

Background: Phox/Bem1p domains are universal domains that organize cellular signaling scaffolds.
Results: Biophysical analyses reveal driving forces and core residues involved in PB1 interaction.
Conclusion: Electrostatic interactions focused around two complementary prongs.
Significance: These results provide the first in-depth analysis of the factors driving self-interaction of a type I/II PB1 domain.
Keywords: nuclear magnetic resonance (NMR), plant biochemistry, plant hormone, protein structure, protein-protein interaction
Abstract
Phox/Bem1p (PB1) domains are universal structural modules that use surfaces of different charge for protein-protein association. In plants, PB1-mediated interactions of auxin response factors (ARF) and auxin/indole 3-acetic acid inducible proteins regulate transcriptional events modulated by the phytohormone auxin. Here we investigate the thermodynamic and structural basis for Arabidopsis thaliana ARF7 PB1 domain self-interaction. Isothermal titration calorimetry and NMR experiments indicate that key residues on both the basic and acidic faces of the PB1 domain contribute to and organize coordinately to stabilize protein-protein interactions. Calorimetric analysis of ARF7PB1 site-directed mutants defines a two-pronged electrostatic interaction. The canonical PB1 interaction between a lysine and a cluster of acidic residues provides one prong with an arginine and a second cluster of acidic residues defining the other prong. Evolutionary conservation of this core recognition feature and other co-varying interface sequences allows for versatile PB1-mediated interactions in auxin signaling.
Introduction
The phytohormone auxin regulates nearly every aspect of plant growth and development (1). To date, all studied plants contain indole 3-acetic acid, the most common active auxin, as well as its biosynthetic precursors, inactive forms, and conjugates (2). Because auxin is the master regulator of plant growth and development, plants defective in auxin production and/or perception display defects in cell division and elongation (3–6). Therefore, tight regulation of auxin signaling is necessary to promote correct plant growth and development.
In Arabidopsis thaliana (thale cress), auxin signaling primarily occurs through three protein families: auxin perceiving F-box proteins, auxin/indole 3-acetic acid inducible (Aux/IAA)2 repressor proteins, and auxin response factor (ARF) transcription factors (7). Under low local auxin concentrations, Aux/IAA proteins interact with ARF proteins through two C-terminal regions of homologous amino acid sequence, termed domains III/IV (8, 9). This protein-protein interaction results in repression of auxin-regulated gene transcription by the ARF protein family (9). When local auxin levels increase, indole 3-acetic acid serves as the “molecular glue” to form a co-receptor with one of the six auxin perceiving F-box proteins and an Aux/IAA (10). This interaction results in polyubiquitination and degradation of the Aux/IAA repressor proteins (11), thus freeing ARF proteins to regulate transcription of target genes. Dimerization of ARF proteins at their N-terminal B3 domain is required for recognition of auxin response elements that control gene expression (12).
Recent structural studies revealed the presence of a Phox/Bem1p (PB1) domain that comprises the C-terminal III/IV interaction sequence motif in ARF proteins (13, 14) and Aux/IAA proteins (15). PB1 domains are conserved throughout all kingdoms and often confer interaction specificity in highly redundant protein scaffolds to facilitate signaling events (16). PB1 domains adopt a ubiquitin-like β-grasp fold that can present two oppositely charged faces on the protein surface. The positive face bears an invariant lysine residue, and the negative face contains a cluster of aspartate and glutamate residues (DX(D/E)XDXnD) known as the OPCA motif. PB1 domains may possess a negative face (type I), a positive face (type II), or both faces (type I/II) with protein-protein interactions mediated by oriented binding of the negative face of one PB1 domain to the positive face of another PB1 domain protein. Amino acid sequence alignments (17) and structural studies (13–15) suggest that most Arabidopsis ARF and Aux/IAA PB1 domains are type I/II to allow for higher order protein multimerization. In addition, the PB1 domains of ARF and Aux/IAA proteins provides a means for guiding interactions between various members of these protein families to control auxin responses in plants (18).
Although PB1 domain interactions involve electrostatic pairing of the invariant lysine with the OPCA motif (16), thermodynamic characterization of PB1-mediated interaction has been limited to determining binding constants (15). Furthermore, predicted PB1 interface residues show various levels of conservation in both ARF and Aux/IAA, leading to the hypothesis that these residues may provide clues for interaction specificity (13, 15, 18). To better understand PB1 domain-mediated protein-protein interactions, we used the PB1 domain of Arabidopsis ARF7 to investigate the thermodynamic basis for self-interaction. Subsequent site-directed mutagenesis of residues forming the interaction interface and isothermal titration calorimetry (ITC) identifies a two-pronged hot spot required for protein-protein interaction. Finally, we investigate the protein dynamics of the ARF7 PB1 domain using NMR spectroscopy. Our results indicate that electrostatic forces drive PB1 domain binding and identify core interface residues that stabilize protein-protein interaction across the ARF and Aux/IAA protein families.
EXPERIMENTAL PROCEDURES
Construct Generation
The constructs used for bacterial expression of the Arabidopsis ARF7 PB1 domain (Met-1037–Asn-1131) with either the K1042A (ARF7PB1K1042A) or D1092A/D1096A (ARF7PB1opca) mutations were previously described (13). Site-directed mutants of residues in the protein interaction interface were generated using the QuikChange Lightning site-directed mutagenesis kit (Agilent) with the appropriate template vector.
Protein Expression and Purification
All constructs were cloned into in Escherichia coli (DE3) Rosetta (Invitrogen) for protein expression. For ITC experiments, cells were grown in Terrific Broth. For three- and two-dimensional-NMR experiments, cells were grown in minimal media supplemented with [15N]ammonium chloride (Sigma) and/or d-[13C6]glucose (Sigma). Proteins were purified as described previously (13). For ITC experiments, purified protein was dialyzed overnight at 4 °C against 25 mm Tris, pH 8.0, 100 mm NaCl, 5% glycerol, and 3 mm 2-mercaptoethanol. For the salt dependence experiments, proteins were dialyzed overnight at 4 °C against 25 mm Tris, pH 8.0, 5% glycerol, and 3 mm 2-mercaptoethanol supplemented with 50–500 mm NaCl. For NMR spectroscopy experiments, size-exclusion chromatography fractions containing purified protein were pooled and dialyzed overnight at 4 °C against 25 mm MOPSO, pH 7.0, 100 mm NaCl. Protein concentrations were determined by UV/visible spectroscopy (ϵ280 nm = 16,560 cm−1 m−1).
