length C20) inhibits exocytosis in yeast (15). Similarly, replacement of the Vam3 TMD by a prenyl anchor reduced fusion activity of vacuoles (16). Dimerization of the TMDs of the R-SNARE Vamp2 was observed in intact cells, but it was shown not to have an important functional role (26). C-terminal extension of the Vamp2 TMD inhibited exocytosis, leading to the proposal that the TMD contributes to the resolution of the hemifusion intermediate by being pulled into the inner membrane leaflet (51). None of these studies had addressed the roles of interactions between the different TMDs in the trans-SNARE complex nor a requirement of changes in TMD conformation. Such roles should depend on a specific TMD sequence. Accordingly, replacement by non-SNARE TMDs should interfere with SNARE function. We addressed this aspect with isolated yeast vacuoles. Vacuoles carry all major fusion factors associated with them and allow the accumulation and assay of various reaction stages, such as lipid and content mixing and trans-SNARE pairing (4, 9, 11, 13, 31, 52, 53). Lipid mixing is measured continuously, and content mixing is quantified at the end of a phase of a fairly constant increase in signal. Nevertheless, these approaches do not provide a high time resolution comparable with electrophysiological measurements, raising the caveat that they might fail to detect more subtle kinetic differences.
We observed that the fusion activity of vacuoles remained intact if any one of the vacuolar SNAREs carried a non-SNARE TMD. Also the pairwise exchange of two out of three membrane anchors for unrelated TMDs could be performed in different combinations without compromising fusion activity. Because the vacuolar SNARE Vam7 does not carry a TMD, the simultaneous exchange of two of the three TMDs on Vam3, Nyv1, and Vti1 eliminates the possibility that a specific TMD-TMD interaction remains. Thus, our observations argue strongly against a role for TMD-TMD interactions in fusion. They also argue against a sequence-specific role of the SNARE TMDs in this reaction. This conclusion is valid irrespective of the topology of the trans-SNARE complex. A recent study explored the topology and assembly requirements of the vacuolar trans-SNARE complex and led to the astonishing finding that the preferred distribution of the subunits does not follow the generally assumed model of a Qabc complex in one fusion partner and an R-SNARE in the other (38, 54). Instead, the preferred trans-SNARE topology is QbcR-Qa (Fig. 8) (55), i.e. the Qa-SNARE acts as a single subunit on one of the fusion partners. The pairwise exchanges of TMDs that we performed exclude a role for specific TMD-TMD interactions in both models. However, our results on the effects of lipid anchoring are more compatible with the QbcR-Qa model because lipid anchoring of Vam3 (Qa), which in this model acts alone in one fusion partner, can be expected to bear more severe consequences than replacing the TMDs of Vti1 (Qb) or Nyv1 (R). Vti1 and Nyv1 act together in a QbcR complex, which then would retain at least one proteinaceous TMD that could ameliorate the effects of a loss of the Nyv1 TMD.
FIGURE 8.
