

Background: Protein interactions play an important role in the pyrroloquinoline quinone biosynthetic pathway.
Results: PqqD binds PqqA, the peptide substrate, with a submicromolar KD, and they form a ternary complex with PqqE.
Conclusion: PqqD is a peptide chaperone that interacts with PqqE.
Significance: Orthologous PqqDs may play similar roles in other peptide modification pathways.
Keywords: peptide biosynthesis, peptide interaction, protein complex, quinone, small-angle x-ray scattering (SAXS), PqqD, PqqE
Abstract
Pyrroloquinoline quinone (PQQ) is a product of a ribosomally synthesized and post-translationally modified pathway consisting of five conserved genes, pqqA-E. PqqE is a radical S-adenosylmethionine (RS) protein with a C-terminal SPASM domain, and is proposed to catalyze the formation of a carbon-carbon bond between the glutamate and tyrosine side chains of the peptide substrate PqqA. PqqD is a 10-kDa protein with an unknown function, but is essential for PQQ production. Recently, in Klebsiella pneumoniae (Kp), PqqD and PqqE were shown to interact; however, the stoichiometry and KD were not obtained. Here, we show that the PqqE and PqqD interaction transcends species, also occurring in Methylobacterium extorquens AM1 (Me). The stoichiometry of the MePqqD and MePqqE interaction is 1:1 and the KD, determined by surface plasmon resonance spectroscopy (SPR), was found to be ∼12 μm. Moreover, using SPR and isothermal calorimetry techniques, we establish for the first time that MePqqD binds MePqqA tightly (KD ∼200 nm). The formation of a ternary MePqqA-D-E complex was captured by native mass spectrometry and the KD for the MePqqAD-MePqqE interaction was found to be ∼5 μm. Finally, using a bioinformatic analysis, we found that PqqD orthologues are associated with the RS-SPASM family of proteins (subtilosin, pyrroloquinoline quinone, anaerobic sulfatase maturating enzyme, and mycofactocin), all of which modify either peptides or proteins. In conclusion, we propose that PqqD is a novel peptide chaperone and that PqqD orthologues may play a similar role in peptide modification pathways that use an RS-SPASM protein.
Introduction
Ribosomally synthesized and post-translationally modified peptides constitute a major class of natural products consisting of bioactive compounds such as antibiotics (1–3), quorum sensing molecules (4, 5), metal chelators (6), and redox cofactors (7). Certain ribosomally derived products rely on radical S-adenosylmethionine enzymes (herein referred to as RS proteins) to catalyze the formation of α-carbon thioether bonds or carbon-carbon bonds (8, 9). In the subtilosin and the pyrroloquinoline quinone (PQQ)2 pathways, the RS protein also contains a C-terminal extension referred to as a SPASM domain (subtilosin, PQQ, anaerobic sulfatase maturating enzyme, and mycofactocin). RS-SPASM proteins are typically found in peptide or protein modification pathways (10, 11). In some cases, the RS-SPASM protein is fused to a ∼90 amino acid protein (e.g. AlbA) or, alternatively, the RS-SPASM gene is located adjacent to a gene encoding for a ∼90 amino acid protein (e.g. PqqE). The function of the small protein and its relationship to RS-SPASM proteins remains unknown and, its role in the PQQ biosynthetic pathway is the focus of this paper.
PQQ (4,5-dihydro-4,5-dioxo-1H-pyrrolo[2,3-f] quinolone-2,7,9-tricarboxylic acid) is a redox active tricyclic o-quinone that serves as a cofactor, predominantly, for prokaryotic alcohol and sugar dehydrogenases (12, 13). Biosynthesis of PQQ relies on five strictly conserved gene products, pqqABCDE (8, 9, 14–16). In all cases, pqqA encodes for a short peptide, typically 20–30 amino acids in length, containing a conserved glutamate and tyrosine that serve as the backbone in PQQ biogenesis (Fig. 1). The glutamate and tyrosine undergo post-translational modifications to form the intermediate 3a-(2-amino-2-carboxyethyl)-4,5-dioxo-4,5,6,7,8,9-hexahydroquinoline-7,9-dicarboxylic acid (AHQQ). Of the pqq gene products, PqqC is the most characterized and has been shown to catalyze the eight-electron oxidation and ring cyclization of AHQQ to form PQQ (17, 18). The functions of the remaining pqq gene products remain elusive, although, a recent bioinformatic analysis proposed that PqqB, a metallo-β-lactamase-fold member, oxidizes some form of intermediate that leads to the formation of AHQQ (15). PqqE is an RS-SPASM protein and has been shown to cleave S-adenosylmethionine to methionine and 5′-deoxyadenosine in an uncoupled reaction (19). PqqE is proposed to catalyze the C-C bond formation between the glutamate and tyrosine; however, efforts to couple S-adenosylmethionine cleavage to the cross-linking have thus far been unsuccessful (19).
FIGURE 1.

Abbreviated PQQ biosynthesis. Formation of PQQ from conserved PqqA glutamate and tyrosine residues.
PqqD is a small protein (∼90 amino acids) with no detectable cofactor and few strictly conserved residues. PqqD can be found as an individual protein, as a C-terminal gene fusion to PqqC (e.g. Methylobacterium extorquens AM1 PqqCD) or as a N-terminal gene fusion to PqqE (e.g. Methylocystis sp. SC2 PqqDE). A crystal structure has been solved for Xanthomonas campestris PqqD (XcPqqD, PDB number 3G2B). Analysis of the structure indicates that XcPqqD forms a saddle-like dimer under crystallization conditions (Fig. 2) but little functional information could be derived (20). A bioinformatic analysis on PqqD could not identify functionally relevant residues (15), adding to the ambiguity of the role that PqqD plays in PQQ biogenesis. However, recent electron paramagnetic resonance (EPR) and hydrogen-deuterium exchange mass spectrometry experiments have shown that Klebsiella pneumonia PqqE (KpPqqE) and PqqD (KpPqqD) interact, suggesting that the formation of a macromolecular complex may lead to function of one or more enzymes (21).
FIGURE 2.
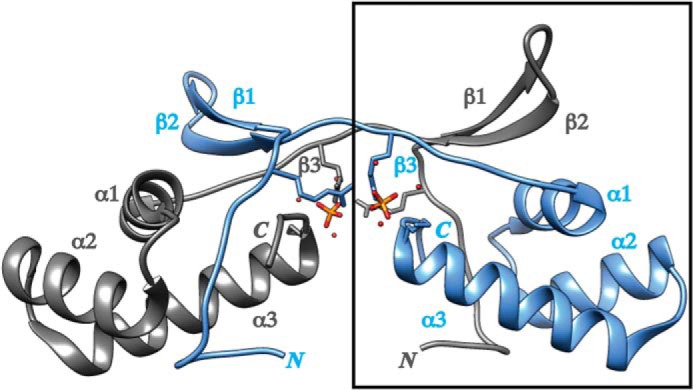
Structure of XcPqqD. The XcPqqD (PDB code 3G2B) dimer is depicted in ribbon form with each monomer differentiated by color. Each elongated monomer consists of a three α-helix bundle and a β-turn-β domain connected by a β-strand. Phosphate ligands from crystallization buffer are shown in orange. The box indicates the portion of the crystal structure used to model a compact XcPqqD monomer.
