

Abstract
All Qinghai-Tibetan Plateau (QTP) endemic species are assumed to have originated recently, although very rare species most likely diverged early. These ancient species provide an excellent model to examine the origin and evolution of QTP endemic plants in response to the QTP uplifts and the climate changes that followed in this high altitude region. In this study, we examined these hypotheses by employing sequence variation from multiple nuclear and chloroplast DNA of 239 individuals of Juniperus microsperma and its five congeners. Both phylogenetic and population genetic analyses revealed that J. microsperma diverged from its sister clade comprising two species with long isolation around the Early Miocene, which corresponds to early QTP uplift. Demographic modeling and coalescent tests suggest that J. microsperma experienced an obvious bottleneck event during the Quaternary when the global climate greatly oscillated. The results presented here support the hypotheses that the QTP uplifts and Quaternary climate changes played important roles in shaping the evolutionary history of this rare juniper.
Because of the complex geological setting of the Qinghai-Tibetan Plateau (QTP), the effects of historical orogenesis and climatic oscillations on the evolutionary histories of plants in this region remain controversial1,2,3,4. The QTP, covering approximately 2.5 million km2, with an average altitude above 4000 m (a.s.l.), is the highest and one of the most extensive plateaus on Earth5. The QTP uplifts began in the Early Eocene (ca. 50 million years ago, (Mya)), when the Indian subcontinent collided into Eurasia; but the extensive uplifts of the plateau did not start until the latest Oligocene. These uplifts occurred at least four times from the latest Oligocene through the Early Miocene, Middle Miocene, Late Miocene, Late Pliocene and early Pleistocene6,7. Additionally, geological evidence suggests that three or four Quaternary glaciations have occurred on the QTP6,7. The largest glaciation (the Naynayxungla Glaciation) began ca. 1.2 Mya and reached its maximum between 0.8 and 0.5 Mya7,8. Several cycles of climatic oscillations producing montane glaciations appear to have occurred well into the Holocene6,7.
As a topographically complex region, the QTP contains 9000 to 12000 species of vascular plants in ca. 1500 genera. At least 20% of these species and ca. 50 genera are endemic3,9. The origin and current distribution of these endemic taxa are the products of the continual interplay between evolutionary processes, such as speciation and gene flow, and environmental changes in this region, such as phased uplifts of the QTP and the subsequent climate changes3,9. The evolutionary histories of most QTP flora can be partitioned roughly into three stages. During the earliest stage, spanning from the Eocene to the Early Miocene, QTP uplift began and extended to its southern territories4, and many genera or major lineages of some genera that have high species diversity in the QTP diverged from their sister species or lineages3,9,10. In the second stage, from the Middle Miocene to the Pliocene, the QTP experienced several extensive uplifts that resulted in most mountain ranges in this region. These orogenic events created intense habitat fragmentation and triggered the diversification of many genera or lineages10,11,12,13 as well as spectacular rapid radiations of several species-rich genera14,15,16. It appears that most of the diversity of endemic species in this region originated during this period3,9. From the Pleistocene to the present (the third stage), the QTP has undergone Quaternary climate oscillations as experienced in many temperate regions in the Northern Hemisphere. However, the magnitude of the climate changes were heterogeneous in different parts of the QTP because of its complex topology3,4. Although some species were driven to extinction, most species experienced a dynamic demography (e.g., glacial retreat to eastern declivities of the QTP and postglacial recolonization, glacial in situ survival and postglacial local expansion)1,2. Other species may have experienced intraspecific divergence3,17. Most early phylogenetic research on QTP plant species has focused on the second stage and the earliest stage3,9. By contrast, recent phylogeographic and population genetics studies have examined changes that have occurred in the latest stage1,2.
So far, there has been very little research on the correlation between orogenic and evolutionary events across all three stages in a single species, despite that most QTP endemic species originated after the Middle Miocene. Juniperus microsperma (W. C. Cheng and L. K. Fu) R. P. Adams provides an excellent model to address this topic. Juniperus L. is an important element of arid and semi-arid ecosystems throughout the Northern Hemisphere18,19, but J. microsperma is an extremely rare conifer that is currently known only from the Parlung Zangbo Valley, Bomi County, southeastern QTP19. In our previous phylogenetic study based on chloroplast DNA sequence variation, we found that J. microsperma is basal to phylogenetic clades that contain J. semiglobosa, J. sabina and North American smooth-leaf junipers10. Juniperus microsperma appears to have diverged from its closely related species around the Early Miocene10. Although J. microsperma clusters with a group of allopatric species that grow far away, it shares morphological traits and leaf terpenoid components with two sympatric species, J. convallium and J. saltuaria, indicating possible gene flow from J. microsperma to the latter two species or vice versa20. Considering the current narrow distribution and small population size of J. microsperma, and the much wider distribution and larger population size of its sister species, J. sabina and J. semiglobosa, it is highly likely that this rare juniper experienced a dynamic demographic history. These hypotheses merit further validation using bi-parental inherited nuclear makers and modern population genetic approaches.
Recent developments in population genetic and phylogenetic approaches based on multiple nuclear loci have greatly improved our ability to infer speciation and demographic histories. *BEAST estimates species trees by sampling multiple loci from multiple individuals of each species and adopting a Bayesian framework, and it enhances phylogenetic estimation approaches by directly modeling intraspecific polymorphism and incomplete lineage sorting21. The latest IM method (IMa2)22 is capable of estimating splitting time, population size and migration parameters for an IM model with multiple populations under a known phylogeny22. Additionally, various ABC methods (e.g., DIYABC)23 can provide flexible models to test and compare a wide range of scenarios concerning dynamic population size, migration and splitting time parameters for a group of closely related species, and can even test population size changes through time for a single species24.
In this study, we employed multiple nuclear loci and two cpDNA markers to investigate genetic variation across populations of J. microsperma and its congeners, J. semiglobosa, J. sabina, J. convallium, J. saltuaria and J. tibetica. Then, by utilizing a series of phylogenetic and population genetic approaches, including *BEAST21, IMa222 and DIYABC23, the speciation history of J. microsperma and its close relatives as well as its demographic history were inferred based on genetic variation among and within species. We addressed the following questions: (1) What is the speciation history of J. microsperma and closely related species? (2) What is the demographic history of J. microsperma? (3) How did geological and climatic histories on the QTP effect the speciation and demographic histories of J. microsperma? Understanding these questions will not only shed light on the evolutionary history of QTP flora but will also facilitate the conservation of rare endemics in this topographically complex region.