Isothermal Titration Calorimetry
ITC experiments were carried out using a VP-ITC (Malvern) instrument at the temperatures indicated in the figure legends. For all ITC experiments, syringe protein concentration was 100 μm, and the cell protein concentration was 10 μm. Thermodynamic analysis of ARF7PB1 interaction was performed using titrations of ARF7PB1K1042A into ARF7PB1opca and vice versa. Titrations were carried out with the interface alanine mutant in the syringe titrated into either ARF7PB1K1042A or ARF7PB1opca. All ITC experiments consisted of 29 consecutive 10-μl titrations, each separated by a 600-s interval. The first injection of each experiment was limited to 6 μl. Results were analyzed using Origin 7.0 with data fit to a single-site binding model. Values for the change in Gibbs free energy (ΔG) were calculated using ΔG = −RTln(Keq), where R is the gas constant (1.9872 cal K−1 mol−1), and T is temperature in Kelvin. Entropy changes (ΔS) were calculated using ΔG = ΔH − TΔS. Kd was calculated as 1/Keq.
NMR Spectroscopy
Standard two-dimensional 1H,15N HSQC (19) spectra were collected for ARF7PB1K1042A, ARF7PB1opca, and ARF7PB1K1042A,opca at 30 °C on a Bruker Avance III 600 MHz spectrometer equipped with a cryogenic probe. All samples were dissolved in 25 mm MOPSO, pH 7.0, 100 mm NaCl, and 10% D2O. Protein concentrations in the three samples were 40, 60, and 445 μm for ARF7PB1K1042A, ARF7PB1opca, and ARF7PB1K1042A,opca, respectively. Spectra were acquired using 128 (ARF7PB1K1042A), 64 (ARF7PB1opca), or 16 (ARF7PB1K1042A,opca) scans and 128 × 1024 complex points in t1 and t2, respectively. The NMR data were processed using NMRPipe (20) and analyzed using NMRView (21). For backbone assignments of ARF7PB1K1042A,opca, NMR data were collected on a 425 μm 13C,15N uniformly labeled sample dissolved in 25 mm MOPSO, pH 7.0, 100 mm NaCl, and 10% D2O. Two-dimensional 1H,15N HSQC (22), three-dimensional-HNCACB (23), and three-dimensional CBCA(CO)NH (24) experiments were collected at the National Magnetic Resonance Facility at Madison (NMRFAM) on an Agilent DDR spectrometer operating at 900 MHz and equipped with a z axis pulsed field gradient triple-resonance cryogenic probe. The temperature of the sample was regulated at 30 °C throughout the experiments. A longitudinal-relaxation-enhanced pulse program (BEST) (25, 26) was used to record the three-dimensional-HNCACB spectrum with 128 repetitions for each scan and a delay of 0.4 s in between scans. Non-uniform sampling (27) was used to record both three-dimensional HNCACB and three-dimensional CBCA(CO)NH spectra with a sampling rate of 25 and 50%, respectively. The programs istHMS (27) and NMRPipe (20) were used to reconstruct and process three-dimensional NUS data. Processed spectra were analyzed using NMRFAM-Sparky (28). Backbone assignments were made automatically with the PINE-NMR software (29) and confirmed manually using NMRFAM-Sparky (28). The HSQC spectra of ARF7PB1K1042A and ARF7PB1opca closely resembled that of ARF7PB1K1042A,opca. Seventy-one backbone peaks in ARF7PB1K1042A and 60 backbone peaks in ARF7PB1opca could be directly traced to the corresponding assigned peaks in ARF7PB1K1042A,opca. These “transferred” assignments were used to analyze results of NMR chemical shift mapping. NMR chemical shift mapping was carried out using 15N uniformly labeled ARF7PB1K1042A at 75 μm protein concentration mixed with unlabeled ARF7PB1opca at molar ratios of 1:0, 1:1, 1:2, and 1:3. Reciprocal titrations were performed using 100 μm 15N-labeled ARF7PB1opca mixed with unlabeled ARF7PB1K1042A at 1:0, 1:1, 1:2, and 1:3 molar ratios. Identical two-dimensional-HSQC experiments with 64 scans (15N-ARF7PB1K1042A) or 32 scans (15N-ARF7PB1opca) and 128 × 2048 complex points were collected at 30 °C. Data were processed with NMRPipe and visualized using NMRView.
RESULTS
Thermodynamic Analysis of ARF7 PB1 Domain Interaction
We performed ITC experiments on the ARF7-ARF7 interaction to examine the thermodynamic basis for ARF7 PB1 domain association. To limit the interaction to dimerization rather than higher order multimerization, we used the ARF7PB1K1042A and ARF7PB1D1092A/D1096A(opca) mutants. Each ARF7PB1 variant is monomeric in solution as observed with size-exclusion chromatography, and dimerization occurs upon mixing the two variants (13). Combination of the two ARF7PB1 variants leads to an oriented interaction of the positive face (Lys-1042) of one PB1 domain with the negative face (OPCA) of the other molecule. Our initial ITC studies revealed that ARF7PB1K1042A and ARF7PB1opca interact with a Kd = 0.18 μm at 25 °C (Fig. 1a; Table 1). Similar results were observed when ARF7PB1K1042A was titrated into ARF7PB1opca and when ARF7PB1opca was titrated into ARF7PB1K1042A. The observed binding constant for association of ARF7 PB1 domains is comparable to the Kd value (0.87 μm) determined for ARF5 PB1 domain interaction (15). The stoichiometry (n = 1.05) of the ARF7PB1 interaction is also consistent with dimer formation, suggesting that we are interrogating a single interaction interface.
FIGURE 1.

Thermodynamic analysis of ARF7 PB1 domain interaction. a, titration of ARF7PB1K1042A into ARF7PB1opca. The top panel shows the heat signal (μcal s−1) plotted versus time (min). The experiment consisted of 30 sequential injections of 10 μl of ARF7PB1K1042A (100 μm) into a cell solution of ARF7PB1opca (10 μm) at 25 °C. The bottom panel shows the integrated heat response per injection from the upper panel plotted as a function of moles of injectant. The solid line represents the fit of the data to a single-site binding model. b, temperature dependence of ΔH (squares), ΔG (open circles), and −TΔS (filled circles) determined by ITC for ARF7PB1 interaction. c, salt dependence of ARF7PB1 interaction. The Keq (1/Kd) values determined by ITC at varied NaCl concentrations are shown.
TABLE 1.