Although the formation of a KpPqqD-KpPqqE complex has been reported (19), a detailed biophysical characterization of the interaction has not yet been completed. To learn more about the interaction, we determined the stoichiometry of the KpPqqD-KpPqqE complex using native mass spectrometry experiments. We expanded our investigations into M.extorquens AM1 (Me) and determined the stoichiometry and dissociation constant (KD) for the MePqqD-MePqqE interaction. We also determined, through various biophysical techniques, that MePqqD binds the peptide MePqqA with a submicromolar KD independent of the presence of MePqqE. Additionally, we were able to capture the MePqqADE ternary complex by mass spectrometry and establish the KD for the MePqqAD-MePqqE interaction. Last, we use small angle x-ray scattering to demonstrate that the KpPqqD adopts a different oligomerization and conformational state than was found for the XcPqqD crystal.
EXPERIMENTAL PROCEDURES
Materials
All restriction enzymes and T4 DNA ligase were purchased from New England BioLabs (Ipswich, MA). Polymerase was purchased from Agilent (Santa Clara, CA). Oligonucleotides were purchased from Eurofins (Huntsville, AL). DNA sequencing was conducted by the University of California DNA Sequencing Facility (Berkeley, CA). The peptide MePqqA (Uniprot number Q49148) was synthesized and purified to >80% purity by CPC Scientific (Emeryville, CA) and used without further purification. The peptide sequence, KWAAPIVSEISVGMEVTYSESAEIDTFN, incorporated Ser-11 in place of the wild-type Cys to eliminate dimer formation.
Preparation of Recombinant PqqB
Wild-type PqqB-His6 (UniProt number Q49149) was prepared by using a PCR-based strategy. Accordingly, the gene was amplified by PCR using genomic DNA from M. extorquens AM1 (ATCC, Manassas, VA) as template, commercial oligonucleotides as primers, and Pfu Turbo as the polymerase. The PCR product was digested by the restriction enzymes NdeI and XhoI and then purified by agarose gel electrophoresis. The gene was ligated to an NdeI- and XhoI-digested pET24b vector (EMD Millipore) using T4 DNA ligase. The cloned gene was verified by DNA sequencing and then used to transform competent Escherichia coli BL21(DE3) cells for gene expression. The PqqB/pET24b-transformed E. coli BL21(DE3) cells were grown aerobically at 37 °C in LB media containing 50 μg/ml of kanamycin. Production of PqqB-His6 was induced by the addition of 1 mm isopropyl β-d-galactopyranoside and 30 mg/liter of ZnCl2 once the cell density had reached A600 ∼0.6. Following a 12-h induction period at 19 °C, the cells were harvested by centrifugation at 6,500 rpm for 10 min. The cells were suspended in five times the mass of cell paste of 50 mm Tris buffer (pH 7.9), 50 mm imidazole, and 200 mm NaCl (lysis buffer). The cells were lysed by sonication and the lysate was centrifuged at 20,000 rpm for 15 min. The supernatant was loaded onto a 5-ml HisTrap FF column (GE Healthcare) and the column was washed at 4 °C with lysis buffer to remove non-tagged protein and then with 50 mm Tris (pH 7.9), 300 mm imidazole, and 200 mm NaCl (elution buffer) to elute the tagged protein. The desired fractions were combined, concentrated, and buffer exchanged over PD-10 columns (GE Healthcare) equilibrated with 50 mm Tris (pH 7.9), 150 mm NaCl. Fractions containing PqqB-His6 were pooled and concentrated prior to being flash frozen. Column fractions were monitored by solution absorbance at 280 nm and by carrying out SDS-PAGE analysis. Yield was 60 mg/liter of culture.
Preparation of Recombinant PqqCD
Wild-type MePqqCD (UniProt number Q49150) was cloned using procedures similar to PqqB. The amplified PCR products were digested with NdeI and XhoI and purified by agarose electrophoresis. The restricted digested PCR product was ligated to an NdeI- and XhoI-digested pET28a vector (EMD Millipore) using T4 DNA ligase. The cloned gene was sequence verified and used to transform E. coli BL21(DE3) for gene expression. The His-tagged encoded MePqqCD/pET28a-transformed E. coli BL21(DE3) cells were grown aerobically at 37 °C in LB media containing 50 μg/ml of kanamycin. Production and purification of His6-MePqqCD followed the same procedures listed for MePqqB-His6 except that ZnCl2 was not added upon induction and 1 mm TCEP was added to all buffers as a precaution. The desired fractions were combined, concentrated, and buffer exchanged over PD-10 columns equilibrated with 50 mm Tris (pH 7.9), 150 mm NaCl, and 1 mm TCEP prior to being flash frozen. The homogeneity was confirmed by SDS-PAGE analysis. Yield for His6-MePqqCD was 24 mg/liter of culture.
Preparation of Recombinant PqqD
For experiments using only native MePqqD protein (amino acids 280–372), a MePqqD/pET24b was constructed using the NdeI and XhoI restriction sites and expressed as described for His6-MePqqCD. For native MePqqD, cells were suspended in five times the mass of cell paste of 50 mm HEPES buffer (pH 6.8). The cells were lysed by sonication and clarified by centrifugation at 20,000 rpm for 15 min. The clarified lysate was loaded onto a pre-equilibrated 20-ml DEAE-Sepharose FF column (GE Healthcare), washed with lysis buffer, and eluted with a 0–0.5 m NaCl gradient in 50 mm HEPES buffer (pH 6.8) over 25 column volumes. MePqqD eluted between 100 and 200 mm NaCl. The MePqqD containing fractions were pooled, equilibrated with 17% saturated NH4SO4, and loaded onto a pre-equilibrated HiPrep 16/10 Phenyl FF column (GE Healthcare). The column was washed with 50 mm HEPES (pH 6.8) and 17% saturated NH4SO4 and MePqqD was eluted with a 17–0% NH4SO4 gradient over 15 column volumes. MePqqD eluted between 9 and 12% saturated NH4SO4. MePqqD containing fractions were pooled, concentrated in a 10-kDa MWCO spin column (Millipore), and further purified over a 26/60 Sephacyl S-200 size exclusion column equilibrated with 50 mm HEPES (pH 6.8) and 150 mm NaCl. Fractions containing MePqqD were pooled and concentrated prior to being flash frozen. Column fractions were monitored by solution absorbance at 280 nm and by SDS-PAGE analysis. Yield was 6 mg/liter. The His6-MePqqD protein was prepared from a MePqqD/pET28a construct following the described procedures for the His6-MePqqCD protein. Yield for His6-MePqqD was 8 mg/liter of culture.
Preparation of Recombinant PqqE
MePqqE (UniProt number P71517) was cloned into pET28a using the NdeI and XhoI restriction sites from genomic DNA. The cloned gene was sequence verified and used to transform E. coli BL21(DE3) harboring the suf operon plasmid pPH149, for gene expression. The MePqqE/pET28a and pPH149-transformed E. coli BL21(DE3) cells were grown aerobically at 37 °C in TB media containing 50 μg/ml of kanamycin and 35 μg/ml of chloramphenicol. Once the cell density reached A600 ∼0.6, 5 mm fumarate and 50 μm Fe(III) citrate were added and the flasks were stoppered to transition from aerobic to anaerobic conditions. After 30 min of anaerobic growth at 37 °C, production of His6-MePqqE was induced with 0.4 mm isopropyl β-d-galactopyranoside. Following a 12-h induction period at 19 °C, the cells were harvested by centrifugation at 6,500 rpm for 10 min and frozen. The cells were transferred to an anaerobic chamber suspended in five times the mass of cell paste of degassed 50 mm Tris buffer (pH 7.9), 50 mm imidazole, 1 mm TCEP, and 200 mm NaCl (lysis buffer). BugBuster (Novagen) supplemented with Benzonase (Novagen) was used to lyse the cells following the manufacturer's guidelines. The lysate was transferred to sealed tubes and centrifuged at 20,000 rpm for 15 min. The sealed tubes containing clarified lysate were transferred to the anaerobic chamber and the supernatant was loaded onto a 5-ml HisTrap FF column (GE Healthcare) by a peristaltic pump. The column was washed at 22 °C with lysis buffer to remove non-tagged protein and then with degassed 50 mm Tris (pH 7.9), 300 mm imidazole, 1 mm TCEP, and 200 mm NaCl (elution buffer) to elute the tagged protein. The desired fractions were combined, concentrated, and buffer exchanged over PD-10 columns equilibrated with degassed 50 mm Tris (pH 7.9), 100 mm NaCl, 5% glycerol, and 1 mm TCEP prior to being flash frozen in sealed vials. The homogeneity was confirmed by SDS-PAGE analysis. Yield for His6-MePqqE was 18 mg/liter of culture. For native MePqqE, thrombin was used to cleave the N-terminal His6 tag, which was removed by passage through a 1-ml nickel-nitrilotriacetic acid column. The C-terminally His-tagged MePqqE-His6 was purified as described above from E. coli BL21(DE3) harboring the plasmid pPH149 and the plasmid MePqqE/pET24b where MePqqE was cloned into the NdeI and XhoI restriction sites of pET24b. Yield for MePqqE-His6 was 18 mg/liters of culture.