Results
Genetic variation and interspecific divergence
We analyzed DNA sequence data of eight nuclear regions (CC1147, CC2920, HemA, Pgi, CC1333, CC2241, Maldehy and LHCA4; for details, see Methods) and two chloroplast fragments (trnT–trnL and trnL–trnF; for details, see Methods) for 239 individuals from 56 populations of J. microsperma (60 individuals from seven populations) and five of its congeners, J. sabina, J. semiglobosa, J. convallium, J. saltuaria and J. tibetica (Table S1). Among these, DNA sequences of 83 individuals belonging to the former three species were generated here (GenBank accession numbers: KP198658-KP199941), and the other sequences were collected previously25.
Two cpDNA fragments were sequenced across all individuals of J. microsperma, J. sabina and J. semiglobosa, and 1504 bp of aligned sequence data were obtained. Five substitutions and two indels were detected (Table S2), and these polymorphisms identified a single haplotype in J. microsperma, and two haplotypes each in the other two species; no haplotype was shared among these three species. We also built a chloroplast haplotype network, which showed that chloroplast haplotypes of the six species were grouped in two distinct groups (Fig. 1).
Figure 1.

Geographic distributions (A), networks (B) and Maximum Likelihood tree (C) of the 10 chlorotypes that were identified in Juniperus microsperma, J. sabina, J. semiglobosa and J. tibetica complex (J. convallium, J. saltuaria and J. tibetica). In (A) and (B), each circle represent a population, the color of the circle outline represents the species attribute of each population, and the color that fills the circles is proportional to the frequency of each chlorotype in each population. In (C), bootstrap values above 0.5 are shown on the branch next to each node. Part (A) was drawn by H.Y.S. using CorelDraw X4 (Corel Corporation, Ottawa, Canada).
The sequencing of eight nuclear loci resulted in 4504 bp of aligned sequence data, and 51, 58, 43, 40, 62, and 47 segregating sites were identified in J. microsperma, J. sabina, J. semiglobosa, J. convallium, J. saltuaria and J. tibetica, respectively (Table S3). Nuclear DNA sequence diversity was detected at all eight loci in each of the six species (Table S3), although the amount of nucleotide polymorphism varied greatly among loci. Considering silent nucleotide diversity (πs) and total nucleotide diversity (πt) averaged over eight loci, the nucleotide diversity of J. microsperma (0.00458, 0.00245) is moderate among the six junipers (Fig. 2; Table S3).
Figure 2.

Box plots of the summary statistics for Juniperus microsperma, J. sabina, J. semiglobosa and the J. tibetica complex (J. convallium, J. saltuaria and J. tibetica). Bars represent the median values, boxes represent the interquartile ranges and whiskers extend to 1.5 times of each interquartile range.
Significant population differentiation (FST) and net sequence divergence (Da) among species were detected at nearly all nuclear DNA loci (Table S4). FST values showed that genetic differentiation between J. microsperma and the other five species were significant at seven of the eight loci or all loci. Results for Da were similar to those for FST (Table S4). The ΦST values among species estimated by AMOVA also showed that divergence between each species pair was highly significant (Table S5). The estimated divergences between J. microsperma and J. sabina (ΦST = 0.52257, P < 0.001), and between J. microsperma and J. semiglobosa (ΦST = 0.78665, P < 0.001), were comparatively lower than between J. microsperma and each of J. convallium, J. saltuaria and J. tibetica (Table S5). The median joining genotype networks constructed for each nuclear locus (Fig. 3) also showed that J. microsperma is most closely related to J. sabina and J. semiglobosa.
Figure 3.

Networks for all detected haplotypes at each of the eight nuclear loci. Each circle represents a haplotype, and each color represents a species; the size of each circle is proportional to the sample size of each population; and in each circle that comprises more than one species, the area of each color is proportional to number of individuals attributed to each species. The black circles represent missing haplotypes, and mutational steps are the black bars.
Median joining networks for the genotypes at each of the eight nuclear loci also showed that J. microsperma is most closely related to J. sabina and J. semiglobosa. As shown in Fig. 3, nuclear genotypes are generally divided into two groups: one group comprises genotypes from J. microsperma, J. sabina and J. semiglobosa, whereas another group includes genotypes from J. convallium, J. saltuaria and J. tibetica. The PCA plot employing identical data generated a similar pattern, in which individuals of J. microsperma, J. sabina and J. semiglobosa, and the remaining three species were clustered into three distinct groups (Fig. 4).
Figure 4.

A principal component analysis (PCA) plot for representative individuals of Juniperus microsperma and its five congeners. Each circle represents an individual, and the circle color represents the species attribute of each individual.
Additionally, the Bayesian clustering algorithm (STRUCTURE, version 2.3) revealed that the most likely number of clusters was K = 2 when using eight nuclear loci from all individuals of the six junipers (Fig. 5A). For K = 2, the first cluster comprised J. microsperma, J. sabina and J. semiglobosa, whereas the second cluster comprised J. convallium, J. saltuaria and J. tibetica; for K = 3, J. microsperma became an independent cluster; and for K = 4, individuals of J. convallium, J. saltuaria and J. tibetica were further assigned to two clusters. It has been proposed that intraspecific genetic structure is among the factors that play a role in generating false bottleneck signals26. Therefore, we further conducted Bayesian clustering analysis for J. microsperma but found no detectable population structure in this species (Fig. S1). A deltaK plot suggested that K = 2 is the best cluster number (Fig. S1C), we also found that the mean LnPD is greatest when K = 2 (Fig. S1B); notably, the mean LnPD is slightly smaller but much less variable when K = 1 (Fig. S1B). Nevertheless, as shown in Fig. S1A, whenever K = 2, K = 3 or K = 4, the genetic composition of any pair of populations is similar to one another.
Figure 5.

(A) Bayesian clustering for individuals of the six junipers based on eight nuclear loci; two, three and four clusters (K = 2, 3, 4) were assumed, (B) Delta K values of eight runs that assumed two to nine clusters (K = 2–9), and (C) LnPD values of nine runs that assumed two to ten clusters (K = 10). For each K value, results of the run with the highest value of LnPD were used. Dashed vertical lines in (A) represent boundaries among species.
Inference of the species tree
We employed *BEAST to estimate a species tree using a Bayesian framework based on multiple nuclear loci from multiple individuals of each species. The species tree topology that was generated by *BEAST provided strong support values to cluster J. microsperma, J. semiglobosa and J. sabina (PP = 1.0), and to cluster J. convallium, J. saltuaria and J. tibetica (PP = 1.0), as well as to cluster these two clades; additionally, the sister relationship between J. semiglobosa and J. sabina received moderate support (PP = 0.9554; Fig. 6).
Figure 6.