ITC analysis of the temperature dependence of ARF7 PB1 domain interaction
Titrations were performed at the indicated temperature.
| Temperature | n | Kd | ΔG | ΔH | −TΔS |
|---|---|---|---|---|---|
| μm | kcal mol−1 | kcal mol−1 | kcal mol−1 | ||
| 10 | 1.05 ± 0.01 | 0.06 ± 0.01 | −9.37 ± 1.59 | −12.57 ± 0.16 | 3.20 |
| 15 | 0.97 ± 0.01 | 0.07 ± 0.01 | −9.42 ± 1.31 | −12.91 ± 0.15 | 3.49 |
| 20 | 0.98 ± 0.01 | 0.09 ± 0.02 | −9.43 ± 1.59 | −13.74 ± 0.20 | 4.31 |
| 25 | 1.05 ± 0.02 | 0.18 ± 0.03 | −9.21 ± 1.73 | −14.19 ± 0.29 | 4.98 |
| 30 | 0.99 ± 0.01 | 0.28 ± 0.04 | −9.09 ± 1.15 | −14.82 ± 0.25 | 5.73 |
| 35 | 1.00 ± 0.02 | 0.36 ± 0.06 | −9.09 ± 1.45 | −15.22 ± 0.39 | 6.13 |
| 40 | 1.04 ± 0.03 | 0.63 ± 0.10 | −8.88 ± 1.39 | −15.83 ± 0.48 | 6.95 |
Analysis of the temperature dependence of ARF7PB1 interaction by ITC was used to determine the enthalpic (ΔH) and entropic (ΔS) contributions to the free energy of binding (ΔG) (Fig. 1b; Table 1). The Kd values increased ∼10-fold from 0.06 to 0.63 μm with temperature (10–40 °C). The overall modest 0.49-kcal mol−1 change in ΔG with increasing temperature results from compensatory changes in the ΔH and ΔS of interaction. As temperature increased, the enthalpic contributions to binding decreased, and the entropic component increased. Extrapolation of the specific heat capacity at constant pressure (ΔCp) from the measured ΔH values (Fig. 1b) yields a ΔCp = −110.6 kcal mol−1 K−1, suggesting that the ARF7PB1 interaction is driven primarily by electrostatic interactions (30). Furthermore, the relatively low magnitude of ΔCp indicates that association of the ARF7PB1 may not be particularly dynamic; e.g. the domain is largely globular and does not undergo major structural rearrangement upon binding (31), which is further supported by a linear van't Hoff plot of ARF7PB1 interaction (Fig. 2).
FIGURE 2.

van't Hoff plot of ARF7 PB1 domain interaction. A plot of Keq versus temperature for ARF7 PB1 domain interaction is shown. The line shows a linear fit to the data.
To test the role of electrostatic interaction on ARF7PB1 interaction, ITC was used to examine the salt dependence of interaction. Binding between ARF7PB1K1042A and ARF7PB1opca was analyzed in conditions of increasing ionic strength (Fig. 1c; Table 2). Changing the NaCl concentration from 50 to 500 mm led to a 17-fold increase in the Kd of interaction, consistent with the interaction being driven by electrostatic interactions rather than by hydrophobic effects. In summary, these results indicate that ARF7PB1 self-interaction is energetically favorable with submicromolar affinity and driven by electrostatic forces.
TABLE 2.
ITC analysis of the salt dependence of ARF7 PB1 domain interaction
Titrations were performed at 25 °C and the indicated NaCl concentrations.
| [NaCl] | Kd |
|---|---|
| m | μm |
| 0.05 | 0.07 ± 0.01 |
| 0.1 | 0.21 ± 0.04 |
| 0.25 | 0.55 ± 0.09 |
| 0.5 | 1.22 ± 0.22 |
Site-directed Mutagenesis of the ARF7 PB1 Domain Interaction Interface
Structural studies of the ARF7 PB1 domain show that oligomer formation is oriented with the basic (Lys-1042) face of one chain packed against the acidic (OPCA) face of another chain (Fig. 3a) (13). The interaction interface buries 497 Å2 of protein surface and places the invariant lysine (Lys-1042 in ARF7) opposite from residues of the OPCA motif (Asp-1092, Glu-1094, and Asp-1096 in ARF7). Analysis of the protein-protein interface using PISA (32) suggests a total of 27 amino acids form contacts across the interaction interface with nine residues specifically contributing either ionic or hydrogen bond interactions (13). Four of these residues are on the basic face (Thr-1039, Gly-1050, Arg-1051, and Ser-1052) and five on the acidic face (Ile-1097, Leu-1099, Asp-1102, Asp-1103, Glu-1107). In addition to these residues, a protein-protein interaction role for a conserved tryptophan (Trp-1105 in ARF7) in a rice Aux/IAA was recently described (33). Previous mutagenesis studies of PB1 domains from ARF5, ARF7, and IAA17 as well as other proteins have targeted the conserved lysine and main cluster of OPCA residues (13–16), but have not examined the contribution of other amino acids to PB1-mediated protein interaction.
FIGURE 3.
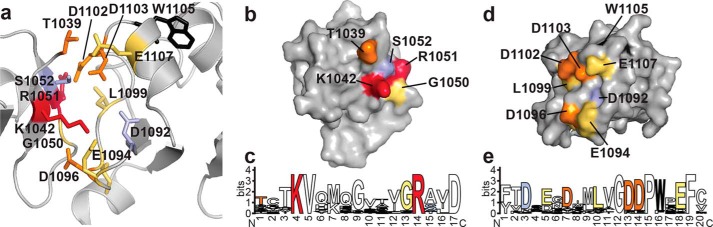
Binding contributions of ARF7 PB1 domain interface residues. a, structure of the ARF7 PB1 domain interface. The view shows the interface between two ARF7 PB1 domains (Ref. 13; PDB code 4NJ6). Residues contributed by the basic face of one PB1 domain (left) and by the OPCA face of a second PB1 domain (right) are shown as stick models. Colors correspond to the effect of alanine mutations on Kd determined by ITC experiments, as follows: light purple, <2-fold; yellow, 2–10-fold; orange, >10-fold. Residues that abolished detectable interactions are red. The structurally important tryptophan is black. The same color scheme is used in panels b–e. b, surface representation of the ARF7 PB1 domain basic face. Positions of residues with colors corresponding to changes in binding affinity are indicated. c, amino acid sequence logo showing conservation of residues on the ARF7 PB1 domain basic face of all 22 Arabidopsis ARF proteins. d, surface representation of the ARF7 PB1 domain OPCA face. Positions of residues with colors corresponding to changes in binding affinity are indicated. e, amino acid sequence logo showing conservation of residues on the ARF7 PB1 domain OPCA face of all 22 Arabidopsis ARF proteins. Panels c and e were generated using WebLogo (39).
To determine the contribution of key interface residues to ARF7PB1 interaction, we performed alanine-scanning mutagenesis and assessed protein binding by ITC. Because ARF7PB1opca displays a disrupted acidic face, we used this variant to query basic face interface residues. Likewise, we used the ARF7PB1K1042A variant to query acidic face residues. Single alanine substitutions (T1039A, K1042A, G1050A, R1051A, and S1052A) were made to the basic face of ARF7PB1opca. Similar changes (D1092A, E1094A, D1096A, I1097A, L1099A, D1102A, D1103A, W1105A, and E1107A) were introduced to the acidic face of ARF7PB1K1042A. We performed ITC experiments by titrating the interface alanine mutant into the cell containing either ARF7PB1K1042A or ARF7PB1opca, as appropriate (Table 3).
