

Abstract
Background
Arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome is a multisystem autosomal-recessive disorder caused by defects in the VPS33B and VIPAR genes, involved in localization of apical membrane proteins. Affected children usually die by 1 year of age, often secondary to infective complications. The classic renal manifestation previously described in ARC syndrome is proximal–tubular dysfunction. The aim of this study is to gain further insight into the renal manifestations of this syndrome.
Methods
Clinical review of three cases of ARC syndrome presenting to a tertiary centre. Together with measurement of VPS33B and VIPAR protein expression in the human glomerulus.
Results
The cases demonstrated severe failure to thrive and in addition to commonly described features profound proteinuria and albuminuria, together with hypoalbuminaemia, suggesting glomerular involvement of this syndrome. Western blotting of conditionally immortalized human glomerular cells and ex vivo immunofluorescent analysis of the human glomerulus revealed that VPS33B and VIPAR were highly expressed in glomerular endothelium, and podocytes, but not in the mesangium.
Conclusions
ARC syndrome affects the glomerulus as well as the proximal tubule in the kidney. Our molecular studies suggest that both cell types that constitute the glomerular filtration barrier are affected in this condition, providing an explanation for the albuminuria that we have observed in our cases.
Keywords: albuminuria, ARC syndrome, glomerulus, VIPAR, VPS33B
Introduction
Arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome is a rare autosomal-recessive multisystem disorder which is usually fatal by 1 year of age. Wide phenotypic variability is recognized [1] and multiple associated clinical features are reported in the literature, including severe failure to thrive, poor feeding, gastro-oesophageal reflux, developmental delay, ichthyosis, dysmorphic features, corpus callosum dysgenesis, recurrent infections, abnormal platelets, bleeding tendency, diarrhoea, pathological fractures, sensorineural deafness, structural cardiac defects and hypothyroidism [1, 2].
The classically recognized renal manifestation of ARC syndrome is proximal renal tubular dysfunction [2] accompanied by glycosuria, phosphaturia, generalized aminoaciduria, renal tubular acidosis and hypernatremic dehydration. The severity of renal tubular acidosis and hypernatremic dehydration commonly worsens during episodes of intercurrent illness. Proteinuria has previously been noted in a small number of cases [1–7] but not investigated further.
Approximately 75% of cases of ARC syndrome are caused by mutations in VPS33B [8], which is a gene involved in the regulation of vesicle to target SNARE complex formation and membrane fusion [9]. In addition, mutations have recently been identified in VIPAR (VPS33B Interactor protein apical-basolateral polarity regulator), in cases without VPS33B mutations. VIPAR has been shown to form a complex with VPS33B, which together has diverse functions in pathways regulating apical-basolateral polarity in the liver and kidneys [10].
This manuscript describes the clinical details of three cases of ARC syndrome presenting to a tertiary Children's Hospital over 4 years, focussing on renal manifestations. We present evidence that renal involvement extends to the glomerulus, and postulate that proteinuria secondary to a glomerular problem may underlie some of the clinical complications of ARC syndrome.
Materials and methods
Patients
Clinical data for three patients that presented consecutively to a single tertiary paediatric centre were obtained retrospectively from case notes and pathology records. A clinical diagnosis of ARC syndrome was based on the presence of athrogryposis, renal tubular dysfunction and cholestasis.
Genotyping
Genetic analysis for both VPS33B and VIPAR mutations was performed as previously described [9, 10].
VPS33B and VIPAR western blotting in human glomerular cells
Conditionally immortalized cells were derived from the three cell types that are present in the glomerulus, podocytes [11], glomerular endothelial cells (GEnC) [12] and mesangial cells [13]. This was achieved using a temperature sensitive large T antigen SV40 construct as previously described. The advantage of this technique is that when the cells are thermo-switched to 37°C they exit the cell cycle and are able to differentiate, expressing cellular markers of differentiation. We studied podocytes after 14 days of thermo-switching and GEnC and mesangial cells after 5 days. We also studied a proximal tubular cell line that was generated in the same manner [14].