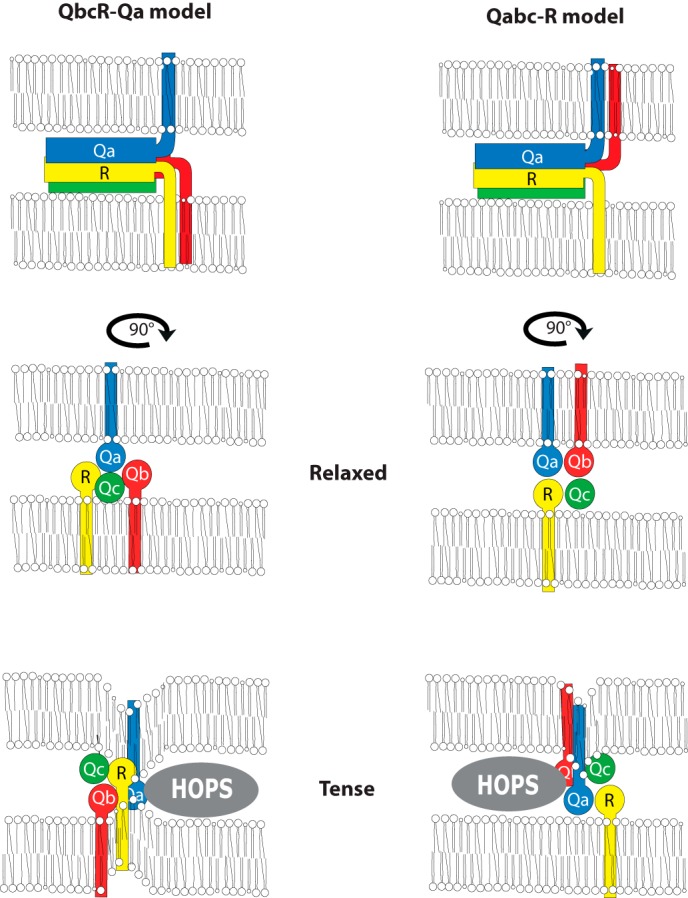
Two models for topology and orientation of the trans-SNARE complex in homotypic vacuole fusion. The generally assumed model of a Qabc complex in one fusion partner and an R-SNARE in the other (38, 54) and an alternative model, suggesting that the preferred trans-SNARE topology is QbcR-Qa, i.e. the Qa-SNARE acts as a single subunit on one of the fusion partners (55). The lower panels show the SNARE complexes seen from the juxta-membrane regions. They illustrate how SNARE complexes could exist in a relaxed or tense state. Association of the SNARE complex with dedicated assembly factors, such as SM proteins and Rab effectors, which form part of the HOPS complex, could dictate assembly of the SNARE complex in a twisted orientation and lead to the tense state. Twisting of the coiled-coil domains can augment the mechanical stress they can exert on the membrane via their TMDs.
Our analyses showed differential effects of Vti1-LA, Nyv1-LA, and Vam3-LA on lipid and content mixing. The TMDs of Vam3 and Nyv1 cannot be replaced by lipid anchors without losing function. This is consistent with the interpretation that these SNAREs can be force-transducing molecules and that this force transduction may be particularly important for opening the fusion pore (31, 56, 57). However, Vam3-LA inhibited content and lipid mixing to similar degrees, whereas Nyv1-LA permitted efficient lipid but not content mixing. This suggests that the induction of hemifusion requires only the Vam3 TMD, whereas fusion pore opening requires proteinaceous anchors on both the Qa and the R-SNARE.
A further interesting aspect is that the TMD of Vti1 could be replaced by a prenyl anchor without compromising lipid or content mixing. In the QbcR-Qa model, Vti1 acts in a complex with Nyv1, and in the Qabc-R model, it acts in complex with Vam3. In both models, loss of the Vti1 TMD is less severe than the loss of the TMD of the other SNARE with which it acts in a cis-complex. This suggest that the TMDs of the two SNARE subunits in the same membrane cannot replace each other. A possibility for explaining this phenomenon is offered by a model that takes into account SNARE complex twisting, i.e. the positioning of the coiled-coil domains relative to the two membranes (31). This model assumes that the coiled-coil region is not acting freely and might not be able to assume a relaxed conformation that allows the largest possible distance between the TMDs anchored in the opposing membranes. The trans-SNAREs complex interacts with numerous other proteins, such as the Rab-GTPase-dependent HOPS complex on vacuoles (31, 53). This interaction is expected to restrain the positioning, particularly the rotation of the coiled-coil region of the complex relative to the membrane, leading to a tense state. Such twisting should have consequences for the pulling strain that the different TMDs can exert onto the membrane (Fig. 8). Twisting of the complex could position the Vti1 TMD such that it can exert significantly less strain on the membrane than the TMDs of Vam3 and Nyv1. This model can give a satisfying explanation for the observed difference. Nevertheless, the data remain open to other interpretations. Irrespective of the model, our data suggest that vacuole fusion is an asymmetric process with unequal contributions of Qa-, Qb, and R-SNARE TMDs to lipid and content mixing. This is remarkable because hemifusion intermediates are usually depicted as symmetric structures, with Q- and R-SNARE TMDs making equal contributions to their formation and their resolution. Also, simulations of fusion have not revealed unequal contributions of the TMDs (58). However, these simulations have been based on exocytotic SNARE complexes with only two TMDs. A restriction of the rotation of the coiled-coil region of the complex by SNARE-associated proteins such as Rab-GTPases and tether complexes had not been taken into account.