TABLE 3.
ITC analysis of interface mutations on ARF7 PB1 domain interaction
Titrations were performed at 25 °C. ND = heat signature was not detected.
| Proteins |
Kd | ΔG | ΔH | −TΔS | |
|---|---|---|---|---|---|
| Cell | Titrant | ||||
| μm | kcal mol−1 | kcal mol−1 | kcal mol−1 | ||
| ARF7PB1K1042A | ARF7PB1opca | 0.18 ± 0.03 | −9.21 ± 1.73 | −14.2 ± 0.3 | 4.98 |
| ARF7PB1K1042A | ARF7PB1opca,T1039A | 53.4 ± 1.3 | −5.77 ± 7.45 | −935 ± 24 | 929 |
| ARF7PB1K1042A | ARF7PB1opca,K1042A | ND | ND | ND | ND |
| ARF7PB1K1042A | ARF7PB1opca,G1050A | 0.68 ± 0.13 | −8.33 ± 1.62 | −9.36 ± 0.31 | 1.02 |
| ARF7PB1K1042A | ARF7PB1opca,R1051A | ND | ND | ND | ND |
| ARF7PB1K1042A | ARF7PB1opca,S1052A | 0.41 ± 0.11 | −8.63 ± 2.24 | −12.4 ± 0.4 | 3.80 |
| ARF7PB1opca | ARF7PB1K1042A,D1092A | 0.31 ± 0.02 | −8.80 ± 1.59 | −6.97 ± 0.12 | −1.83 |
| ARF7PB1opca | ARF7PB1K1042A.E1094A | 0.58 ± 0.08 | −8.42 ± 1.10 | −12.7 ± 0.3 | 4.25 |
| ARF7PB1opca | ARF7PB1K1042A,D1096A | 6.33 ± 1.67 | −7.02 ± 1.74 | −11.9 ± 1.8 | 4.90 |
| ARF7PB1opca | ARF7PB1K1042A,I1097A | 0.58 ± 0.11 | −8.42 ± 1.53 | −13.4 ± 0.4 | 5.02 |
| ARF7PB1opca | ARF7PB1K1042A,L1099A | 1.69 ± 0.23 | −7.80 ± 1.06 | −19.6 ± 0.7 | 11.8 |
| ARF7PB1opca | ARF7PB1K1042A,D1102A | ND | ND | ND | ND |
| ARF7PB1opca | ARF7PB1K1042A,D1102N | ND | ND | ND | ND |
| ARF7PB1opca | ARF7PB1K1042A,D1103A | ND | ND | ND | ND |
| ARF7PB1opca | ARF7PB1K1042A,D1103N | ND | ND | ND | ND |
| ARF7PB1opca | ARF7PB1K1042A,E1107A | 0.92 ± 0.14 | −8.15 ± 1.27 | −11.2 ± 0.4 | 3.96 |
The basic face of ARF7PB1 bears two residues essential for interaction: Lys-1042 and Arg-1051 (Fig. 3b; Table 3). Substitution of either residue with an alanine completely abrogated interaction between monomers. Amino acid sequence comparison of the 22 Arabidopsis ARF PB1 domains indicates that these two basic residues are invariant (Fig. 3c). Mutation of Thr-1039 also severely affects binding with a >200-fold increase in Kd. The G1050A and S1052A mutants displayed only modest (i.e. <3-fold) effects on the ARF7 PB1 interaction (Kd = 0.6 μm and 0.4 μm, respectively). The residue corresponding to Thr-1039 is either a threonine or serine in ARF proteins with a glycine favored at position 1050.
On the acidic face of the ARF7 PB1 domain, alanine substitutions identify differential contributions by residues of the OPCA motif and a new structural feature for interaction (Fig. 3d; Table 3). The OPCA motif is invariant across the Arabidopsis ARF (Figs. 3e and 4). ITC analysis of mutants that disrupt the OPCA motif (D1092A, E1094A, and D1096A) indicates that Asp-1096 provides the largest contribution to binding energy, as the D1096A mutant results in a 35-fold decrease in binding affinity. Although the D1092A mutant displays a 3-fold change in Kd, the entropic contribution to binding increases, and the enthalpic contribution decreases. This suggests the possible loss of a hydrogen bond responsible for stabilizing the β3-β4 loop that defines the OPCA motif. Substitution of Glu-1094 with an alanine had minimal effects on the interaction energetics.
FIGURE 4.

Multiple sequence alignment of PB1 domains from A. thaliana ARF and Aux/IAA. The secondary structure of the Arabidopsis ARF7 PB1 domain (Ref. 13; PDB code 4NJ6) is shown at the top of the alignment. Sequences of the PB1 domains of Arabidopsis ARF and Aux/IAA were aligned using MegAlign (DNASTAR). Colors used to highlight key positions correspond to the effect of alanine mutations on Kd determined by ITC experiments (see Table 3), as follows: light purple, <2-fold; yellow, 2–10-fold; orange, >10-fold. Residues that abolished detectable interactions are red. The structurally important tryptophan is black.
In addition to the main cluster of OPCA residues, ITC analysis of ARF7PB1 identifies a distal set of acidic residues (Asp-1102 and Asp-1103) as critical for interaction. Both aspartates are nearly invariant in Arabidopsis ARF PB1 domains (Figs. 3e and 4). Unlike experiments with K1042A and R1051A that showed a complete loss of interaction, minor heat signature changes could be detected for the D1102A and D1103A mutants; however, the signal was insufficient for accurate data fitting. Based on this result, the Kd values for these mutants likely exceed 100 μm.
Because Asp-1102 and Asp-1103 interact with Arg-1051, we examined the effect of subtler asparagine mutations of each residue. ITC analysis of the D1102N mutant showed a complete lack of interaction. Mutation of Asp-1103 to asparagine severely reduced interaction with insufficient heat signal changes for accurate quantification, as observed with the alanine substitution. Together these results suggest that the presence of negative charge at both Asp-1102 and -1103 is crucial to ARFPB1 interaction.
Alanine mutations of Ile-1097, Leu-1099, and Glu-1107 resulted in 3–9-fold increases in Kd (Fig. 3d; Table 3). In Arabidopsis ARF proteins, position 1097 is variable, but the presence of a leucine and a glutamate at positions 1099 and 1107, respectively, is generally conserved (Figs. 3e and 4).
Interestingly, substitution of an alanine for Trp-1105, a residue conserved in all but one Arabidopsis ARF (Figs. 3e and 4), yielded unstable protein that precipitated upon purification. A previous study identified a mutation of this residue in an intragenic suppressor line bearing an Aux/IAA gain-of-function mutation (33). Our results indicate that this residue is required for globular stability and is not necessarily a determinant of protein-protein interaction. Overall, these results identify key core and peripheral residues required for stabilization of the interaction of the ARF7 PB1 domain.