Western blotting for VPS33B and VIPAR was performed on whole-cell lysate and equal amounts of protein analysed after estimating with a Bradford assay and checking with the actin housekeeping protein as previously described [8, 15]. The VPS33B antibody was purchased from Proteintech, Manchester, UK, and VIPAR antibody was from Sigma, Pool, UK.
VIPAR glomerular immuno-fluorescence analysis
This was performed on human glomeruli from transplant kidneys, which had been obtained with full ethical permission, that were unsuitable for transplantation due to their vasculature. Techniques as previously described were used to do this [16]. The same VIPAR antibody as described above was used at a concentration of 1:100 with a GFP tagged anti-rabbit secondary (Jackson, West Grove). Nephrin was stained using a guinea pig (Acris, Germany) antibody with an anti-guinea pig TRITC labelled secondary (Jackson, Westgrove). We also tried VPS33B but this antibody was poor for immuno-fluorescence of the kidney (data not shown).
Results
Case reports
Additional clinical features of the three cases are summarized in Table 1.
Table 1.
Selected clinical features of the three cases of ARC syndrome
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Birth weight (kg) | 1.9 kg (2nd centile) |
2.6 kg (9th centile) |
2.6 kg (2nd centile) |
| Last weight (kg) | At 4 months–2.3 kg (<0.4th centile) |
At 9 months–5.6 kg (<0.4th centile) |
At 19 months-5.6 kg (<0.4th centile) |
| Additional features | |||
|
Proteinuria/haematuria Protein:creatinine ratio range (normal <45 mg/mmol in adults) |
+ 1326–3051 mg/mmol No haematuria |
+ 547–2750 mg/mmol No haematuria |
+ 1947–3056 mg/mmol No haematuria |
|
Albuminuria Albumin:creatinine ratio (normal 2.5–3 mg/mmol in adults [22]) |
Not recorded | + 249 mg/mmol |
+ 77 mg/mmol |
| Renal impairment | Intermittent | Intermittent | Intermittent |
| Nephrocalcinosis on renal USS | + | + | + |
|
Metabolic acidosis Lowest serum bicarbonate (normal 21–34 mmol/L) |
+ 9 mmol/L |
+ 7 mmol/L |
+ 10 mmol/L |
|
Hypoalbuminaemia lowest serum albumin (normal 29–55 g/L) |
+ 17 g/L |
+ 12 g/L |
+ 17 g/L |
| Hypothyroidism | + | Transient | − |
| Vitamin D deficiency | + | + | + |
| Secondary hyperparathyroidism | + | + | + |
| Pathological fractures | + Left femur |
+ Left 7th and 8th ribs |
+ Right femur, tibia, radius and ulna |
|
Recurrent infection Pathogens isolated |
+ Enterococcus (×2), Candida (×2), Pseudomonas, Beta haemolytic Streptococcus, Extended spectrum Beta lactamase, Coagulase negative Staphylococcus |
+ Staphylococcus aureus Gram-negative diplococci (type not specified) Respiratory Syncytial virus (×2) Human herpes virus type 6 |
+ Pneumococcus, Enterococcus, Candida (×3) |
| Immunological findings | Low immunoglobulin levels | Not assessed | Normal immunoglobulin levels |
| Haematological findings | Normocytic anaemia Normal platelet count | Normocytic anaemia Normal haematinics Normal platelet count |
Normocytic anaemia Normal haematinics Normal platelet count with large platelets noted on blood film |
+ = present.