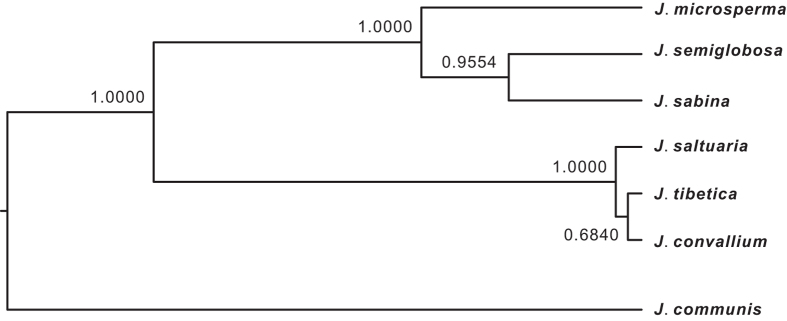
A species tree for Juniperus microsperma and six of its congeners based on 8 nuclear genes that was constructed using *BEAST. The tree was rooted with J. communis according to previous phylogenetic studies10,32. The posterior probabilities are reported above the nodes.
Neutral and demographic tests
We calculated a set of indices including Tajima’s D, Fu and Li’s D* and F* and Fay and Wu’s H (Fig. 2; Table S3) to detect departures from the standard neutral model of molecular evolution at each nuclear locus. Among J. microsperma and its two most closely related species, the mean Fay and Wu’s H value was negative (−1.43659) for J. microsperma, the mean Tajima’s D and Fu and Li’s D* were negative (−0.10658, −0.24666) for J. sabina, and the mean Fu and Li’s D* and F* and Fay and Wu’s H value were all negative (−0.49369, −0.26376, −1.90953) for J. semiglobosa. Next, we performed HKA tests to test departure from neutrality at individual locus. All six species were tested in turn with J. communis as the outgroup, and we found no significant deviation from the neutral model (Table S6). Additionally, the MFDM test indicated that there was a significant probability (P = 0.0174) of selection at the CC1333 locus in J. microsperma. Additionally, at CC1333 (P = 0.057) and PGI (P = 0.057) in J. semiglobosa, the results indicated near significance for selection. Finally, the LAMARC analysis suggested that J. microsperma has undergone large changes in population size. The population growth rate parameter g was estimated at −301.05, providing evidence for recent population size declines or bottlenecks.
Modeling isolation and migration among species
We performed two-population IM simulation runs to estimate demographic parameters: effective population size, divergence time and migration rate (Fig. S2; Table S7). The results showed an estimate of 26,900–45,900 for the effective population size of J. microsperma. The mean divergence time was estimated at 14 to 21.6 Mya between J. microsperma and the J. convallium-J. saltuaria-J. tibetica clade, 13.8 Mya between J. microsperma and J. sabina, and 4.45 Mya between J. microsperma and J. semiglobosa. However, the migration rate between J. microsperma and the other five juniper species was approximately zero, suggesting that gene flow was very low during or soon after the species divergence. Simultaneously, we conducted an IM simulation that involves three species, J. microsperma and its two most closely related species; a significant but minor migration from J. sabina to J. microsperma, and significant and bi-directional migration between J. semiglobosa and J. sabina were detected (Fig. 7A; Table S8); and it was estimated that J. microsperma and the MRCA of its two most closely related species may have diverged 19 Mya. Because both PCA and STRUCTURE suggested that the six juniper species were clustered into three groups, we therefore also performed a three-group IM simulation, and each group was treated as a population. The results showed that J. microsperma and its two most closely related species might have diverged with the MRCA of J. convallium, J. saltuaria and J. tibetica (“J. tibetica complex”, Fig. 7B) at ca. 25 Mya, and J. microsperma diverged from its two most close relatives at ca. 16.1 Mya; significant but minor gene flow was detected from its most close relatives to J. microsperma (Fig. 7B). We preferred divergence timescale of multi-population to two-population IM analyses in the following discussions because the former have better sampling coverage.
Figure 7.
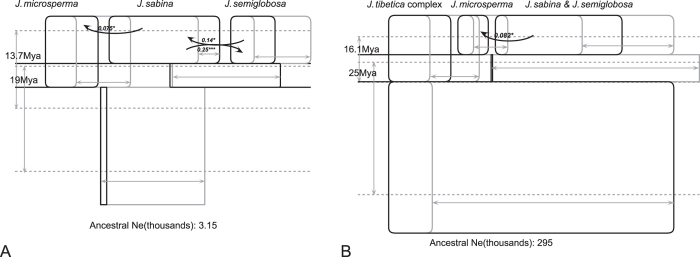
(A) Histories for populations of Juniperus microsperma, J. sabina and J. semiglobosa, and (B) histories for populations of Juniperus microsperma, J. sabina plus J. semiglobosa, and the J. tibetica complex are represented as boxes (for both sampled and ancestral populations). Boxes spanning from the top border of each subfigure represent living populations, whereas other boxes represent ancestral populations. For each box, its horizontal and vertical widths represent effective population size and time, respectively. Curved arrows linking boxes represent migration between populations. Time is represented on the vertical axis in each figure, with the sampled species names provided at the top of each figure at the most recent time point. For all figures, the 95% highest posterior density intervals are shown with arrows in gray for population sizes (i.e., box widths) and splitting times (dotted lines). Only those population migration rates that were found to be statistically significant using a likelihood-ratio test are shown, in which case the estimated value of 2Nm is provided as well as the significance level. *P < 0.05, **P < 0.01, ***P < 0.001.
Testing demographic scenarios
To further understand the demographic history of J. microsperma, we employed an approximate Bayesian computation (ABC) framework in DIYABC 2.0.423 to test five plausible scenarios of demographic changes based on nuclear loci (Fig. 8, Table S9). The DIYABC analyses provide the strongest support for a scenario of ancient population growth and a recent population bottleneck (scenario 1, Fig. 8). The posterior probability of scenario 1 (62.64%) is significantly higher than that of scenario 2 (11.42%), scenario 3 (2.87%), scenario 4 (11.33%) and scenario 5 (11.91%). The average type I error rate (that is, the probability that the data sets simulated under the other scenarios were assigned to the best scenario) of scenario 1 was 6.15% and considered reasonable. The type II error rate (that is, the probability that the data sets simulated under the other scenarios were assigned to the best scenario) was 12.6%. Marginal posterior probability densities for the demographic parameters of the best-supported demographic scenario are provided in the supplementary material (Table S10). The parameter estimate values indicate that J. microsperma populations have undergone a population expansion from Ne to N3 during the Early Miocene, and the population decreased to N1 after a relatively recent bottleneck event that may have occurred during the Late Quaternary.
Figure 8.