Construction of the MePqqDE Fusion
A MePqqDE fusion was constructed through a PCR-based strategy using commercial oligonucleotides as primers and MePqqE/pET28a and MePqqD/pET24b as templates. The MePqqD gene was amplified with a 5′-3′ oligonucleotide encoding a NdeI restriction site and a 3′-5′ oligonucleotide encoding for a (GGGGS)2 repeat and a BamHI restriction site. The MePqqE gene was amplified with a 5′-3′ oligonucleotide encoding for a (GGGGS) linker and a BamHI restriction site and the original 3′-5′ oligonucleotide encoding a XhoI restriction site. The PCR products were digested with the respective enzymes and a triple ligation strategy was used to ligate the products to each other and to pET28a digested with NdeI and XhoI. The purification of His6-MePqqDE was identical to the procedures listed for His6-MePqqE. Yield for His6-MePqqDE was 23 mg/liter of culture.
Analytical Size Exclusion Chromatography (SEC)
Analytical SEC was performed on a Superdex 75 10/300 GL (GE Healthcare) outfitted to an AKTA FPLC (GE Healthcare) with a flow rate of 0.5 ml/min at 4 °C. The mobile phase was 50 mm HEPES (pH 7.5) and 150 mm NaCl buffer and 100 μg of protein was injected for each run. The elution volume was monitored by a change in absorbance at 280 nm. A calibration curve was constructed from aprotonin (6.5 kDa, eluted at 15.1 ml), ribonuclease A (13.7 kDa, eluted at 13.5 ml), myoglobin equine from skeletal muscle (17 kDa, eluted at 12.3 ml), carbonic anhydrase (29 kDa, eluted at 12.3 ml), and albumin (66 kDa, eluted at 13.5 ml) (Sigma). The dead volume was determined by blue dextran (Sigma).
Isothermal Titration Calorimetry Analysis
Recombinant MePqqD, His6-MePqqCD, and His6-MePqqDE were buffer exchanged into PBS buffer (Corning) over a PD-10 (GE Healthcare) column. MePqqA was dissolved into PBS to make a 0.5 mm stock solution. Calorimetric analysis was performed in triplicate on a MicroCal AutoCal ITC200 (GE Healthcare). The reference cell was filled with water. In a typical experiment, the sample cell was loaded with a 20 μm solution of protein. The cell solution was titrated with an initial 1.2-μl injection followed by 12 aliquots of 3.4 μl of a 200 μm MePqqA solution. The obtained heat signals from the ITC were integrated using the Origin software package supplied by GE Healthcare to obtain the binding isotherms. The data, except from the first injection, were fitted to a single site-binding model by regression routines programmed into Origin defined by Equation 1. The expression to calculate the binding constant K from the number of binding sites occupied by the ligand (n, set to 1), change in enthalpy (ΔH), bulk concentration (Xt) of ligand, and bulk concentration (Mt) of macromolecule in a volume Vo is shown in Equation 1.
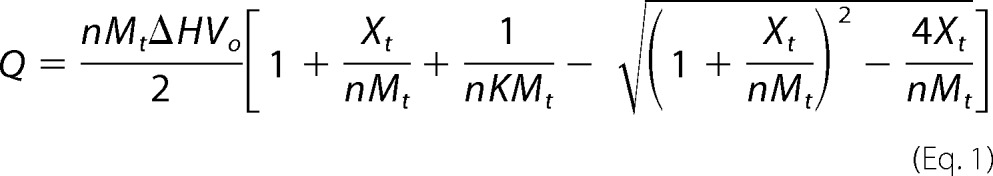 |
Nanoelectrospray Ionization Mass Spectrometry (NanoESI-MS) of Native Proteins
Recombinant His6-KpPqqE, KpPqqD, MePqqE-His6, and MePqqD were buffer exchanged into 25 mm ammonium bicarbonate (pH 7.9) using pre-equilibrated PD-10 columns (GE Healthcare). Solid MePqqA was dissolved directly into the ammonium bicarbonate buffer. Mass spectra of native proteins and complexes were acquired using a quadrupole time-of-flight (Q-TOF) mass spectrometer equipped with a nano-Z-spray nanoESI source (Q-TOF Premier, Waters, Beverly, MA). Acquisition of spectra was performed following previously described procedures (22). Mass spectra of the intact proteins and complexes were processed using MassLynx software (version 4.1, Waters). The KpPqqE and KpPqqD experiments contained 10 μm His6-KpPqqE with or without 40 μm KpPqqD. The MePqqE and MePqqD experiments contained 10 μm MePqqE-His6 with and without 50 μm MePqqD. To examine the ternary complex, experiments contained 10 μm MePqqE-His6, 50 μm MePqqD, and 50 μm MePqqA.
Surface Plasmon Resonance (SPR) Measurements
Binding studies were conducted in triplicate on a Biacore T100 (Biacore, GE Healthcare). His6-tagged proteins were immobilized to a Series NTA chip (Biacore, GE Healthcare) and peptide or purified native PqqE was injected as analyte. Flow cell 1 was not immobilized with protein and served as the reference channel. Ligands and analyte were diluted into HBS-P buffer (GE Healthcare). The His6-tagged proteins were injected at 10 nm and the immobilization level ranged between 100 and 500 response units depending on the analyte. Seven to 9-fold serial dilutions of analyte were injected for 30 s at a flow rate of 30 μl/min followed by a 60-s dissociation phase. After each cycle the surface was regenerated by injecting 300 mm EDTA followed by 500 μm NiCl2 and a 3 mm EDTA wash. Binding affinities were determined from steady state analysis of the maximum steady state response. Final data analysis, plotting and curve fitting were performed by Prism 5 software (Prism).
Small Angle X-ray Scattering (SAXS) Analysis
KpPqqD, used for all SAXS experiments, was frozen and hand delivered to the work site. KpPqqD was prepared as previously described (21) and buffer exchanged into 50 mm Tris (pH 7.9), 100 mm NaCl, 1 mm TCEP, and 2.5% glycerol by SEC. Samples were diluted to 1, 2, 3, 4, and 5 mg/ml at 4 °C. Synchrotron radiation x-ray scattering data were collected using standard procedures on the 4-2 Beamline at the Stanford Synchrotron Radiation Lightsource (SSRL). SAXS data were collected on a MarCCD225 detector at wavelength of 1.3 Å. Fifteen individual 1-s samples were collected at each concentration and buffer scans were collected before each experiment.