The fusogenic activity of SNARE TMDs had been explored in a series of liposome-based studies using synthetic TMD peptides, which did not contain any SNARE domains (25, 50, 59). In these assays, the amino acid composition of TMD peptides strongly affected fusion. Fusogenicity of the TMD peptides increased with decreased stability of the α-helix, suggesting that a conformational flexibility of the TMD may be needed to drive fusion. It was stated that β-branched residues and glycine residues are enriched within SNARE TMDs (60). In our study, replacement of the TMDs of three vacuolar SNAREs by the TMD from ALP, or by a synthetic poly-LV peptide, supported fusion well. This demonstrates the lack of requirement for a specific sequence in the TMD. It might still be consistent with the postulate that global amino acid composition may be the only relevant factor. However, the content of β-branched residues in different SNAREs is highly variable. For the Vti1, it lies at and for Nyv1 far below the average observed in all tail-anchored proteins (23). Thus, a bias for these amino acids in SNARE TMDs may not reflect a fundamental requirement of β-branched residues for SNARE function in fusion. It might be related to other aspects, such as biosynthesis, transport, and sorting of SNAREs to their destination, or to their enrichment in certain membrane domains (57, 61). In line with this assumption, we observed that simultaneous substitutions of two TMDs reduced SNARE abundance on the vacuoles in certain combinations, e.g. for Vam3-ALP/Nyv1-ALP (data not shown).
Might vacuolar SNAREs and their independence of a specific SNARE-TMD represent a special case? We consider this as unlikely because several modifications of vacuolar SNAREs yielded results comparable with those observed in other systems. This is supported by the high sensitivity of vacuole fusion to an elongation of the SNARE sequence in the juxta-membrane region (Fig. 7) (48). Similarly, chromaffin granule exocytosis was shown to be crucially dependent on a short molecular distance between the complex-forming SNARE motif and the transmembrane anchor of the vesicular SNARE protein synaptobrevin II (62). Insertions in this region are expected to increase the distance between the coiled-coil domains and the membrane surface, thereby interfering with the generation of pulling forces or the effective transmission of bending forces. We confirmed this result by testing fusion of vacuoles bearing Vam3 variants with a glycine-serine (GGS) motif insertion in their juxta-membrane region, which considerably decreased fusion activity. Disruption of helical continuity between the SNARE domain and the TMD of Vam3 was also detrimental for fusion activity. These data agree with the deleterious effect of proline insertions in the exocytic yeast SNARE Sso1p, but they partially contradict results obtained in liposome studies. Here, helix-breaking proline residues in the juxta-membrane region showed no effect (synaptobrevin/Vamp) or only a mild effect (syntaxin 1A, Sso1) (47, 63). Furthermore, vacuole fusion is impaired by replacements of the TMDs of Vam3 or Nyv1 by lipid anchors (16, 17), resembling the effect of lipid anchoring of Snc2 on exocytosis (18, 20). Thus, the properties of vacuolar SNAREs are in many respects comparable with those of SNAREs in other membrane trafficking reactions.