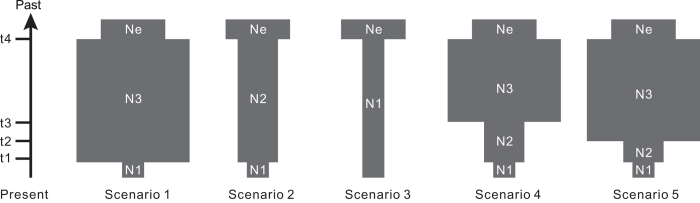
Schematic representation of the five demographic scenarios including model parameters of Juniperus microsperma tested by approximate Bayesian computation (ABC). For the demographic parameters, see Table S9. Times and effective population sizes are not strictly to scale.
Discussion
Our population genetic survey revealed that the evolutionary history of J. microsperma is characterized by two crucial events. First, the divergence of J. microsperma from its two most closely related species, J. sabina and J. semiglobosa, occurred during the Early Miocene, supporting the hypothesis that J. microsperma was isolated following the early uplifts of the QTP. Second, J. microsperma experienced drastic population decreases during the Late Quaternary. The speciation and demographic histories of J. microsperma provide another case in which evolutionary histories of relict species in the QTP are closely related to geological and climatic changes in this sensitive region1,2,3,9.
Our Bayesian species tree inferences (*BEAST) generated a well-supported topology that suggested J. microsperma is sister to the common ancestor of J. semiglobosa and J. sabina; simultaneously, J. convallium, J. saltuaria and J. tibetica formed another group (Fig. 6). Genetic divergence (Table S4) and network analyses (Fig. 3) at eight nuclear loci, PCA plotting (Fig. 4) and Bayesian clustering (Fig. 5A) provided further support of these relationships. This speciation topology is consistent with two earlier phylogenetic studies based on multiple chloroplast fragments10,27.
Under this speciation topology (Fig. 6), our IM analyses, which consider multiple populations, estimated splitting time parameters among these six species and found that the divergence between the MRCAs of J. convallium-J. saltuaria-J. tibetica and J. microsperma-J. semiglobosa-J. sabina, between J. microsperma and the MRCA of J. semiglobosa and J. sabina, and between J. semiglobosa and J. sabina may have occurred 25 Mya (CI: 19.2-73.8 Mya), 16.1 Mya (CI: 8.75-27.4 Mya) or 19 Mya (CI: 14.4-44.6 Mya), and 13.7 Mya (CI: 4.13-26.5 Mya), respectively (Fig. 7). Note that coalescent analyses perform best when sampling size is large enough28; we therefore warn the readers that the results presented here should be treated with caution given our limited sampling of J. semiglobosa and J. sabina. However, age estimation for divergence events that were estimated by IM analyses and previous molecular dating10,27 are consistent. According to previous molecular dating studies based on nine chloroplast fragments and multiple fossils, the above divergence events occurred 30 to 47 Mya, 10 to 28 Mya, and 8.5 to 24.5 Mya, respectively10,27. Because our IM analyses estimated divergence time based on population genetic data and considered gene flow between/among species, whereas previous molecular dating integrated multiple carefully-selected fossils and genetic divergence between three quarters of the species in the genus Juniperus10,27, the convergence of age estimation provides a reliable timeframe for the speciation history of J. microsperma.
The current distribution pattern of these species and splitting times among them suggested that the divergence between J. microsperma and the common ancestor of its two most closely related species was most likely caused by the formation of geological barriers caused by early QTP uplift. Although J. microsperma is known only from Bomi County in the southeastern QTP along the Parlung Zangbo Valley19, J. semiglobosa occurs along the northwestern border of China and the eastern part of central Asia19; and J. sabina is widespread across northern China, Mongolia, Far Eastern Russia, central Asia and Europe18,19. Juniperus semiglobosa and J. sabina have overlapping distributions in Kyrgystan, Tajikistan and the northwestern-most part of China, but the distribution of J. microsperma is distant from these two species (closest ca. 1400 and 800 km, respectively) and numerous mountains are in between, e.g., Bayan Har, Tangula, Nianqing Tangula, Kunlun and Kara Kunlun and others. The QTP has undergone at least four periods of extensive uplifts from the latest Oligocene to the Early Miocene, Middle Miocene, Late Miocene, Late Pliocene to Early Pleistocene3,4,6. The dated divergence between J. microsperma and its two most closely related species coincides with the early timescale of QTP uplifts, when these mountains, which act as geographic barriers among both groups, were formed. This hypothesis is supported by IM analyses because no significant gene flow was detected between J. microsperma and J. semiglobosa or from J. microsperma to J. sabina; nevertheless, low gene flow from the widespread J. sabina to the endemic J. microsperma was detected, indicating the strong dispersal ability of J. sabina over geological barriers or shared ancestral polymorphisms between J. microsperma and J. sabina. The unique leaf terpenoid composition of J. microsperma compared to its two close relatives20 also suggests a long history of isolation and adaptation since separation. Thus, the speciation pattern of J. microsperma is similar to many QTP endemic species, in which geological barriers cut off or largely blocked gene flow between small, isolated populations, and this has led to the formation of these new species3,29.
Additionally, we did not detect gene flow or shared genetic polymorphisms between J. microsperma and its sympatric or parapartic congeners, J. convallium and J. saltuaria, despite that they are phenotypically similar to the former. When J. microsperma was described, this taxon was treated as a variety of J. convallium because of their shared morphological traits, e.g., slender leaves, small seed cones, and one seed per cone30. Additionally, previous studies suggested that J. microsperma shared several leaf terpenoid components with J. saltuaria, such as pregeijerene B, elemol, α- and β-eudesmols20. Our results, although based on limited data, suggested that these shared phenotypic similarities likely arose from convergent evolution considering that divergence between J. microsperma and either J. convallium or J. saltuaria had occurred as early as 30 to 47 Mya (Mao et al., 2010, 2012) or 25 Mya (CI: 19.2–73.8 Mya; Fig. 7B).
After the origin of J. microsperma, our results suggested that it appears to have experienced a dynamic demographic history. Given the currently narrow distribution of J. microsperma (i.e., small population size), one would expect low genetic diversity for this species because inbreeding and genetic drift in small populations usually results in a decrease of genetic diversity31. However, as a rare endemic, the mean silent and total nucleotide diversity of J. microsperma are comparable to five relatively widespread QTP species, J. semiglobosa, J. convallium, J. saltuaria, J. tibetica (Fig. 2; Table S3), J. przewalskii32, and the Mexican endemic J. blancoi33. Additionally, nucleotide diversity of J. microsperma is low-to-moderate among conifer species25,34,35. In addition to the above, IM analyses revealed that the current effective population size of J. microsperma is similar to those of its five congeners, J. tibetica, J. saltuaria, J. convallium, J. przewalskii and J. semiglobosa. Such a combination of moderate nucleotide diversity, small population size and moderate effective population size indicates that this rare juniper most likely has undergone a very recent decrease in distributional range and population size. This hypothesis is supported by LAMARC analysis, with an exponential population growth rate parameter of −301.05.