NMR Analysis of the ARF7 PB1 Domain Interaction Interface
To validate the ARF7PB1 interaction interface and to determine the effect of partner binding in solution at the amino acid level, we performed NMR chemical shift mapping. To investigate the chemical shift perturbations upon binding, we collected three-dimensional 13C,1H,15N spectra and assigned the backbone peaks of ARF7PB1K1042A,opca (Fig. 5). Subsequently, we collected two-dimensional 1H,15N HSQC spectra for 15N-ARF7PB1K1042A,opca, 15N-ARF7PB1K1042A, and 15N-ARF7PB1opca. Overlay of these three spectra (Fig. 6) revealed that the majority of the peak distribution patterns in ARF7PB1K1042A,opca were conserved in ARF7PB1K1042A and ARF7PB1opca. Therefore, the backbone assignments of ARF7PB1K1042A,opca (Fig. 6) were qualitatively transferred to both ARF7PB1K1042A and ARF7PB1opca. In subsequent NMR titration experiments, 15N-ARF7PB1K1042A was mixed with increasing molar ratios of ARFPB1opca to examine the effects of partner binding on the acidic face. The converse experiments with ARF7PB1K1042A titrated with 15N-ARF7PB1opca were also performed to examine binding at the basic face. Chemical shift perturbations as a result of binding were documented and mapped onto the structure of ARF7PB1 (Fig. 7). Perturbations were binned into one of three categories: shifts exhibiting slow exchange, intermediate exchange, or fast exchange regimes.
FIGURE 5.

Backbone assignments of ARF7PB1K1042A,opca. Backbone peaks were assigned (as described under “Experimental Procedures”) for the ARF7PB1K1042A,opca mutant variant. The two-dimensional 15N HSQC spectrum of ARF7PB1K1042A,opca is displayed. Assigned peaks are labeled with residue identity and number according to the three-dimensional data.
FIGURE 6.
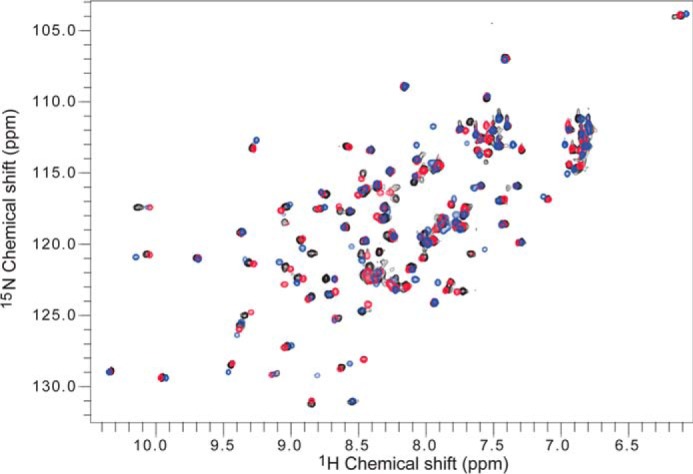
Overlay of two-dimensional HSQC Spectra for ARF7PB1K1042A, ARF7PB1opca, and ARF7PB1K1042A,opca. The two-dimensional 1H,15N HSQC spectra were collected and subsequently overlaid using NMRView (30) for ARF7PB1K1042A (blue), ARF7PB1opca (red), and ARF7PB1K1042A,opca (black).
FIGURE 7.

Effects of ARF7 PB1 domain interaction on NMR peak shifts. a, the results of titrating unlabeled ARF7PB1K1042A into 15N-ARF7PB1opca are mapped onto a ribbon diagram of the ARF7 PB1 domain. Residues shaded red, yellow, and orange participate in slow, intermediate, and fast exchange, respectively. b, two-dimensional 1H,15N HSQC spectra show chemical shift data for the titration of unlabeled ARF7PB1K1042A into 15N-labeled ARF7PB1opca. Molar ratios of unlabeled ARF7PB1K1042A:15N-ARF7PB1opca are indicated by black (0:1), blue (1:1), green (2:1), and red (3:1) colors. Peaks that move upon titration are labeled with a residue number as well as an arrow displaying the direction of peak movement (fast exchange). Peaks that disappear upon titration are labeled with a residue number (slow exchange). Peaks that broaden upon titration (intermediate exchange) are labeled with a residue number and an asterisk (‡). New peaks that appear after titration and cannot be assigned to a residue are labeled with an *. These peaks correspond to the bound state for those residues that are in slow exchange. c, the results of titrating labeled ARF7PB1opca into 15N-ARF7PB1K1042A are mapped onto a ribbon diagram of the ARF7 PB1 domain. Residues shaded red and orange participate in slow and fast exchange, respectively. d, two-dimensional 1H,15N HSQC spectra show titration chemical shift data for the titration of unlabeled ARF7PB1opca into 15N-ARF7PB1K1042A. Molar ratios of unlabeled ARF7PB1opca:15N-ARF7PB1K1042A are indicated by black (0:1), blue (1:1), green (2:1), and red (3:1) colors. Labels and symbols are as in panel b.
For both faces, a mixture of slow and fast exchange predominates at the predicted site of the interface and surrounding the residues previously known to be important for binding (13–15) as well as new core binding residues discovered in ITC analysis of interface mutants (Fig. 3; Table 3). For example, on the basic face, the largest changes were observed in amino acid residues surrounding the essential Lys-1042 and Arg-1051, namely Thr-1041, Val-1043, Gln-1044, Val-1049, and Ile-1053, that displayed slow exchange, whereas Lys-1042 itself, along with Asn-1056, Arg-1057, and Tyr-1058, show peak shifts in a fast exchange regime indicative of smaller conformational changes. Although ITC analysis indicates Lys-1042 and Arg-1051 are essential for binding, the residues surrounding these basic amino acids undergo a relatively larger change in local chemical environment compared with Lys-1042 itself (Fig. 7, a and b). Similarly, on the acidic face, residues that drive binding and the amino acids surrounding them are affected upon protein-protein interaction (Fig. 7, c and d). For example, Asp-1085, Asp-1092, Leu-1099, Gly-1101, Asp-1102, Asp-1103, and Trp-1105 all display slow exchange (Fig. 7, c and d). Mutation of many of these residues, such as L1099A, D1102A, D1103A, and D1092A, all negatively affected the binding affinity to varying degrees in ITC experiments. A number of surrounding residues also show fast exchange upon binding: Lys-1087, Tyr-1090, Val-1100, Glu-1106, Gln-1113, Lys-1116, and Leu-1118 (Fig. 7, c and d). Thus, the chemical shift changes at these locations correspond to crystallographic and thermodynamic analysis of ARF7 PB1 domain interaction. These NMR data are not only consistent with ITC data, but also provide additional information on the PB1 domain interaction interface that suggests the importance of the neighboring residues to the overall binding.