A recent study demonstrated that lipid-anchored syntaxin-1 and synaptobrevin-2 support spontaneous fusion of synaptic vesicles, suggesting that proteinaceous SNARE TMDs are dispensable for membrane fusion during spontaneous neurotransmitter release (21). These data differ from our observations with vacuolar SNAREs. Why might lipid-anchored SNAREs fully support the nontriggered release of neurotransmitter vesicles (21) but not the fusion of yeast vacuoles or the fusion of secretory vesicles at the yeast plasma membrane (15)? One possibility is that SNARE-catalyzed fusion follows different pathways on different compartments, a scenario that seems not very likely in light of the evolutionary conservation of SNAREs and of their associated proteins, such as SM proteins, Rabs and Rab effectors. Another possibility is that synaptic vesicle fusion shows a reduced dependence on transmembrane domains due to the specific properties of the membranes and proteins involved. Neurotransmitter vesicles are particularly small and highly curved, which might render the membranes more fusogenic and more sensitive to perturbations by SNARE proteins, even if those are transmitted only by lipid anchors, which are inserted only into the outer membrane leaflet. A more important factor might be that the neuronal exocytic SNARE complex differs from its homologs acting in nontriggered fusion events by its association with several proteins unique to regulated exocytosis, such as synaptotagmin (64). Synaptotagmins are Ca2+ sensors for regulated exocytosis, which directly interact with the ternary SNARE complex (65, 66). They carry a transmembrane domain and C2 domains, which bind phosphoinositides in the plasma membrane. Synaptotagmin could thus provide a lipid-anchored SNARE complex with a missing TMD and increase the strain on the apposed membrane from the other fusion partner by binding its lipids directly. In this way, it might decrease the dependence of neuronal exocytosis on proteinaceous SNARE TMDs.
In sum, our results support the view that SNARE TMDs serve as force-transducing membrane anchors. They are inconsistent with a sequence-specific function, either in the form of SNARE-zippering into the transmembrane domains or via specific interactions of SNARE-TMDs with other proteins or lipids. Furthermore, we propose that the contributions of the Qa, Qb, and R-SNARE to fusion pore opening are different and that in this sense vacuolar membrane fusion is an asymmetric process.
Acknowledgments
We thank Dirk Fasshauer for discussion and Christian Ungermann for strains and plasmids.
This work was supported by the Roche Research Foundation and the Swiss National Science Foundation.
- NSF
- N-ethylmaleimide-sensitive factor
- TMD
- transmembrane domain
- ALP
- alkaline phosphatase
- LA
- lipid anchor.
REFERENCES
- 1. Ostrowicz C. W., Meiringer C. T., Ungermann C. (2008) Yeast vacuole fusion: a model system for eukaryotic endomembrane dynamics. Autophagy 4, 5–19 [DOI] [PubMed] [Google Scholar]
- 2. Wickner W. (2002) Yeast vacuoles and membrane fusion pathways. EMBO J. 21, 1241–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haas A., Conradt B., Wickner W. (1994) G-protein ligands inhibit in vitro reactions of vacuole inheritance. J. Cell Biol. 126, 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reese C., Heise F., Mayer A. (2005) Trans-SNARE pairing can precede a hemifusion intermediate in intracellular membrane fusion. Nature 436, 410–414 [DOI] [PubMed] [Google Scholar]