Data were integrated, averaged, and buffer subtracted using SasTool (23). PRIMUS was used to calculate a Guinier approximation, where a plot of ln[I(q)] and q2 is linear, as well as to calculate a radius of gyration (Rg) and distribution of distances (23). GNOM was used to generate the pair distribution function |P(r) and the maximum particle distribution (Dmax) (23, 24). DAMMIF and PDB2VOL were used to generate the ab initio shape reconstruction employing the parameters defined by GNOM (25, 26). The predicted SAXS profile for the XcPqqD dimer and monomer (PDB code 3G2B) was generated and fitted to experimental data by FOXS (27). Chimera was used to fit the atomic structure of XcPqqD into the KpPqqD envelope generated by SITUS (26, 28). SAXSMOW was used to determine the relative molecular weight from the SAXS measurement (29).
Construction of the SPASM Domain Sequence Similarity Network
The Interpro SPASM domain designation (IPR023885) was used to generate a sequence similarity network using the EFI-EST tool (30–33). The network was parsed by an E value of 10−45 and the subsequent sequences, with >60% identity, were combined into nodes. The sequence similarity network was visualized and annotated using Cytoscape (32). The gene context of representative node sequences was further analyzed on the EnsemblBacteria website or the NCBI gene website. The gene context of representative sequences for each node was searched within a five gene sliding window on the same DNA strand for a 90–120-amino acid PqqD-like gene (annotated as PqqD or as unknown) and a peptide up to 70 amino acids. The length of the peptide was chosen based on the extreme cases of PqqA peptides with sequence lengths annotated as ∼70 amino acids (e.g. Uniprot numbers I0G208, G7DC35, and A9W3R3).
RESULTS
Analysis of the PqqD Oligomeric Structure from K. pneumoniae
SEC was employed to estimate the molecular weight, and thereby the oligomerization state, for KpPqqD. MePqqD was not analyzed due to the lack of tryptophan in the protein, making detection by absorbance difficult. The SEC elution profile for KpPqqD showed a single peak (Fig. 3A) with an estimated molecular mass of 14.9 kDa (KpPqqD monomer, 10,435 Da) calculated from the standard curve (Fig. 3B). The calculated Mr and the presence of a single elution peak suggested that KpPqqD could be a monomer in solution. Upon finding that the solution oligomerization state for KpPqqD may be different from the dimer found in a crystal structure of the homologous XcPqqD (30 and 44% sequence identity to KpPqqD and MePqqD respectively), we turned to small angle x-ray scattering (SAXS) to explore further the solution structure.
FIGURE 3.

SEC and SAXS analysis of KpPqqD. A, the analytical size exclusion chromatogram of purified KpPqqD monitoring tryptophan absorbance at 280 nm shows a single peak with an elution volume that corresponds to a 14.9-kDa protein, calculated from the standard curve in B. B, the standard curve of the molecular weight standards shows a linear relationship with elution volume (black points and line). The calculated molecular weight for KpPqqD is shown by the red triangle. C, the similarity of the SAXS profiles for KpPqqD at all concentrations studied (1, 2, 3, 4, and 5 mg/ml: black, violet, dark blue, blue, and cyan, respectively) is independent of protein concentration. D, the plot of the radius of gyration as a function of KpPqqD shows its independence of protein concentration. E, experimental SAXS profile of KpPqqD (○) does not agree with the XcPqqD SAXS profiles calculated from the dimer (cyan line) or monomer (blue line) crystal structures. However, the SAXS profile for KpPqqD does agree with the compact model of XcPqqD (red line), constructed as described in the text and illustrated in Fig. 4B. F, the Guinier plot shows the linearity of the low q region of the experimental KpPqqD SAXS data extrapolated to zero scattering angle (red line), indicating that the protein has not aggregated.
Various concentrations of KpPqqD (1, 2, 3, 4, and 5 mg/ml) were analyzed by SAXS (Fig. 3C). Inspection of the low q Guinier region is useful to evaluate the presence of sample aggregation or concentration-dependent scattering (34). The Guinier analysis of the low q region of individual SAXS profiles extrapolated to the zero scattering angle qualitatively demonstrates that KpPqqD is not aggregated. The radius of gyration (Rg), determined from the Guinier analysis, is the root mean square distance from the center of mass to each electron. The Rg can be used as an indicator of protein compactness because it has an approximate relationship with the length of the protein (35, 36). The Rg values were determined for each SAXS profile and plotted against concentration (Fig. 3D). The Rg values were fitted to a linear equation. The near zero slope (m = 0.15) indicates that the protein conformation and oligomerization state are homogeneous and concentration independent for the range analyzed.
The SAXS profiles for each concentration were averaged to generate a combined SAXS profile (Fig. 3E). The averaged SAXS profile was used for the remaining data analysis. The Rg determined from the averaged SAXS profile was found to be 15.43 ± 0.04 Å. This experimental Rg does not agree with the theoretical Rg for the monomer or dimer (20.48 and 21.76 Å, respectively) calculated from the XcPqqD crystal structure using CRYSOL (37). Moreover, the present data provided a calculated maximum length for the protein (Dmax), and this was found to be 55.6 Å, dissimilar from the theoretical Dmax for the XcPqqD monomer or dimer (66.7 and 74.3 Å, respectively). The SAXS profile for KpPqqD overlaid with the monomeric or dimeric calculated SAXS profile derived from the crystal structure by CRYSOL shows (Fig. 3E, blue and cyan, respectively) how the solution structure and the crystal structure are distinct. The radius of gyration and the volume of correlation, calculated from the SAXS profile, can be used to estimate the mass of KpPqqD (38). The experimentally estimated KpPqqD molecular mass was determined to be 10.8 kDa, in agreement with the predicted 10.4 kDa for the monomer of KpPqqD.
To visualize the discrepancies between the experimental and the calculated values for Rg, Dmax, and Mr we fitted the experimental SAXS data to generate a solution structure for KpPqqD. The envelope for KpPqqD was generated by DAMMIF using the experimental parameters defined by GNOM and P1 symmetry as constraints (24–26). DAMMIF uses an initial beaded model that is subjected to simulated annealing and compacting until the shape of the model has a scattering pattern that fits the experimental data with the imposed constraints (25). The DAMMIF derived and experimentally fit model for KpPqqD was shown to be globular in shape (Fig. 4A). The overlay of the XcPqqD monomeric crystal structure in the solution structure (Fig. 4A) further depicts the dissimilarity between the XcPqqD crystal structure and the KpPqqD solution structure.
FIGURE 4.