DISCUSSION
Recent structural studies begin to shed light on the interaction of ARF and Aux/IAA through their PB1 domains to modulate auxin responses (13–15). To date, structural studies establish that the C-terminal III/IV sequence motifs of ARF and Aux/IAA proteins adopt a PB1 domain fold that allows for versatile interactions (i.e. ARF-ARF, Aux/IAA-Aux-IAA, and ARF-Aux/IAA). Moreover, the oriented basic to acidic face interaction of these type I/II PB1 domains has led to a model for protein multimerization in the classic auxin signaling model (13). Here, we use a combination of ITC and NMR to analyze the energetics and determinants of protein-protein interaction using the ARF7 PB1 domain. Our results indicate that the ARF and Aux/IAA use a core set of conserved residues in the PB1 domain to allow for a two-pronged interaction that drives association of these proteins in auxin responses that control plant growth and development.
Thermodynamic analyses of the ARF7 PB1 domain interaction show compensatory enthalpic and entropic changes generally maintain binding energy across a range of temperatures (Fig. 1). As suggested by structural studies showing the oriented positioning of the basic (lysine) and acidic (OPCA) faces of ARF PB1 domains (13–15), the observed changes in enthalpy, estimated heat capacity of the interaction, and the strong salt dependence of binding are consistent with electrostatic forces as the major driving force in the association of the PB1 domains (30, 31). Structural studies on ARF5, ARF7, and IAA17 PB1 domains also suggest interactions between the invariant lysine and the main cluster of acidic residues in the OPCA motif as the basis for this association (13–15); however, closer examination of protein-protein interface residues reveals a second interaction hot spot focused on a key arginine and the distal cluster of acidic residues.
Across the interaction interface of the ARF7 PB1 domain, 9 of 27 contact residues form either ionic or hydrogen bond interactions (Fig. 3a). Site-directed mutagenesis and ITC analysis of the ARF7PB1 interaction defines contributions of residues from each side of the interface and suggests a conserved two-pronged model for protein binding (Table 3 and Figs. 3, b–e). The first prong consists of the canonical lysine (Lys-1042) to the first cluster of acidic residues, e.g. Asp-1092, Glu-1094, and Asp-1096-in the OPCA motif observed in all PB1 domains (16). In the ARF7PB1 interaction, the invariant lysine is critical, and the energetic contribution of Asp-1096 dominates the main OPCA cluster, with Asp-1092 providing the least binding energy. The effects of mutations to Arg-1051 on the basic face and Asp-1102 and Asp-1103 on the OPCA face of ARF7PB1 reveal a previously undescribed role for these amino acids in providing a second prong that contributes to electrostatic interactions. Mutations of these residues led to either a loss of protein interaction (R1051A, D1102N) or a major disruption of binding (D1102A, D1103A, D1103N). The conservation of residues in both prongs expands the core recognition features for ARF PB1 domain interaction beyond the invariant lysine and main OPCA cluster.
Mutagenesis of interface residues in ARF7PB1 defines interaction hot spots for this domain. As described for multiple protein-protein interaction hot spots (31, 34, 35), the ARF PB1 domain interface contains extensive contact residues but only a handful of energetically significant amino acids required for binding. For example, most mutations in residues outside the two electrostatic prongs (Thr-1039, Gly-1050, Ser-1052, Ile-1097, Leu-1099, and Glu-1107) result in only modest changes in binding affinity (Table 3 and Figs. 3, b–e). Trp-1105 appears to have a structural role and not a direct interaction contribution. Interestingly, mutation of Thr-1039 leads to a 200-fold decrease in binding affinity and altered energetic contributions from enthalpy and entropy, which suggests localized structural changes (36). The conservation of the residue in this position as either a threonine or serine in the Arabidopsis ARF PB1 domains (Figs. 3c and 4) is intriguing because this position is a putative phosphorylation site. Recent work suggests that phosphorylation of the basic face of PB1 domains may be a means of regulating protein-protein interaction by disrupting electrostatic complementarity (37). Because of the critical nature of Thr-1039 for ARF7PB1 interaction, modification of this residue may limit multimerization and/or regulate protein-protein interaction in planta.
Although x-ray crystal structures provide a static model for ARF7PB1 interaction, NMR analysis complements the thermodynamic and mutagenesis studies by providing insight into the localized structural changes that occur during PB1 domain association (Fig. 7). Interaction of ARF7PB1 requires two key positively charged residues on the basic face (i.e. Lys-1042 and Arg-1051) that anchor two distinct clusters of acidic residues (i.e. Asp-1092/Glu-1094/Asp-1096 and Asp-1102/Asp-1103).
Chemical shift data for ARF7PB1 show interactions across both faces of the domain; however, the acidic face displays a greater degree of intermediate exchange (Fig. 7a, yellow). Mapping of the chemical shift data to the three-dimensional structure of ARF7PB1 indicates that the basic face is conformationally less labile than the acidic face. These data also indicate localized structural changes are concentrated around the loop regions between β3-β4 and β4-α2 on the acidic face, whereas the basic face binding contributors are generally localized around Lys-1042 and Arg-1051 on β1, β2, and the intermediate loop region. Taken together, these results suggest that the basic face of ARF7 PB1 provides nucleated positive charge required for interaction. The acidic face of ARF7 PB1 provides a larger patch of negative charges that stabilize the interaction around the two main positive charges on the basic face. Furthermore, the basic face of the protein is much more structured, whereas the predicted interaction residues on the acidic face occur more on loops and regions that were disordered in the x-ray structure (13). NMR data also suggest considerable conformational changes on both the acidic and basic faces experienced by the residues other than the residues identified as essential by ITC experiments. This behavior may suggest local packing interactions that prevail on each subunit upon initial interaction are likely to be important in overall complex formation and stabilization. Overall, these data are consistent with a mode of binding where the interaction of the structured basic face is paired with the organization of the less structured acidic face to stabilize and lock in the two-pronged interaction.
Amino acid sequence homology among the 22 Arabidopsis ARF proteins indicates that the two-prong interaction model of ARF7PB1 and the energetic contribution of key interface residues are also likely conserved (Figs. 5 and 8). Extension of this sequence analysis to include the 34 Arabidopsis Aux/IAA shows that residues forming the two-pronged electrostatic hot spot are also maintained in these proteins (Figs. 4 and 8). Comparison of the basic and acidic faces of the ARF7 (13) and IAA17 (15) PB1 domains reveals core recognition features and regions around the electrostatic prongs that co-vary between the ARF and Aux/IAA.