- 5. Mayer A., Wickner W. (1997) Docking of yeast vacuoles is catalyzed by the Ras-like GTPase Ypt7p after symmetric priming by Sec18p (NSF). J. Cell Biol. 136, 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ungermann C., Wickner W. (1998) Vam7p, a vacuolar SNAP-25 homolog, is required for SNARE complex integrity and vacuole docking and fusion. EMBO J. 17, 3269–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hickey C. M., Wickner W. (2010) HOPS initiates vacuole docking by tethering membranes before trans-SNARE complex assembly. Mol. Biol. Cell 21, 2297–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fasshauer D., Sutton R. B., Brunger A. T., Jahn R. (1998) Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. U.S.A. 95, 15781–15786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Collins K. M., Wickner W. T. (2007) Trans-SNARE complex assembly and yeast vacuole membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 104, 8755–8760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schwartz M. L., Merz A. J. (2009) Capture and release of partially zipped trans-SNARE complexes on intact organelles. J. Cell Biol. 185, 535–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dietrich L. E., Gurezka R., Veit M., Ungermann C. (2004) The SNARE Ykt6 mediates protein palmitoylation during an early stage of homotypic vacuole fusion. EMBO J. 23, 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wickner W. (2010) Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu. Rev. Cell Dev. Biol. 26, 115–136 [DOI] [PubMed] [Google Scholar]
- 13. Mayer A., Wickner W., Haas A. (1996) Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell 85, 83–94 [DOI] [PubMed] [Google Scholar]
- 14. Wang L., Ungermann C., Wickner W. (2000) The docking of primed vacuoles can be reversibly arrested by excess Sec17p (α-SNAP). J. Biol. Chem. 275, 22862–22867 [DOI] [PubMed] [Google Scholar]
- 15. Grote E., Baba M., Ohsumi Y., Novick P. J. (2000) Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J. Cell Biol. 151, 453–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rohde J., Dietrich L., Langosch D., Ungermann C. (2003) The transmembrane domain of Vam3 affects the composition of cis- and trans-SNARE complexes to promote homotypic vacuole fusion. J. Biol. Chem. 278, 1656–1662 [DOI] [PubMed] [Google Scholar]
- 17. Jun Y., Xu H., Thorngren N., Wickner W. (2007) Sec18p and Vam7p remodel trans-SNARE complexes to permit a lipid-anchored R-SNARE to support yeast vacuole fusion. EMBO J. 26, 4935–4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McNew J. A., Weber T., Parlati F., Johnston R. J., Melia T. J., Söllner T. H., Rothman J. E. (2000) Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J. Cell Biol. 150, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu H., Zick M., Wickner W. T., Jun Y. (2011) A lipid-anchored SNARE supports membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 108, 17325–17330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu Y., Zhang F., Su Z., McNew J. A., Shin Y. K. (2005) Hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 12, 417–422 [DOI] [PubMed] [Google Scholar]
- 21. Zhou P., Bacaj T., Yang X., Pang Z. P., Südhof T. C. (2013) Lipid-anchored SNAREs lacking transmembrane regions fully support membrane fusion during neurotransmitter release. Neuron 80, 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Langosch D., Crane J. M., Brosig B., Hellwig A., Tamm L. K., Reed J. (2001) Peptide mimics of SNARE transmembrane segments drive membrane fusion depending on their conformational plasticity. J. Mol. Biol. 311, 709–721 [DOI] [PubMed] [Google Scholar]
- 23. Hofmann M. W., Weise K., Ollesch J., Agrawal P., Stalz H., Stelzer W., Hulsbergen F., de Groot H., Gerwert K., Reed J., Langosch D. (2004) De novo design of conformationally flexible transmembrane peptides driving membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 101, 14776–14781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poschner B. C., Quint S., Hofmann M. W., Langosch D. (2009) Sequence-specific conformational dynamics of model transmembrane domains determines their membrane fusogenic function. J. Mol. Biol. 386, 733–741 [DOI] [PubMed] [Google Scholar]
- 25. Poschner B. C., Fischer K., Herrmann J. R., Hofmann M. W., Langosch D. (2010) Structural features of fusogenic model transmembrane domains that differentially regulate inner and outer leaflet mixing in membrane fusion. Mol. Membr. Biol. 27, 1–10 [DOI] [PubMed] [Google Scholar]