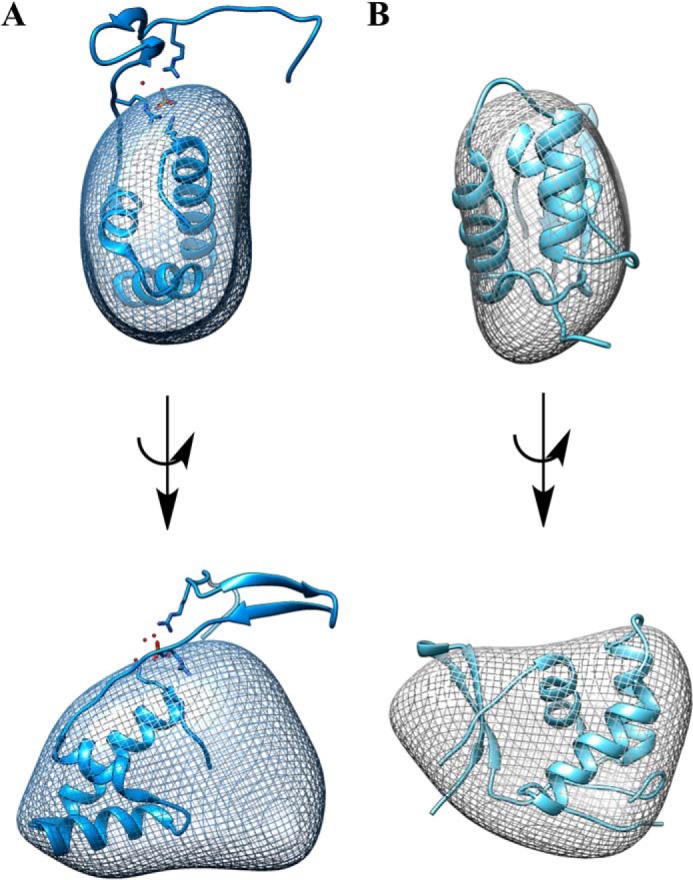
Solution structure of KpPqqD. Ab initio SAXS reconstruction of KpPqqD (mesh) shown as side-on views. A, the ribbon structure of the elongated XcPqqD monomer (PDB code 3G2B) was docked into the SAXS envelope demonstrating that the SAXS-reconstructed envelope and the crystal structure are not in agreement. B, the ribbon structure of the compact XcPqqD model, taken from the β1 and β2 strands from one polypeptide chain and the β3, α1, α2, and α3 motifs from the second polypeptide chain, could be docked into the reconstructed envelope.
Although the oligomerization state is different, the XcPqqD domain-swapped structure may still be informative about the KpPqqD monomer. As suggested by a referee, we generated a model of a compact monomer using the β1 and β2 strands from one polypeptide chain and the β3, α1, α2, and α3 motifs from the second polypeptide chain (highlighted in Fig. 2). The theoretical SAXS profile from the modeled XcPqqD fits the experimental SAXS profile (Fig. 3E, red) reasonably well. Moreover, the theoretical Rg for the model is 14.9 Å, similar to the experimentally derived Rg = 15.4 Å. We conclude that the compact XcPqqD model is able to fit into the SAXS envelope (Fig. 4B).
Examination of the PqqE and PqqD Complex
A previous study (21) from our lab has shown through EPR and hydrogen-deuterium exchange that KpPqqD interacts with KpPqqE; however, the binding affinity and stoichiometry were never determined. To shed light on the binding ratio for the PqqD-PqqE interaction we re-examined the formation of the complex using native nanoflow Q-TOF-ESI-MS. Both KpPqqD (monomer, 10,435 Da) and His6-KpPqqE (monomer, apoprotein, 45,077 Da) were exchanged into ammonium bicarbonate buffer and analyzed individually. Only the 5+ and 6+ 10.4 kDa monomer ions of KpPqqD were detected, confirming that KpPqqD was a monomer in solution (Fig. 5A). The 12–14+ charged 45.9-kDa monomer ions of His6-KpPqqE were also clearly detected (Fig. 5B). Upon analyzing a sample containing 40 μm KpPqqD and 10 μm His6-KpPqqE, new 13+ and 14+ 56.6 kDa ions appeared (Fig. 5C). The mass of the new species is consistent with a KpPqqD-KpPqqE complex with a 1:1 stoichiometry.
FIGURE 5.

Nano-Q-TOF-ESI-MS characterization shows PqqD and PqqE form a 1:1 complex. Native mass spectra of K. pneumoniae (left panels) or M. extorquens AM1 (right panels) PqqE with and without PqqD. A, the spectrum of KpPqqD shows a single population with a molecular mass of 10.4 kDa calculated from the charge state distribution. B, likewise, the spectrum of KpPqqE shows a monomer population with a molecular mass of 45.9 kDa calculated from the charge state distribution. C, in the presence of KpPqqD, a new charge state distribution, corresponding to a mass of 56.6 kDa, is formed consistent with a 1:1 KpPqqD-KpPqqE complex. D, the MePqqD native mass spectrum shows a monomer population with a molecular mass of 10.4 kDa calculated from the charge state distribution. E, the native mass spectrum of MePqqE shows two sets of charge state distributions with masses of 43.6 and 86.9 kDa, indicating that both the monomeric and dimeric species are present. F, in the presence of MePqqD, a 54.0-kDa charge state distribution is formed corresponding to a 1:1 MePqqE-MePqqD complex.
To determine whether the PqqD-PqqE interaction transcends a single species, native mass spectrometry was applied to the M. extorquens AM1 PQQ biosynthetic pathway. MePqqD (monomer, 10,409 Da) and MePqqE-His6 (monomer, apoprotein, 42,612 Da) were buffer exchanged into ammonium bicarbonate and subjected to the same analysis as KpPqqD and KpPqqE. The 5–7+ 10.4 kDa ions of MePqqD were evident from the mass spectra (Fig. 5D). The presence of only the monomeric form of MePqqD supports the notion that PqqD, in general, is a monomer in solution. The 11–13+ 43.6 kDa ions of the MePqqE-His6 monomer and, to a lesser extent, the 18–20+ 86.9 kDa ions of the MePqqE-His6 dimer were clearly detected in the mass spectra (Fig. 5E). Analysis of the 80 μm MePqqD and 20 μm MePqqE mixture led to the identification of new 13+ and 14+ charged 54.0 kDa ions (Fig. 5F) indicating that MePqqD and MePqqE form a 1:1 complex and that the interaction between PqqD and PqqE is not isolated to K. pneumoniae.
SPR experiments were used to determine the dissociation constant (KD) between MePqqD and MePqqE. Due to the nonspecific adsorbing of native MePqqD onto the SPR chip, we used the His6-MePqqD as the ligand and native MePqqE as the analyte. At 25 °C, the KD, determined from steady-state analysis, was found to be 12.5 ± 1.5 μm (Table 1). The KD of ∼12 μm is supported, in part, by un-optimized isothermal calorimetry (ITC) experiments that estimated a KD of ∼10 μm (data not shown). The relatively high KD prevented optimization of ITC experiments due to the necessity of unreasonably high protein concentrations.
TABLE 1.
Dissociation constants for various protein-ligand or protein-protein complexes determined by SPR or ITC
| Complexes examined |
KD |
|
|---|---|---|
| SPR | ITC | |
| μm | ||
| His6-MePqqD + MePqqE | 12.5 ± 1.5 | ∼10a |
| His6-MePqqD + MePqqA | 0.39 ± 0.08 | 0.13 ± 0.03 |
| His6-MePqqCD + MePqqA | 0.18 ± 0.02 | 0.19 ± 0.04 |
| His6-MePqqDEfus + MePqqA | 0.20 ± 0.04 | 0.16 ± 0.04 |
| MePqqB-His6 + MePqqA | NDb | –c |
| His6-MePqqE + MePqqA | ND | –c |
| MePqqA/His6-MePqqD + MePqqEd | 4.5 ± 1.5 | –c |
a The estimated value was obtained through unoptimized ITC experiments.
b ND, not detectable.
c Dash, not determined.
d MePqqA and His6-MePqqD were preformed prior to the addition of MePqqE. Errors are standard deviations of three independent KD determinations.