FIGURE 8.
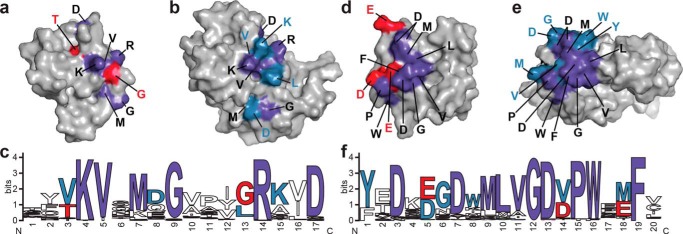
Core and family-specific PB1 domain interface residues. The x-ray crystal structure of ARF7PB1 (Ref. 13; PDB code 4NJ6, chain A) and the NMR solution structure of IAA17 (Ref. 15; PDB 2MUK) are represented as space-filling models with highlighted areas of sequence conservation. Residues conserved in ARF (red), Aux/IAA (blue), and in both ARF and Aux/IAA (purple) are shaded as indicated. a, surface view of the ARF7PB1 basic face. b, surface view of the IAA17PB1 basic face. c, amino acid sequence logo showing conservation of residues on the ARF and Aux/IAA PB1 domain basic faces. d, surface view of the ARF7PB1 acidic face. e, surface-view of the IAA17PB1 acidic face. f, amino acid sequence logo showing conservation of residues on the ARF and Aux/IAA PB1 domain acidic faces. Panels e and f were generated using WebLogo (39).
On the basic face of both the ARF (Fig. 8a) and Aux/IAA (Fig. 8b) families, the critical lysine and arginine of the basic face are invariant in 50 of the 51 Arabidopsis ARF and Aux/IAA proteins (Fig. 8c). Only IAA33 substitutes a threonine and a glutamine, respectively, for these basic residues (Fig. 4). Other surface contact residues highly conserved in the basic faces of the ARF and Aux/IAA PB1 domains correspond to Val-1043, a methionine corresponding to Lys-1045, Gly-1047, and Asp-1054. Co-variation occurs with the residue corresponding to Thr-1039 of ARF7PB1, which is generally either a threonine or serine in ARFs, but is variable in the Aux/IAA family. Similarly, the ARF PB1 domains favor a glycine at position 1050, which is either a glycine or leucine in the Aux/IAA family, and a threonine at position 1041, which is usually a valine in Aux/IAA proteins. The OPCA motif is found in all ARF and Aux/IAA (Fig. 8f) proteins with either glutamate or aspartate occurring in the second acidic position. In the aspartate motif of the second prong, the first aspartate is nearly invariant with an acidic residue in the second position found in ARF PB1 domains; this position is generally a valine in the Aux/IAA PB1 domains (Fig. 8f). Residues corresponding to Leu-1098, Leu-1099, Val-1100, Gly-1101, Pro-1104, Trp-1105, and Phe-1108 are conserved across the ARF and Aux/IAA families (Fig. 8, d–f). In ARFs, position 1097 is variable but is a highly conserved tryptophan in Aux/IAA proteins. The residue corresponding to Glu-1107 is maintained in ARFs but is a methionine in most Aux/IAAs.
The structurally conserved two-pronged recognition feature in the ARF and Aux/IAA PB1 domains appears to provide the electrostatic energy that allows for the biologically versatile interaction of ARF-ARF, Aux/IAA-Aux/IAA, and ARF-Aux/IAA in the auxin response of plants. Surrounding this core set of interactions, residues that co-vary between ARF and Aux/IAA proteins likely provide additional complementary contacts that further tune protein interaction. For example, recent studies indicate that binding of the ARF5 and IAA17 PB1 domains (Kd = 0.07 μm) is 8-fold more favorable than self-interaction of the ARF5 PB1 domains (Kd = 0.87 μm) and 90-fold tighter than interaction between IAA17 PB1 domains (Kd = 6.6 μm) (16). Modest changes in the contact surfaces of the ARF and Aux/IAA PB1 domains could easily lead to favorable energetics of interaction between cognate pairs of proteins (38). Distinguishing between structural elements that drive PB1 domain recognition, contact regions that co-vary between ARF and Aux/IAA PB1 domains, and additional changes that fine-tune ARF and Aux/IAA interactions are a critical step toward understanding the complicated network of ARF and Aux/IAA that regulate multiple components in auxin-regulated plant growth.
The systematic analysis of key residues in the ARF7 PB1 interface described herein reveals a new feature missed in the structural studies and leads to a fuller understanding of this molecular and cellular process. With regard to the larger biological system of auxin responses, how specific ARF and Aux/IAA proteins interact remains to be understood. The combined structural, functional, and sequence analysis presented here establishes the core determinants of protein-protein interaction in this system but also points to residues that co-vary and may contribute to interaction specificity in the ARF and Aux/IAA network, which leads to biological outcomes. Ultimately, the new molecular insights inform future inquiry into this system using in vitro and in vivo approaches.
Acknowledgments
This work made use of the National Magnetic Resonance Facility at Madison, which is supported by National Institutes of Health Grant P41GM103399 (NIGMS; old number: P41RR002301). Equipment was purchased with funds from the University of Wisconsin-Madison, National Institutes of Health Grants P41GM103399, S10RR02781, S10RR08438, S10RR023438, S10RR025062, and S10RR029220, National Science Foundation Grants DMB-8415048, OIA-9977486, and BIR-9214394, and by the United States Department of Agriculture.
This work was supported, in whole or in part, by National Institutes of Health Grant R00 GM089987-03 (to L. C. S.). This work was also supported by National Science Foundation (NSF) Graduate Research Fellowship Program 2011101911 (to D. A. K.), the USDA-NIFA (United States Department of Agriculture-National Institute of Food and Agriculture) Fellowship Program (MOW-2010-05240 (to C. S. W.) and MOW-2014-01877 (to D. A. K.)), and NSF Grant MCB-1157771 (to J. M. J.).
- Aux/IAA
- auxin/indole 3-acetic acid inducible
- ARF
- auxin response factor
- PB1
- Phox/Bem1p
- ITC
- isothermal titration calorimetry
- HSQC
- heteronuclear single quantum correlation
- MOPSO
- 3-morpholino-2-hydroxypropanesulfonic acid.