- 26. Fdez E., Martínez-Salvador M., Beard M., Woodman P., Hilfiker S. (2010) Transmembrane-domain determinants for SNARE-mediated membrane fusion. J. Cell Sci. 123, 2473–2480 [DOI] [PubMed] [Google Scholar]
- 27. Tong J., Borbat P. P., Freed J. H., Shin Y. K. (2009) A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc. Natl. Acad. Sci. U.S.A. 106, 5141–5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hofmann M. W., Peplowska K., Rohde J., Poschner B. C., Ungermann C., Langosch D. (2006) Self-interaction of a SNARE transmembrane domain promotes the hemifusion-to-fusion transition. J. Mol. Biol. 364, 1048–1060 [DOI] [PubMed] [Google Scholar]
- 29. Sutton R. B., Fasshauer D., Jahn R., Brunger A. T. (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347–353 [DOI] [PubMed] [Google Scholar]
- 30. Stein A., Weber G., Wahl M. C., Jahn R. (2009) Helical extension of the neuronal SNARE complex into the membrane. Nature 460, 525–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pieren M., Schmidt A., Mayer A. (2010) The SM protein Vps33 and the t-SNARE H(abc) domain promote fusion pore opening. Nat. Struct. Mol. Biol. 17, 710–717 [DOI] [PubMed] [Google Scholar]
- 32. Haas A., Wickner W. (1996) Homotypic vacuole fusion requires Sec17p (yeast α-SNAP) and Sec18p (yeast NSF). EMBO J. 15, 3296–3305 [PMC free article] [PubMed] [Google Scholar]
- 33. Thorngren N., Collins K. M., Fratti R. A., Wickner W., Merz A. J. (2004) A soluble SNARE drives rapid docking, bypassing ATP and Sec17/18p for vacuole fusion. EMBO J. 23, 2765–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kunkel T. A., Roberts J. D., Zakour R. A. (1987) Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154, 367–382 [DOI] [PubMed] [Google Scholar]
- 35. Nichols B. J., Ungermann C., Pelham H. R., Wickner W. T., Haas A. (1997) Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature 387, 199–202 [DOI] [PubMed] [Google Scholar]
- 36. Boeke J. D., Trueheart J., Natsoulis G., Fink G. R. (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 154, 164–175 [DOI] [PubMed] [Google Scholar]
- 37. Scott J. H., Schekman R. (1980) Lyticase: endoglucanase and protease activities that act together in yeast cell lysis. J. Bacteriol. 142, 414–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weber T., Zemelman B. V., McNew J. A., Westermann B., Gmachl M., Parlati F., Söllner T. H., Rothman J. E. (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 [DOI] [PubMed] [Google Scholar]
- 39. Chernomordik L. V., Kozlov M. M. (2003) Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72, 175–207 [DOI] [PubMed] [Google Scholar]
- 40. Cheever M. L., Sato T. K., de Beer T., Kutateladze T. G., Emr S. D., Overduin M. (2001) Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat. Cell Biol. 3, 613–618 [DOI] [PubMed] [Google Scholar]
- 41. Gao J., Liao J., Yang G. Y. (2009) CAAX-box protein, prenylation process and carcinogenesis. Am. J. Transl. Res. 1, 312–325 [PMC free article] [PubMed] [Google Scholar]
- 42. Ungermann C., Nichols B. J., Pelham H. R., Wickner W. (1998) A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J. Cell Biol. 140, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sharpe H. J., Stevens T. J., Munro S. (2010) A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cordes F. S., Bright J. N., Sansom M. S. (2002) Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323, 951–960 [DOI] [PubMed] [Google Scholar]
- 45. Orzáez M., Salgado J., Giménez-Giner A., Pérez-Payá E., Mingarro I. (2004) Influence of proline residues in transmembrane helix packing. J. Mol. Biol. 335, 631–640 [DOI] [PubMed] [Google Scholar]
- 46. Milovanovic D., Honigmann A., Koike S., Göttfert F., Pähler G., Junius M., Müllar S., Diederichsen U., Janshoff A., Grubmüller H., Risselada H. J., Eggeling C., Hell S. W., van den Bogaart G., Jahn R. (2015) Hydrophobic mismatch sorts SNARE proteins into distinct membrane domains. Nat. Commun. 6, 5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McNew J. A., Weber T., Engelman D. M., Söllner T. H., Rothman J. E. (1999) The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol. Cell 4, 415–421 [DOI] [PubMed] [Google Scholar]