PqqD Binds the Peptide PqqA
To determine the function of MePqqD, we used SPR to screen for binding with the peptide MePqqA. Maximum steady state SPR responses were plotted versus peptide concentration and fitted to derive KD values (Fig. 6, A and B, represent typical datasets). MePqqD was found to bind MePqqA with a KD of 390 ± 80 nm (Table 1). We probed the effects of PqqC being fused to PqqD and found that the MePqqCD fusion bound MePqqA with a KD of 180 ± 20 nm (Table 1). Evidence for a PqqDE fusion protein can be found in Methylocystis sp. SC2, which warranted the engineering of a MePqqDE fusion protein with a flexible (GGGGS)4 linker to determine whether PqqE interferes with PqqA binding. Similar to MePqqCD and MePqqD, the KD for MePqqDE and MePqqA interaction was found to be 200 ± 30 nm. The similar KD values for all constructs suggest that the presence of either PqqC or PqqE does not impact the overall ability of MePqqD to bind the peptide. In their as-isolated forms, neither MePqqB nor MePqqE was found to bind MePqqA. ITC was used to independently verify the KD values for the binding of MePqqA to MePqqD, MePqqCD, and MePqqDE; the values were found to be 130 ± 30, 190 ± 40, and 160 ± 40 nm, respectively (Table 1). Moreover, the ITC experiments estimated the molar ratio between MePqqD constructs and MePqqA to be 1:1 (Fig. 6C), consistent with native mass spectrometry experiments (data not shown).
FIGURE 6.

Binding of PqqCD and PqqA observed by SPR and ITC. A representative dataset (His6-MePqqCD as ligand and MePqqA as analyte) showing that: A, the SPR response increases as MePqqA concentration increases (blue to violet) and B, the steady state fits were used to calculate the dissociation constant (error bars represent standard deviation of three independent experiments. C, ITC was used to independently verify the dissociation constants and determine the molar ratio. The representative raw isotherm traces (top panel) and the integral data points (bottom panel) are shown for an experiment with MePqqA and His6-MePqqCD (black) and a control experiment lacking His6-MePqqCD (blue). As discussed under “Experimental Procedures,” the first injection for each experiment was omitted in data analysis. The single-site model fit curve generated for the experimental data is shown in red.
A dual injection SPR technique was used to determine the KD of the MePqqAD complex to MePqqE. The first injection consisted of 10 μm MePqqA over the SPR chip with bound His6-MePqqD. The second injection consisted of 10 μm MePqqA and various concentrations of native MePqqE. The KD of the MePqqADE complex was determined to be 4.5 ± 1.5 μm (Table 1). To verify that a ternary MePqqADE complex was formed, native mass spectra were obtained. Analysis of a sample containing 50 μm MePqqD, 50 μm MePqqA, and 10 μm MePqqE-His6 led to the formation of new 13–15+ 57.0 kDa ions (Fig. 7) consistent with a 1:1:1 ternary MePqqA, MePqqD, and MePqqE complex.
FIGURE 7.
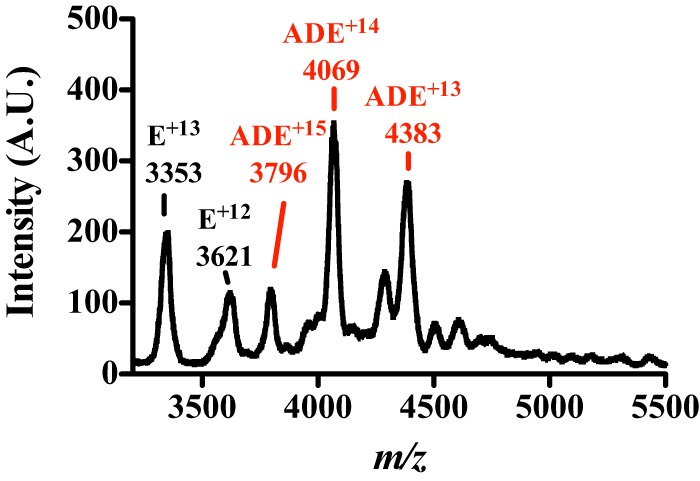
Nano-Q-TOF-ESI-MS characterization of the MePqqA-MePqqD-MePqqE complex. The native mass spectrum of a solution containing 50 μm MePqqA, 50 μm MePqqD, and 10 μm MePqqE shows two sets of charge state distributions. The 12–13+ charged 43.6 kDa ions correspond to the MePqqE monomer and the new 13–15+ charged 57.0 kDa ions correspond to a ternary 1:1:1 MePqqA-MePqqD-MePqqE complex.
PqqD Can Be Found with Peptide Modifying RS-SPASM Proteins
A bioinformatic approach was used to examine the biological range of PqqD in relationship to RS-SPASM proteins. From the 21,353 sequences listed under the SPASM domain Interpro identifier (IPR023885), the generation of the sequence similarity network yielded 1,882 nodes (Fig. 8). Each node represents a set of related protein sequences and contains a unique representative sequence encoding for a SPASM domain protein. The representative sequence, in essence, is most similar to all other proteins within the node. In principle, the representative sequence can be used to generalize for all sequences contained within the node. Therefore, the gene context of representative sequences for nodes that were found in a three-node cluster or more (1,476 nodes) was manually inspected for the presence of the PqqD homologue and a peptide. We found that a PqqD homologue was fused to an RS-SPASM protein in 582 representative sequences. Moreover, a PqqD homologue was found nearby an RS-SPASM protein in 179 representative sequences (761 total representative sequences). Of the remaining 715 representative sequences, 547 did not have a PqqD homologue annotated and 168 did not have gene information available. The presence of a peptide was found in the gene context of 292 of 761 (∼38%) representative sequences with a PqqD homologue, whereas a peptide could only be found in the gene context of 45 of 547 (∼8%) representative sequences without a PqqD homologue.
FIGURE 8.
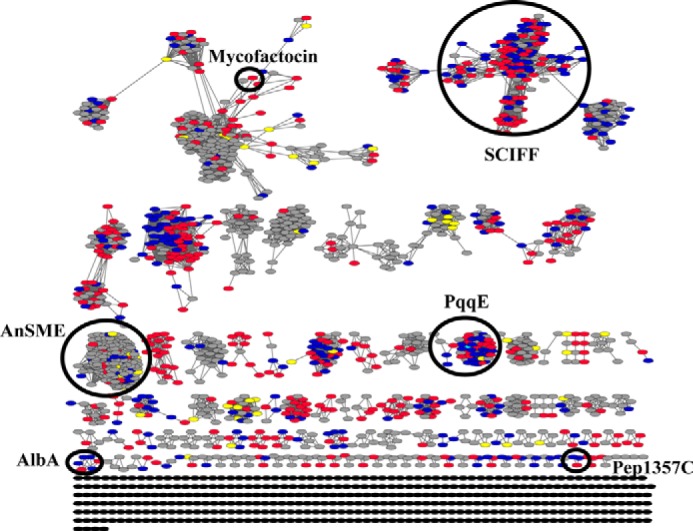
Representative sequence similarity network for the SPASM domain family. Each node (oval) represents a group of sequences where each sequence in the node is at least 60% identical to the representative sequence that defines the node (computed by the CD-HIT program (33)). The 1,882 nodes shown in this network represent over 20,000 sequences. Each edge (lines) that connects two nodes represents a BLAST similarity score of an -log(E-value) of 45 or greater, alignments of lengths between 340 and 450 amino acids (typical length of RS-SPASM proteins) and an average sequence identity of 35%. Cytoscape was used to visualize the network yFiles in an organic layout. The gene contexts of representative sequences were examined for the presence of a PqqD homologue and a peptide within a ±5-gene region on the same DNA strand. A node is colored blue if the representative sequence was fused to or found nearby a PqqD orthologue and a peptide was present in the gene context. A node is colored red if the representative sequence was fused to or found nearby a PqqD orthologue but a peptide was not found in the gene context. A node is colored yellow if the representative sequence was not fused to or found nearby a PqqD orthologue and a peptide was found in the gene context. A node is colored in gray if the representative sequence is not fused to or found nearby a PqqD orthologue and a peptide was not found in the gene context or the gene information was unavailable. Nodes colored in black were not analyzed.