However, multiple lines of evidence suggest that J. microsperma may have experienced population expansion in its early evolutionary history. DIYABC statistics prefer a scenario in which population expansion of this species occurred during the Middle Miocene (ca. 20-17 Mya). IM analyses produced a significantly smaller effective population size estimate for the common ancestor of J. microsperma and its two most closely related species compared to the effective population size estimate of J. microsperma (Fig. 7A). A similar pattern was detected in another IM analysis when all six species were considered as three groups (Fig. 7B). Neutrality tests suggest ancient expansions in J. microsperma and its two most closely related species. A positive but near zero value of Tajima’s D and a negative value of Fay and Wu’s H revealed no skew towards low-frequency variants (small positive D) and skew towards high-frequency variants (negative H), respectively. This situation has generally been shown to reflect the presence of an ancient expansion event25, which is the case for J. microsperma and J. semiglobosa (Table S3). Simultaneously, a negative Tajima’s D (skew towards low-frequency variants) and a positive Fay and Wu’s H was detected in J. sabina, which suggested that this species most likely also has experienced a relatively ancient expansion event because Fay and Wu’s H becomes positive as new mutations accumulate25.
When did the distribution of J. microsperma shrink? Our DIYABC statistics and model simulations suggested that this bottleneck event most likely occurred in the Middle-Late Quaternary when the most extensive glaciation occurred and continued during this stage5,6,8. This estimation indicates that orogenic events since the Late Miocene may have not caused a significant effect on the demography of this rare juniper, but the Quaternary climate oscillations did. Because J. microsperma grows in relatively warm and xeric river valleys in the eastern QTP, the Late Quaternary population bottleneck event would indicate a drastic decrease of similar habitat if we assume niche conservatism for this species. However, congeners of J. microsperma, such as J. saltuaria, J. tibetica, and J. convallium and J. przewalskii, which occur in adjacent areas, may have experienced different demographic histories. These species appear to have experienced population expansion25,32 as other cold-preferring conifers during this stage36,37.
Contrasting demographic histories between J. microsperma and the other conifer species in the QTP underscore the urgent necessity to conserve this rare endemic. Although this species has a moderate effective population size and moderate genetic diversity, its distribution and population size are fairly small. Therefore, this juniper would face high extinction risk in the short term. Nevertheless, J. microsperma was conservatively treated as J. convallium var. microsperma in the latest version of Flora of China30 as well as on the IUCN red list38; in addition to its taxonomic status, the geographic distribution of this taxon was previously unclear. Thus, this taxon was assigned to the Data Deficiency category in the IUCN red list38, and it was NOT listed as an endangered species in China. In this study, our population genetic data of both chloroplast and nuclear DNA regions rejected the previous morphological hypothesis that this taxon is a variety of J. convallium, and instead confirmed the taxonomic treatment of J. microsperma as an independent species39. In light of this evidence, we advocate that more attention should be given to this rare juniper, and the first step should be listing it as an independent species that is ‘Endangered’ in the Red List of plants in both the IUCN and China.
In summary, our study is a unique case study that focused on testing the correlation between evolutionary history of an ancient species and the historical environmental changes in the QTP, and our findings suggested that QTP uplifts and Quaternary climate changes may have fundamentally shaped the evolutionary history of plants in this region.
Methods
Sampling and data collection
Throughout the known natural distribution of J. microsperma in the QTP, leaf samples were collected from seven natural populations, and four and 11 representative populations of J. sabina and J. semiglobosa were also collected. Additionally, we also compiled a set of DNA sequence data from 19, seven, and eight populations of J. tibetica, J. convallium, and J. saltuaria on the QTP, as generated in a previous study25. Thus, DNA sequences from a total of 239 individuals were analyzed in this study (Table S1). We sampled one to ten individuals per population, resulting in 11 to 60 individuals per species. The sampled individuals within each population were more than 100 m apart. Fresh leaves were sampled and then dried immediately using silica gel. The latitude, longitude and altitude of each sampling site was measured by Extrex GIS (Garmin, Taiwan).
For each sample, approximately 20 mg of silica gel dried leaves were used to extract total genomic DNA according to a modified cetyl trimethyl ammonium bromide (CTAB) procedure40. Previous studies of Cupressaceae suggested that chloroplast DNA (cpDNA) is haploid and paternally inherited in this family41, whereas nuclear DNA is bi-parentally inherited. Thus, we amplified and sequenced two cpDNA fragments (trnT-trnL and trnL-trnF: see Table S11 for primers and references) and eight nuclear DNA fragments (CC1147, CC2241, CC1333, CC2920, HemA, LHCA4, Maldehy and PGI; see Table S11 for primers and references) to study the evolutionary history of the six juniper species on the QTP. The polymerase chain reaction (PCR) was performed in total volume of 25 μL, which contained 10–40 ng plant DNA, 50 mM Tris-HCl, 1.5 mM MgCl2, 0.5 mM dNTPs, 2 mM of each primer and 0.75 unit of Taq polymerase. All reactions were performed using the following temperature profile: 5 min at 94 °C, 36 cycles of 50 s at 94 °C, 50 s of annealing at 56 °C for the two chloroplast DNA fragments and 53 °C – 58 °C for the 8 nuclear DNA fragments, and 1 min 10 s at 72 °C, with a final 8 min extension at 72 °C.
Sequencing reactions and successive purifications were performed using an ABI Prism BigDye Terminator Cycle V3.1 Sequencing Kit (Applied Biosystems, Foster City, CA, USA), and capillary analyses were run on an ABI 3130XL gene analyzer (Lanzhou University, Lanzhou, China) following the manufacturers’ protocols. All DNA sequences were aligned with ClustalW as implemented in MEGA 5.142 and double-checked manually. All insertions/deletions (indels) were excluded.
Phylogenetic analyses and species tree reconstruction
For both cpDNA haplotypes and nuclear genotypes, phylogenetic relationships among were established using NETWORK version 4.2.0.143 (available at http://www.fluxus-engineering.com). Note that because a previous study had conducted a population survey for cpDNA variation of J. convallium, J. saltuaria and J. tibetica in a large sampling size44, five frequent chloroplast haplotypes were adopted to represent these three species. We used MrBayes 3.2.145 to construct the phylogenetic tree of cpDNA haplotypes. Additionally, we performed principal component analysis (PCA) of the genetic polymorphisms using the software R project 3.0.2 ( http://www.r-project.org/).