- 48. Wang Y., Dulubova I., Rizo J., Südhof T. C. (2001) Functional analysis of conserved structural elements in yeast syntaxin Vam3p. J. Biol. Chem. 276, 28598–28605 [DOI] [PubMed] [Google Scholar]
- 49. Kee Y., Scheller R. H. (1996) Localization of synaptotagmin-binding domains on syntaxin. J. Neurosci. 16, 1975–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yassine W., Taib N., Federman S., Milochau A., Castano S., Sbi W., Manigand C., Laguerre M., Desbat B., Oda R., Lang J. (2009) Reversible transition between α-helix and β-sheet conformation of a transmembrane domain. Biochim. Biophys. Acta 1788, 1722–1730 [DOI] [PubMed] [Google Scholar]
- 51. Ngatchou A. N., Kisler K., Fang Q., Walter A. M., Zhao Y., Bruns D., Sørensen J. B., Lindau M. (2010) Role of the synaptobrevin C terminus in fusion pore formation. Proc. Natl. Acad. Sci. U.S.A. 107, 18463–18468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Conradt B., Haas A., Wickner W. (1994) Determination of four biochemically distinct, sequential stages during vacuole inheritance in vitro. J. Cell Biol. 126, 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Collins K. M., Thorngren N. L., Fratti R. A., Wickner W. T. (2005) Sec17p and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J. 24, 1775–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parlati F., McNew J. A., Fukuda R., Miller R., Söllner T. H., Rothman J. E. (2000) Topological restriction of SNARE-dependent membrane fusion. Nature 407, 194–198 [DOI] [PubMed] [Google Scholar]
- 55. Alpadi K., Kulkarni A., Comte V., Reinhardt M., Schmidt A., Namjoshi S., Mayer A., Peters C. (2012) Sequential analysis of trans-SNARE formation in intracellular membrane fusion. PLoS Biol. 10, e1001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chernomordik L. V., Kozlov M. M. (2008) Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 15, 675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jahn R., Lang T., Südhof T. C. (2003) Membrane fusion. Cell 112, 519–533 [DOI] [PubMed] [Google Scholar]
- 58. Risselada H. J., Grubmüller H. (2012) How SNARE molecules mediate membrane fusion: recent insights from molecular simulations. Curr. Opin. Struct. Biol. 22, 187–196 [DOI] [PubMed] [Google Scholar]
- 59. Langosch D., Brosig B., Pipkorn R. (2001) Peptide mimics of the vesicular stomatitis virus G-protein transmembrane segment drive membrane fusion in vitro. J. Biol. Chem. 276, 32016–32021 [DOI] [PubMed] [Google Scholar]
- 60. Neumann S., Langosch D. (2011) Conserved conformational dynamics of membrane fusion protein transmembrane domains and flanking regions indicated by sequence statistics. Proteins 79, 2418–2427 [DOI] [PubMed] [Google Scholar]
- 61. Jahn R., Südhof T. C. (1999) Membrane fusion and exocytosis. Annu. Rev. Biochem. 68, 863–911 [DOI] [PubMed] [Google Scholar]
- 62. Kesavan J., Borisovska M., Bruns D. (2007) v-SNARE actions during Ca2+-triggered exocytosis. Cell 131, 351–363 [DOI] [PubMed] [Google Scholar]
- 63. Van Komen J. S., Bai X., Rodkey T. L., Schaub J., McNew J. A. (2005) The polybasic juxtamembrane region of Sso1p is required for SNARE function in vivo. Eukaryot. Cell 4, 2017–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Südhof T. C. (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Choi U. B., Strop P., Vrljic M., Chu S., Brunger A. T., Weninger K. R. (2010) Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat. Struct. Mol. Biol. 17, 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim J. Y., Choi B. K., Choi M. G., Kim S. A., Lai Y., Shin Y. K., Lee N. K. (2012) Solution single-vesicle assay reveals PIP2-mediated sequential actions of synaptotagmin-1 on SNAREs. EMBO J. 31, 2144–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jones E. W., Zubenko G. S., Parker R. R. (1982) PEP4 gene function is required for expression of several vacuolar hydrolases in Saccharomyces cerevisiae. Genetics 102, 665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]