REFERENCES
- 1. Enders T. A., Strader L. C. (2015) Auxin activity: past, present, and future. Am. J. Bot. 102, 180–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Korasick D. A., Enders T. A., Strader L. C. (2013) Auxin biosynthesis and storage forms. J. Exp. Bot. 64, 2541–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lavenus J., Goh T., Roberts I., Guyomarc'h S., Lucas M., De Smet I., Fukaki H., Beeckman T., Bennett M., Laplaze L. (2013) Lateral root development in Arabidopsis: fifty shades of auxin. Trends Plant Sci. 18, 450–458 [DOI] [PubMed] [Google Scholar]
- 4. Zhao Y. (2010) Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 61, 49–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vanneste S., Friml J. (2009) Auxin: a trigger for change in plant development. Cell 136, 1005–1016 [DOI] [PubMed] [Google Scholar]
- 6. Teale W. D., Paponov I. A., Palme K. (2006) Auxin in action: signalling, transport. and the control of plant growth and development. Nat. Rev. Mol. Cell Biol. 7, 847–859 [DOI] [PubMed] [Google Scholar]
- 7. Wang R., Estelle M. (2014) Diversity and specificity: auxin perception and signaling through the TIR1/AFB pathway. Curr. Opin. Plant Biol. 21, 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim J., Harter K., Theologis A. (1997) Protein-protein interactions among the Aux/IAA proteins. Proc. Natl. Acad. Sci. U.S.A. 94, 11786–11791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ulmasov T., Murfett J., Hagen G., Guilfoyle T. J. (1997) Aux/IAA proteins repress expression of reporter genes containing natural and highly active synthetic auxin response elements. Plant Cell 9, 1963–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tan X., Calderon-Villalobos L. I., Sharon M., Zheng C., Robinson C. V., Estelle M., Zheng N. (2007) Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645 [DOI] [PubMed] [Google Scholar]
- 11. Gray W. M., Kepinski S., Rouse D., Leyser O., Estelle M. (2001) Auxin regulates SCFTIR1-dependent degradation of Aux/IAA proteins. Nature 414, 271–276 [DOI] [PubMed] [Google Scholar]
- 12. Boer D. R., Freire-Rios A., van den Berg W. A., Saaki T., Manfield I. W., Kepinski S., López-Vidrieo I., Franco-Zorrilla J. M., de Vries S. C., Solano R., Weijers D., Coll M. (2014) Structural basis for DNA binding specificity by the auxin-dependent ARF transcription factors. Cell 156, 577–589 [DOI] [PubMed] [Google Scholar]
- 13. Korasick D. A., Westfall C. S., Lee S. G., Nanao M. H., Dumas R., Hagen G., Guilfoyle T. J., Jez J. M., Strader L. C. (2014) Molecular basis for auxin response factor protein interaction and the control of auxin response repression. Proc. Natl. Acad. Sci. U.S.A. 111, 5427–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nanao M. H., Vinos-Poyo T., Brunoud G., Thévenon E., Mazzoleni M., Mast D., Lainé S., Wang S., Hagen G., Li H., Guilfoyle T. J., Parcy F., Vernoux T., Dumas R. (2014) Structural basis for oligomerization of auxin transcriptional regulators. Nat. Commun. 5, 3617. [DOI] [PubMed] [Google Scholar]
- 15. Han M., Park Y., Kim I., Kim E. H., Yu T. K., Rhee S., Suh J. Y. (2014) Structural basis for the auxin-induced transcriptional regulation by Aux/IAA17. Proc. Natl. Acad. Sci. U.S.A. 111, 18613–18618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sumimoto H., Kamakura S., Ito T. (2007) Structure and function of the PB1 domain, a protein interaction module conserved in animals, fungi, amoebas, and plants. Sci. STKE 2007, re6. [DOI] [PubMed] [Google Scholar]
- 17. Guilfoyle T. J., Hagen G. (2012) Getting a grasp on domain III/IV responsible for auxin response factor-IAA protein interactions. Plant Sci. 190, 82–88 [DOI] [PubMed] [Google Scholar]
- 18. Guilfoyle T. J. (2015) The PB1 domain in auxin response factor and Aux/IAA proteins: a versatile protein interaction module in the auxin response. Plant Cell 27, 33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis A. L., Keeler J., Laue E. D., Moskau D. (1992) Experiments for recording pure-absorption heteronuclear correlation spectra using pulsed field gradients. J. Magn. Reson. 98, 207–216 [Google Scholar]
- 20. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 21. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 22. Kay L. E., Keifer P., Saarinen T. (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114, 10663–10665 [Google Scholar]
- 23. Grzesiek S., Bax A. (1992) An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J. Magn. Reson. 99, 201–207 [Google Scholar]
- 24. Grzesiek S., Bax A. (1992) Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114, 6291–6293 [Google Scholar]
- 25. Schanda P., Van Melckebeke H., Brutscher B. (2006) Speeding up three-dimensional protein NMR experiments to a few minutes. J. Am. Chem. Soc. 128, 9042–9043 [DOI] [PubMed] [Google Scholar]
- 26. Lescop E., Schanda P., Brutscher B. (2007) A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson. 187, 163–169 [DOI] [PubMed] [Google Scholar]
- 27. Hyberts S. G., Milbradt A. G., Wagner A. B., Arthanari H., Wagner G. (2012) Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 52, 315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee W., Tonelli M., Markley J. L. (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bahrami A., Assadi A. H., Markley J. L., Eghbalnia H. R. (2009) Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput. Biol. 5, e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prabhu N. V., Sharp K. A. (2005) Heat capacity in proteins. Annu. Rev. Phys. Chem. 56, 521–548 [DOI] [PubMed] [Google Scholar]
- 31. Stites W. E. (1997) Protein-protein interactions: interface structure, binding thermodynamics, and mutational analysis. Chem. Rev. 97, 1233–1250 [DOI] [PubMed] [Google Scholar]
- 32. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 33. Ni J., Zhu Z., Wang G., Shen Y., Zhang Y., Wu P. (2014) Intragenic suppressor of Osiaa23 revealed a conserved tryptophan residue crucial for protein-protein interactions. PLoS ONE 9, e85358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wells J. A., de Vos A. M. (1996) Hematopoietic receptor complexes. Annu. Rev. Biochem. 65, 609–634 [DOI] [PubMed] [Google Scholar]
- 35. Bogan A. A., Thorn K. S. (1998) Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280, 1–9 [DOI] [PubMed] [Google Scholar]
- 36. Pearce K. H., Jr., Ultsch M. H., Kelley R. F., de Vos A. M., Wells J. A. (1996) Structural and mutational analysis of affinity-inert contact residues at the growth hormone-receptor interface. Biochemistry 35, 10300–10307 [DOI] [PubMed] [Google Scholar]
- 37. Christian F., Krause E., Houslay M. D., Baillie G. S. (2014) PKA phosphorylation of p62/SQSTM1 regulates PB1 domain interaction partner binding. Biochim. Biophys. Acta 1843, 2765–2774 [DOI] [PubMed] [Google Scholar]
- 38. Piya S., Shrestha S. K., Binder B., Stewart C. N., Jr., Hewezi T. (2014) Protein-protein interaction and gene co-expression maps of ARFs and Aux/IAAs in Arabidopsis. Front. Plant Sci. 5, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]