We also conducted species tree analyses using *BEAST v. 1.8.021, which uses multi-locus data to simultaneously co-estimate gene trees and divergence times under a coalescent model. The computer program BEAUTI 1.8.0 was employed to transform eight separate nuclear gene alignments in nexus format to the initial XML files for *BEAST species tree analyses. We used the general time reversible plus invariant sites plus gamma correction (GTR+I+G) nucleotide substitution model with four gamma categories. An average mutation rate of 1.94 × 10−10 substitutions per site per year was applied25, and we chose the relaxed lognormal clock models for all loci, a constant population size, and a Yule model species tree prior. We ran the MCMC analysis for 1.5 billion generations sampling every 50000 generations. Tracer v1.5 ( http://tree.bio.ed.ac.uk/software/tracer/) was used to assess convergence and effective sample sizes (ESS) for all parameters. After discarding the first 3000 trees as burn-in, the remaining trees were summarized in a maximum clade credibility tree with TreeAnnotator v1.8.0 with the posterior possibility (PP) limit set to zero and including mean node heights. Finally, the summary tree was visualized with FigTree v1.4.0 ( http://tree.bio.ed.ac.uk/software/figtree/).
Genetic diversity and neutrality tests
Nuclear sequences with additive peaks were phased and separated into two allele sequences by PHASE46 in the software package DnaSP version 5.00.0447. Then, we used the nuclear sequences to estimate the basic population genetic parameters, including the number of segregating sites (S), Watterson’s parameter (θw), nucleotide diversity (π), the minimum number of recombinant events (Rm), haplotype number (Nh) and diversity (He) in the same software package47. We classified the segregating sites (S) into four categories, S1, S2, Ss and Sf. For each locus, S1 and S2 are the numbers of polymorphic sites unique to samples 1 and 2, respectively, Ss is the number of sites with alleles shared between the two samples, and Sf is the number of sites with fixed alleles in either sample.
We also calculated Tajima’s D, Fu and Li’s D* and F* statistics and Fay and Wu’s H using the DnaSP version 5.00.0447 to test for departure from the standard neutral model, and J. communis was used as an outgroup for Fay and Wu’s H neutrality tests. Both D and H are expected to be zero under the standard neutral model. We used the multi-locus Hudson–Kreitman–Aguade test48 to assess the fit of data to the neutral equilibrium mode using J. communis as an outgroup. Finally, we used the recently developed maximum frequency of derived mutations (MFDM) test to examine the likelihood of natural selection acting on individual loci at the species level49.
Population structure and genetic delimitation
To quantify the extent of population genetic structure, first we estimated Wright’s fixation index (FST) and net sequence divergence (Da) at each nuclear DNA locus for each species pair using an analysis of molecular variance as implemented in ARLEQUIN version 3.1.150 and DnaSP version 5.00.0447. Similarly, to assess population differentiation within and among species, ΦST at the multi-locus level was calculated for each species pair. ΦST values were estimated by AMOVA using ARLEQUIN version 3.1.150 on a locus-by-locus basis as well as averaged across all loci. The significance of FST and ΦST were tested based on 10,000 permutations.
Additionally, Bayesian clustering of genotypes were performed in the program STRUCTURE version 2.3.251 to assess the correspondence between geographical groupings and genotypic clustering based on the nuclear DNA sequences; linkage disequilibrium at each site was determined by significant Fisher’s exact test after Bonferroni correction, and only sites under linkage disequilibrium were considered in the STRUCTURE analyses. To infer the structure of the sampled populations, the likelihood of all clustered subdivision schemes (values of K from 1 to 10) were explored using an admixture model. We performed 20 independent runs per K with a burn-in of 200,000 and then 500,000 iterations. Graphics were illustrated using the program DISTRUCT version1.152. The most likely number of clusters was estimated with the original method53 and the ΔK statistic method54 (Figs. 5B, C); both were implemented in Structure Harvester55.
Inference and simulation of demographic histories
We used IMa2 based on the Isolation with Migration (IM) model to investigate current and ancestral effective population sizes, long-term rates of effective migration and splitting time parameters. The IM model assumes that the variation within the data set is neutral, there is no recombination within the gene and random mating in ancestral and descendent populations22,56. First, the IMGC program57 was employed to exclude the recombination loci of the genes. Then, we analyzed species in a pairwise manner using a basic two-population model. We used a geometric heating scheme and a burn-in of 1,000,000 steps and ran MCMC under the HKY model of nucleotide substitution, saving 200,000 genealogies. Parameters were estimated based on an average mutation rate (μ = 1.94 × 10−10 per site per year) according to a previous study focusing on a group of Juniperus species25. The average generation time was set to 50 years according to previous field surveys and studies of other Cupressaceae species58. Finally, we used the IMfig program to generate the figures of an Isolation with Migration model.
To infer the demographic dynamics of J. microsperma, we estimated the exponential population growth rate parameter using LAMARC version 2.1.859 (available at http://evolution.genetics.washington.edu/lamarc/index.html), which can perform Bayesian analysis to account for the genealogical relationships among alleles and allows for population size changes, to assess the population dynamics based on multiple nuclear loci. We conducted two LAMARC runs; each initial chain was performed with 5,000 samples and each final chain was performed with 50,000 samples. The effective sample size (ESS) was used to determine the Bayesian statistical significance of each parameter. Large and positive values of the exponential growth parameter g indicate population expansion, but negative g values indicate population shrinkage, whereas relatively small positive values (g = 10) may indicate little or no growth.
To further understand the demographic history of J. microsperma, we also employed an approximate Bayesian computation (ABC) framework in DIYABC 2.0.423 to test five plausible scenarios of demographic changes based on nuclear DNA sequences (Fig. 8). Because no genetic structure was found in this species (Fig. S1), all scenarios were simulated under the framework of a single population. Furthermore, all scenarios assumed the same initial population size (Ne), whereas scenario 1 assumed ancient population growth (t4), a larger and stable population size (N3) and a recent bottleneck (t1, N1); scenario 2 assumed an ancient bottleneck event (t4), and subsequently moderate but stable population size (N2) and a recent bottleneck (t1, N1); scenario 3 assumed an ancient bottleneck event (t4) and subsequently smaller stable population size (N1); on the basis of scenario 1, both scenarios 4 and 5 assumed one additional bottleneck that occurred at different times (t3 and t2, respectively), which led to the same population size (N2). The prior distributions of demographic parameters are listed in Table S9. The mean mutation rate per nucleotide and per generation were drawn in Uniform [10–9, 10–8] for each sequenced locus, and we simulated 1 × 106 data sets for each of the five competing scenarios.