DISCUSSION
PqqD has been shown, through gene knock-out and bioinformatic studies, to be an essential component for PQQ biosynthesis (8, 14, 15). The crystal structure for XcPqqD indicates that PqqD is a dimer and may form a scaffold for a larger macromolecular complex (15). When we purified KpPqqD, however, we found that the protein eluted from the SEC column later than was expected for the size of the dimer. Upon further analytical SEC analysis we found a molecular weight for KpPqqD that was closer to that of monomer than a dimer. We therefore used SAXS as a means to determine the solution molecular weight and, thereby, the oligomerization state of KpPqqD. SAXS has been used to determine the molecular weight of proteins with an average error of 5% (29). This technique provides strong support for the existence of KpPqqD as a monomer.
The difference in oligomerization states for crystallized XcPqqD and solution KpPqqD warranted a further analysis. In the crystal, XcPqqD adopts a saddle-like dimeric conformation with each monomer, containing an α-helix bundle and an extended β1-β2 hairpin feature, interlocked with the another monomer by H-bonds and hydrophobic interactions in an elongated β3 strand region (20). By contrast, in solution KpPqqD adopts a monomeric globular structure that is quite different from the crystallized dimer or the monomer of XcPqqD. When the XcPqqD monomer is fit into the volume of the KpPqqD solution structure it becomes evident that the β1-β2 hairpin adopts an extended conformation. We postulated that the positions of the β1 and β2 strands are compacted in relationship to the α-helix bundle of KpPqqD and that the dimerization state, or domain swapped state, may either be an innate property to XcPqqD or the result of the crystallographic conditions. We proceeded to generate a model of a compact XcPqqD monomer consisting of the β1 and β2 strands from one polypeptide chain and the β3, α1, α2, and α3 motifs from the second polypeptide chain. The theoretical SAXS profile matches the experimental SAXS profile for KpPqqD quite well. Follow up experiments will be necessary to interrogate whether the crystal structure of XcPqqD has implications for a relevant conformational alteration that could facilitate the formation of a tight complex with PqqA.
As mentioned before, the KpPqqD-KpPqqE interaction had previously been discovered, although the KD, stoichiometry, and how it relates to PQQ biosynthesis were not determined. We re-examined the interaction by using native mass spectrometry to derive the stoichiometric ratios for the KpPqqD-KpPqqE complex and found the molar ratio to be 1:1. We could not probe for other protein-protein interactions within the K. pneumonae PQQ pathway due to solubility issues with KpPqqB and the peptide substrate KpPqqA, prompting the move to the M. extorquens AM1 PQQ pathway. In M. exotorquens AM1, the PQQ biosynthetic pathway consists of the operon pqqAB(CD)E where pqqC and pqqD are a gene fusion. All M. extorquens AM1 pqq genes were cloned into E. coli expression vectors and were found to be soluble.
Native mass spectrometry experiments were used to verify a MePqqD-MePqqE complex and clearly demonstrated that the interaction transcends species. Moreover, the mass spectra confirmed the 1:1 molar ratio of the MePqqD-MePqqE complex. The KD for the MePqqD-MePqqE interaction, determined by SPR, is found to be ∼12 μm. The relatively weak KD may suggest that the MePqqD-MePqqE interaction is transient (39). We attempted to determine the stoichiometry of the complex by analytical SEC but found that the complex did not remain intact under these conditions. Using the as isolated form of MePqqB, no interactions have been detected between MePqqD or MePqqE through native mass spectrometry. We did not probe for any protein interactions with PqqC because gene knock-out studies have shown that in the absence of PqqC, the intermediate AHQQ is still formed (8). The fact that AHQQ can be formed decreases the likelihood that a functionally relevant protein complex with PqqC is necessary. The native mass spectral data suggest that the major protein-protein interaction in the PQQ pathway is between MePqqD and MePqqE.
In a brute force effort to discover the binding partner for the peptide PqqA, we utilized SPR and ITC to screen for binding events between MePqqA and MePqqB, MePqqE, and MePqqD. Surprisingly, we find that MePqqD binds MePqqA tightly (KD ∼ 200–300 nm depending on the construct). In contrast, evidence for peptide binding by MePqqE or MePqqB was not found. Significantly, these experiments provide the first evidence that PqqD is the PqqA binding partner. Moreover, to the best of our knowledge, this is the first instance that a PqqD homologue has been shown to bind a peptide.
Taking into account the interaction between MePqqD and MePqqE, we also probed for a MePqqA-D-E complex. Native mass spectrometry experiments provided evidence for a ternary complex and the molar ratio was determined to be 1:1:1. The KD between the MePqqAD complex and MePqqE is 4.5 μm, by a dual injection SPR technique, about the same as the KD determined for MePqqD and MePqqE. The similar KD values between MePqqE and MePqqD or MePqqAD suggest that the presence of the peptide does not assist in PqqD-PqqE complex formation. The binding data determined here provides a new picture of the first steps in PQQ biosynthesis. Our new hypothesis is that PqqD first binds PqqA to protect the peptide from premature proteolytic cleavage and/or to present the glutamate and tyrosine to PqqE. PqqE then binds the PqqAD complex and catalyzes the formation of the carbon-carbon bond between the glutamate and tyrosine.
The idea that the RS-SPASM protein directly acts on the peptide is consistent with other post-translational pathways such as in subtilosin biosynthesis. In subtilosin biosynthesis, the peptide is directly modified by AlbA (40). Moreover, our bioinformatic analysis supports, in part, the notion that a functionally relevant interaction between a PqqD homologue and an RS-SPASM protein exists in peptide modification pathways. Not only does AlbA contain a N-terminal PqqD orthologue, other RS-SPASM proteins can be found with a PqqD orthologue fused to or near the RS-SPASM gene (e.g. the putative mycofactocin pathway, the Pep1357C pathway, and in the putative 6 cysteines in 45 modification pathway). The exact role of the PqqD homologues in other peptide modification pathways will need to be addressed; however, from the work presented here, we propose that PqqD is a peptide chaperone that forms a stable complex with PqqA.
This work was supported, in whole or in part, by National Institutes of Health Grant GM 039296 (to J. P. K.).
- PQQ
- pyrroloquinoline quinine
- AHQQ
- 3a-(2-amino-2-carboxyethyl)-4,5-dioxo-4,5,6,7,8,9-hexahydroquinoline-7,9-dicarboxylic acid
- ESI
- electrospray ionization
- ITC
- isothermal calorimetry
- Kp
- Klebsiella pneumoniae
- Me
- Methylobacterium extorquens AM1
- RS
- radical S-adenosylmethionine
- SAXS
- small angle x-ray scattering
- SEC
- size exclusion chromatography
- SPASM
- subtilosin, pyrroloquinoline quinone, anaerobic sulfatase maturating enzyme, mycofactocin
- Q-TOF
- quadrupole time-of-flight
- SPR
- surface plasmon resonance
- TCEP
- (tris(2-arboxyethyl)phosphine)
- Xc
- Xanthomonas campestris
- PDB
- Protein Data Bank.