Evaluations of the fitness of each scenario compared to the real data sets were subsequently conducted. First, after simulation of each scenario, the ‘pre-evaluation scenario-prior combinations’ option in DIYABC was employed to check the similarity between the simulated data of each scenario and the observed data. Then, to compare different models, we estimated the posterior probabilities of the competing scenarios by employing polychotomous logistic regression on the 1% of simulated data sets closest to the observed data set; and the scenario with the highest posterior probability was selected as the best. Third, type I and type II errors were evaluated to assess the confidence in choice of scenario60. While type I errors were estimated by counting the proportion of instances in which the chosen scenario did not exhibit the highest posterior probability compared to the competing scenarios, type II errors were estimated by counting the proportion of data sets of the other scenarios that resulted in the highest posterior probability of the chosen scenario. Subsequently, we estimated the posterior distributions of parameters under the best scenario. To evaluate the precision of parameter estimation, we computed the median of the absolute error divided by the true parameter value of the 10,000 pseudo-observed data sets simulated under the best scenario using the median of the posterior distribution as point estimate. Finally, we performed model checking for each studied model to assess the ability of a given scenario to produce data sets similar to the real data set.
Author Contributions
K.S.M. designed the research, K.S.M. collected samples, H.Y.S. and M.D. performed experiments, H.Y.S. and Z.H.L. analyzed data, H.Y.S. and K.S.M. prepared figures and tables, H.Y.S., Z.H.L. and K.S.M. wrote the manuscript, and H.Y.S., Z.H.L., M.D., R.P.A., G.M., L.O. and K.S.M. revised the manuscript.
Additional Information
How to cite this article: Shang, H.-Y. et al. Evolutionary origin and demographic history of an ancient conifer (Juniperus microsperma) in the Qinghai-Tibetan Plateau. Sci. Rep. 5, 10216; doi: 10.1038/srep10216 (2015).
Supplementary Material
Acknowledgments
We are grateful to Prof. Jian-Quan Liu for supervising this work. This work was financially supported by the National Basic Research Program of China (grant 2014CB954100), the National Natural Science Foundation of China (grants 31100488, 31370261), Sichuan Provincial Department of Science and Technology (grant 2015JQO018) and Sichuan University.
References
- Qiu Y.-X., Fu C.-X. & Comes H. P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Molecular Phylogenetics and Evolution 59, 225–244 (2011). [DOI] [PubMed] [Google Scholar]
- Liu J.-Q., Sun Y.-S., Ge X.-J., Gao L.-M. & Qiu Y.-X. Phylogeographic studies of plants in China: Advances in the past and directions in the future. Journal of Systematics and Evolution 50, 267–275 (2012). [Google Scholar]
- Wen J., Zhang J. Q., Nie Z. L., Zhong Y. & Sun H. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Frontiers in genetics 5, 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre A. et al. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biological reviews of the Cambridge Philosophical Society, 10.1111/brv.12107 (2014). [DOI] [PubMed] [Google Scholar]
- Zheng D. & Yao T. D. Uplifting of Tibetan Plateau with its environmental effects. Science Press 2004). [Google Scholar]
- Shi Y. F., Li J. J. & Li B. Y. Uplift and environmental changes of Qinghai-Xizang (Tibetan) Plateau in the late Cenozoic. Guangdong Scirence & Technology Press 1998). [Google Scholar]
- Zheng B. X., Xu Q. Q. & Shen Y. P. The relationship between climate change and Quaternary glacial cycles on the Qinghai-Tibetan Plateau: review and speculation. Quaternary International 97-98, 93–101 (2002). [Google Scholar]
- Zhou S. Z. & Li J. J. The sequence of Quaternary glaciation in the Bayan Har Mountains. Quaternary International 45-46, 135–142 (1998). [Google Scholar]
- Liu J.-Q., Duan Y.-W., Hao G., Ge X.-J. & Sun H. Evolutionary history and underlying adaptation of alpine plants on the Qinghai-Tibet Plateau. Journal of Systematics and Evolution 52, 241–249 (2014). [Google Scholar]
- Mao K. S., Hao G., Liu J. Q., Adams R. P. & Milne R. I. Diversification and biogeography of Juniperus (Cupressaceae): variable diversification rates and multiple intercontinental dispersals. New Phytol. 188, 254–272 (2010). [DOI] [PubMed] [Google Scholar]
- Xu T. et al. Phylogeography and allopatric divergence of cypress species (Cupressus L.) in the Qinghai-Tibetan Plateau and adjacent regions. BMC evolutionary biology 10, 194 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F. S., Qin A. L., Li Y. F. & Wang X. Q. Great genetic differentiation among populations of Meconopsis integrifolia and its implication for plant speciation in the Qinghai-Tibetan Plateau. PLoS One 7, e37196 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Wang A., Wan D., Wang Q. & Liu J. Rapid radiation of Rheum (Polygonaceae) and parallel evolution of morphological traits. Molecular Phylogenetics and Evolution 63, 150–158 (2012). [DOI] [PubMed] [Google Scholar]
- Liu J. Q., Gao T. G., Chen Z. D. & Lu A. M. Molecular phylogeny and biogeography of the Qinghai-Tibet Plateau endemic Nannoglottis (Asteraceae). Mol. Phylogenet Evol. 23, 307–325 (2002). [DOI] [PubMed] [Google Scholar]
- Liu J. Q., Wang Y. J., Wang A. L., Hideaki O. & Abbott R. J. Radiation and diversification within the Ligularia-Cremanthodium-Parasenecio complex (Asteraceae) triggered by uplift of the Qinghai-Tibetan Plateau. Molecular Phylogenetics and Evolution 38, 31–49 (2006). [DOI] [PubMed] [Google Scholar]
- Wang Y. J., Susanna A., Raab-straube E. V., Milne R. I. & Liu J. Q. Island-like radiation of Saussurea (Asteraceae: Cardueae) triggered by uplifts of the Qinghai-Tibetan Plateau. Biological Journal of the Linnean Society 97, 893–903 (2009). [Google Scholar]
- Wang L. et al. History and evolution of alpine plants endemic to the Qinghai-Tibetan Plateau: Aconitum gymnandrum (Ranunculaceae). Mol. Ecol. 18, 709–721 (2009). [DOI] [PubMed] [Google Scholar]
- Farjon A. A monograph of Cupressaceae and Sciadopitys. Royal Botanic Gardens: Kew, 2005). [Google Scholar]
- Adams R. P. Junipers of the World: The genus Juniperus. 4th edn Trafford Publishing 2014). [Google Scholar]
- Adams R. P., Mao K. S. & Liu J. Q. The volatile leaf oil of Juniperus microsperma and its taxonomy. Phytologia 95, 87–93 (2013). [Google Scholar]
- Heled J. & Drummond A. J. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27, 570–580 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. Isolation with migration models for more than two populations. Mol. Biol. Evol. 27, 905–920 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornuet J. M. et al. DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30, 1187–1189 (2014). [DOI] [PubMed] [Google Scholar]
- Beaumont M. A. Approximate Bayesian computation in evolution and ecology. Annual Review of Ecology, Evolution, and Systematics 41, 379–406 (2010). [Google Scholar]
- Li Z. H. et al. Population genetic evidence for complex evolutionary histories of four high altitude juniper species in the Qinghai-Tibetan Plateau. Evolution 66, 831–845 (2012). [DOI] [PubMed] [Google Scholar]
- Chikhi L., Sousa V. C., Luisi P., Goossens B. & Beaumont M. A. The Confounding Effects of Population Structure, Genetic Diversity and the Sampling Scheme on the Detection and Quantification of Population Size Changes. Genetics 186, 983–U347 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K. S. et al. Distribution of living Cupressaceae reflects the breakup of Pangea. Proceedings of the National Academy of Sciences of the United States of America 109, 7793–7798 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson R. R. Gene genealogies and the coalescent process. Oxford Surveys in Evolutionary Biology 7, 1–44 (1991). [Google Scholar]
- Tian X., Luo J., Wang A., Mao K. & Liu J. On the origin of the woody buckwheat Fagopyrum tibeticum (=Parapteropyrum tibeticum) in the Qinghai-Tibetan Plateau. Mol. Phylogenet Evol. 61, 515–520 (2011). [DOI] [PubMed] [Google Scholar]
- Fu L. K., Yu Y. F. & Farjon A. in Flora of China Vol. 4 (eds Wu Z. Y. & Raven P. H. ) 62–77 Science Press & Missouri Botanical Garden Press 1999). [Google Scholar]
- Freeland J. R., Kirk H. & Petersen S. D. Molecular ecology. 2nd edn John Wiley & Sons 2011). [Google Scholar]
- Li Z., Zhang Q., Liu J., Källman T. & Lascoux M. The Pleistocene demography of an alpine juniper of the Qinghai-Tibetan Plateau: tabula rasa, cryptic refugia or something else? Journal of Biogeography 38, 31–43 (2011). [Google Scholar]
- Moreno-Letelier A., Mastretta-Yanes A. & Barraclough T. G. Late Miocene lineage divergence and ecological differentiation of rare endemic Juniperus blancoi: clues for the diversification of North American conifers. New Phytol. 203, 335–347 (2014). [DOI] [PubMed] [Google Scholar]
- Li Y. et al. Demographic histories of four spruce (Picea) species of the Qinghai-Tibetan Plateau and neighboring areas inferred from multiple nuclear loci. Molecular biology and evolution 27, 1001–1014 (2010). [DOI] [PubMed] [Google Scholar]
- Wang B. et al. Extremely low nucleotide polymorphism in Pinus krempfii Lecomte, a unique flat needle pine endemic to Vietnam. Ecology and evolution 4, 2228–2238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L. et al. Mitochondrial and chloroplast phylogeography of Picea crassifolia Kom. (Pinaceae) in the Qinghai-Tibetan Plateau and adjacent highlands. Mol. Ecol. 16, 4128–4137 (2007). [DOI] [PubMed] [Google Scholar]
- Latalowa M. & van der Knaap W. O. Late Quaternary expansion of Norway spruce Picea abies (L.) Karst. in Europe according to pollen data. Quaternary Science Reviews 25, 2780–2805 (2006). [Google Scholar]
- IUCN. The IUCN red list of threatened species, version 2014.2. (2014). Available at: http://www.iucnredlist.org. (Accessed 20th November 2014)
- Adams R. P. Systematics of the one seeded Juniperus of the eastern hemisphere based on leaf essential oils and random amplified polymorphic DNAs (RAPDs). Biochemical Systematics and Ecology 28, 529–543 (2000). [DOI] [PubMed] [Google Scholar]
- Doyle J. J. & Doyle J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19, 11–15 (1987). [Google Scholar]
- Sakaguchi S., Tsumura Y., Crisp M. D., Bowman D. M. J. S. & Isagi Y. Genetic evidence for paternal inheritance of the chloroplast in four Australian Callitris species (Cupressaceae). J Forest Res.-Jpn. 19, 244–248 (2014). [Google Scholar]
- Tamura K. et al. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Molecular Biology and Evolution 28, 2731–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt H.-J., Forster P. & Röhl A. Median-joining networks for inferring intraspecific phylogenies. Molecular biology and evolution 16, 37–48 (1999). [DOI] [PubMed] [Google Scholar]
- Opgenoorth L. et al. Tree endurance on the Tibetan Plateau marks the world’s highest known tree line of the Last Glacial Maximum. New Phytol. 185, 332–342 (2010). [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J. P. & Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001). [DOI] [PubMed] [Google Scholar]
- Stephens M. & Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. The American Journal of Human Genetics 73, 1162–1169 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P. & Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009). [DOI] [PubMed] [Google Scholar]
- Hudson R. R., Kreitman M. & Aguadé M. A test of neutral molecular evolution based on nucleotide data. Genetics 116, 153–159 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A new test for detecting recent positive selection that is free from the confounding impacts of demography. Molecular biology and evolution 28, 365–375 (2011). [DOI] [PubMed] [Google Scholar]
- Excoffier L., Laval G. & Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinform Online 1, 47–50 (2005). [PMC free article] [PubMed] [Google Scholar]
- Hubisz M. J., Falush D., Stephens M. & Pritchard J. K. Inferring weak population structure with the assistance of sample group information. Molecular ecology resources 9, 1322–1332 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N. A. DISTRUCT: a program for the graphical display of population structure. Molecular Ecology Notes 4, 137–138 (2004). [Google Scholar]
- Pritchard J. K., Stephens M. & Donnelly P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G., Regnaut S. & Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular ecology 14, 2611–2620 (2005). [DOI] [PubMed] [Google Scholar]
- Earl D. A. & Vonholdt B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4, 359–361 (2012). [Google Scholar]
- Hey J. & Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167, 747–760 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner A. E., Cox M. P. & Hammer M. F. Recombination-filtered genomic datasets by information maximization. Bioinformatics 23, 1851–1853 (2007). [DOI] [PubMed] [Google Scholar]
- Fujimoto A., Kado T., Yoshimaru H., Tsumura Y. & Tachida H. Adaptive and slightly deleterious evolution in a conifer, Cryptomeria japonica. J. Mol. Evol. 67, 201–210 (2008). [DOI] [PubMed] [Google Scholar]
- Kuhner M. K. LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 22, 768–770 (2006). [DOI] [PubMed] [Google Scholar]
- Cornuet J. M., Ravigne V. & Estoup A. Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). Bmc. Bioinformatics 11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.