REFERENCES
- 1. Babasaki K., Takao T., Shimonishi Y., Kurahashi K. (1985) Subtilosin A, a new antibiotic peptide produced by Bacillus subtilis 168: isolation, structural analysis, and biogenesis. J. Biochem. 98, 585–603 [DOI] [PubMed] [Google Scholar]
- 2. Duquesne S., Destoumieux-Garzón D. (2007) Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat. Prod. Rep. 24, 708–734 [DOI] [PubMed] [Google Scholar]
- 3. Willey J. M., van der Donk W. A. (2007) Lantibiotics: peptides of diverse structure and function. Annu. Rev. Microbiol. 61, 477–501 [DOI] [PubMed] [Google Scholar]
- 4. Bacon Schneider K., Palmer T. M., Grossman A. D. (2002) Characterization of comQ and comX, two genes required for production of ComX pheromone in Bacillus subtilis. J. Bacteriol. 184, 410–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okada M., Sato I., Cho S. J., Iwata H., Nishio T., Dubnau D., Sakagami Y. (2005) Structure of the Bacillus subtilis quorum-sensing peptide pheromone ComX. Nat. Chem. Biol. 1, 23–24 [DOI] [PubMed] [Google Scholar]
- 6. Behling L. A., Hartsel S. C., Lewis D. E., DiSpirito A. A., Choi D. W., Masterson L. R., Veglia G., Gallagher W. H. (2008) NMR, mass spectrometry and chemical evidence reveal a different chemical structure for methanobactin that contains oxazolone rings. J. Am. Chem. Soc. 130, 12604–12605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anthony C. (1998) The pyrroloquinoline quinone (PQQ)-containing quinoprotein dehydrogenases. Biochem. Soc. Trans. 26, 413–417 [DOI] [PubMed] [Google Scholar]
- 8. Toyama H., Chistoserdova L., Lidstrom M. E. (1997) Sequence analysis of pqq genes required for biosynthesis of pyrroloquinoline quinone in Methylobacterium extorquens AM1 and the purification of a biosynthetic intermediate. Microbiology 143, 595–602 [DOI] [PubMed] [Google Scholar]
- 9. Goosen N., Huinen R. G., van de Putte P. (1992) A 24-amino-acid polypeptide is essential for the biosynthesis of the coenzyme pyrrolo-quinoline-quinone. J. Bacteriol. 174, 1426–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grell T. A., Goldman P. J., Drennan C. L. (2015) SPASM and twitch domains in AdoMet radical enzyme structures. J. Biol. Chem. 290, 3964–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haft D. H., Basu M. K. (2011) Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol. 193, 2745–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goodwin P. M., Anthony C. (1998) The biochemistry, physiology and genetics of PQQ and PQQ-containing enzymes. Adv. Microb. Physiol. 40, 1–80 [DOI] [PubMed] [Google Scholar]
- 13. Anthony C. (2001) Pyrroloquinoline quinone (PQQ) and quinoprotein enzymes. Antioxidants Redox. Signal. 3, 757–774 [DOI] [PubMed] [Google Scholar]
- 14. Gliese N., Khodaverdi V., Görisch H. (2010) The PQQ biosynthetic operons and their transcriptional regulation in Pseudomonas aeruginosa. Arch. Microbiol. 192, 1–14 [DOI] [PubMed] [Google Scholar]
- 15. Shen Y.-Q., Bonnot F., Imsand E. M., RoseFigura J. M., Sjölander K., Klinman J. P. (2012) Distribution and properties of the genes encoding the biosynthesis of the bacterial cofactor, pyrroloquinoline quinone. Biochemistry 51, 2265–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Velterop J. S., Sellink E., Meulenberg J. J., David S., Bulder I., Postma P. W. (1995) Synthesis of pyrroloquinoline quinone in vivo and in vitro and detection of an intermediate in the biosynthetic pathway: synthesis of pyrroloquinoline quinone in vivo and in vitro and detection of an intermediate in the biosynthetic pathway. J. Bacteriol. 177, 5088–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonnot F., Iavarone A. T., Klinman J. P. (2013) Multistep, eight-electron oxidation catalyzed by the cofactorless oxidase, PqqC: identification of chemical intermediates and their dependence on molecular oxygen. Biochemistry 52, 4667–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Magnusson O. T., Toyama H., Saeki M., Rojas A., Reed J. C., Liddington R. C., Klinman J. P., Schwarzenbacher R. (2004) Quinone biogenesis: structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone. Proc. Natl. Acad. Sci. U.S.A. 101, 7913–7918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wecksler S. R., Stoll S., Tran H., Magnusson O. T., Wu S.-P., King D., Britt R. D., Klinman J. P. (2009) Pyrroloquinoline quinone biogenesis: demonstration that PqqE from Klebsiella pneumoniae is a radical S-adenosyl-l-methionine enzyme. Biochemistry 48, 10151–10161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsai T. Y., Yang C. Y., Shih H. L., Wang A. H. J., Chou S. H. (2009) Xanthomonas campestris PqqD in the pyrroloquinoline quinone biosynthesis operon adopts a novel saddle-like fold that possibly serves as a PQQ carrier. Proteins Struct. Funct. Bioinforma. 76, 1042–1048 [DOI] [PubMed] [Google Scholar]
- 21. Wecksler S. R., Stoll S., Iavarone A. T., Imsand E. M., Tran H., Britt R. D., Klinman J. P. (2010) Interaction of PqqE and PqqD in the pyrroloquinoline quinone (PQQ) biosynthetic pathway links PqqD to the radical SAM superfamily. Chem. Commun. 46, 7031–7033 [DOI] [PubMed] [Google Scholar]
- 22. Beeson W. T., 4th, Iavarone A. T., Hausmann C. D., Cate J. H., Marletta M. A. (2011) Extracellular aldonolactonase from Myceliophthora thermophila. Appl. Environ. Microbiol. 77, 650–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konarev P. V., Volkov V. V., Sokolova A. V., Koch H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 24. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 25. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wriggers W. (2010) Using Situs for the integration of multi-resolution structures. Biophys. Rev. 2, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schneidman-Duhovny D., Hammel M., Sali A. (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 29. Fischer H., de Oliveira Neto M., Napolitano H. B., Polikarpov I., Craievich A. F. (2009) Determination of the molecular weight of proteins in solution from a single small-angle x-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43, 101–109 [Google Scholar]
- 30. Atkinson H. J., Morris J. H., Ferrin T. E., Babbitt P. C. (2009) Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One 4, e4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barber A. E., 2nd, Babbitt P. C. (2012) Pythoscape: a framework for generation of large protein similarity networks. Bioinformatics 28, 2845–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fu L., Niu B., Zhu Z., Wu S., Li W. (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Putnam C. D., Hammel M., Hura G. L., Tainer J. A. (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 40, 191–285 [DOI] [PubMed] [Google Scholar]
- 35. Lobanov M. I., Bogatyreva N. S., Galzitskaia O. V. (2008) Radius of gyration is indicator of compactness of protein structure. Mol. Biol. (Mosk) 42, 701–706 [PubMed] [Google Scholar]
- 36. Zhang Y., Kolinski A., Skolnick J. (2003) TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Svergun D., Barberato C., Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 38. Mylonas E., Svergun D. (2007) Accuracy of molecular mass determination of proteins in solution by small-angle x-ray scattering. J. Appl. Crystallogr. 40, s245–s249 [Google Scholar]
- 39. Ozbabacan S. E., Engin H. B., Gursoy A., Keskin O. (2011) Transient protein-protein interactions. Protein Eng. Des. Sel. 24, 635–648 [DOI] [PubMed] [Google Scholar]
- 40. Flühe L., Knappe T. A., Gattner M. J., Schäfer A., Burghaus O., Linne U., Marahiel M. A. (2012) The radical S-adenosylmethionine enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat. Chem. Biol. 8, 350–357 [DOI] [PubMed] [Google Scholar]