Case 1
The first case was a male infant born at 36 weeks gestation by spontaneous assisted breech delivery, with a birth-weight on the 2nd centile. It was the first pregnancy of first cousin parents of Pakistani origin, with no known significant family history (although a high degree of consanguinity was noted in the wider family). The mother had received no antenatal care in the UK, but scans in Pakistan demonstrated oligohydramnios, intra-uterine growth restriction (IUGR), reduced movements and flexed posture of both upper and lower limbs. The infant was in a poor condition at birth with low APGAR scores and required 5 min of ventilation. Neonatal examination revealed a small thin baby with dry scaly skin, bilateral lower limb contractures, subluxed hips and talipes calcaneovalgus. A swelling of the left thigh was found to be a femoral shaft fracture. Nasogastric tube feeding was commenced due to poor feeding but was followed by persistent vomiting and the baby had lost 15% of his birth weight by 14 days of age. Further evaluation revealed urosepsis, polyuria, heavy proteinuria and glycosuria, and blood results showing a renal tubular acidosis, hypernatremia, hyperchloraemia, anaemia, high inflammatory markers and low serum albumin levels. He was commenced on bicarbonate and total parental nutrition. By 5 weeks of age, he had developed cholestasis and malabsorption (but importantly liver function tests initially showed normal Bilirubin and alanine transaminase levels when the child was hypoalbuminaemic with extremely high urinary protein:creatinine levels) and blood results subsequently demonstrated conjugated hyper-bilirubinaemia with normal gamma GT levels. Multiple other features emerged over time (Table 1). ARC syndrome was first suggested as a possibility at 1 month of age and genetic analysis subsequently confirmed the presence of a homozygous VPS33B p.Arg438X mutation most common in UK patients of Pakistani origin with ARC [2]. Due to a combination of medical and social issues, he never left hospital and died at the age of 4 months from presumed sepsis although no causative organism could be identified. This was the first patient who presented to our services and we did not formally measure urinary albumin:creatinine levels although we did find extremely high protein:creatinine levels, low serum albumin and low circulating immunoglobulins.
Case 2
This male infant was the second of dizygotic twins, born at 37 + 6 weeks. Birth weight was on the 9th centile and knee contractures were noted on neonatal examination. It was the first pregnancy of White British non-consanguineous parents, with no significant family history. IUGR was identified on antenatal scans, but the pregnancy was otherwise normal and the first twin was unaffected. The index case was heavier than his twin at birth, but developed early problems with vomiting, hypoglycaemia, difficulty establishing feeds and weight loss of 17% by Day 5. In the second week of life, he developed abdominal distension and metabolic acidosis. By 1 month of age he was noted to have deranged liver function tests and pale stools, suggestive of progressive intrahepatic cholestasis. He spent 2 months on the Neonatal Intensive Care Unit where the triad of ARC features was identified, plus multiple other abnormalities (Table 1) including mitral valve regurgitation. Genetic analysis was negative for VPS33B, but confirmed mutations in the VIPAR gene [compound heterozygote for pathogenic frameshift mutation (c.463_464delTG; p.Trp155GlufsX4) and pathogenic nonsense mutation (c.484C>T: p.Arg162X)]. He had highly elevated levels of albuminuria (one hundred times normal reference values) and persistently low serum albumin levels despite improvement in his liver disease. He was admitted at 9 months of age and deteriorated rapidly with multi-organ failure despite management on PICU. At this time, he was found to be respiratory syncytial virus positive.
Case 3
The last case was a male infant, born at term by normal vaginal delivery, with a birth weight on the 2nd centile. It was the second pregnancy of first cousin Pakistani parents, with no known significant family history. No problems were identified during pregnancy. At 17 days old, he was brought to the Accident and Emergency department floppy and unwell, with reduced feeding and weight loss. On examination he was found to be thin, pale and jaundiced, with dry skin, bilateral talipes and fixed contractures of the hips and knees. Urinalysis revealed proteinuria and glycosuria and blood results demonstrated conjugated hyperbilirubinaemia. Investigation of neonatal jaundice was undertaken and a diagnosis of biliary atresia considered. He also had raised urinary albumin:creatinine levels. On further evaluation, the possibility of ARC syndrome was raised, with the relevant genetic tests sent. He was readmitted at 12 weeks old with metabolic acidosis and sepsis secondary to a respiratory tract infection, and further phenotypic characteristics of ARC syndrome were identified. Genetic analysis was negative for VPS33B, but confirmed a mutation in the VIPAR gene [homozygous pathogenic nonsense mutation (c.808C>T; p.Arg270X) identified in exon 12]. He subsequently had a number of short stays in hospital, including admissions with infectious episodes and long bone fractures. At the age of 20 months, he passed away at home after a very short history of fever and increased respiratory rate, with an infection being the likely terminal event.
Further clinical features
In addition to the three cardinal features, further selected features and investigations are shown in Table 1. Poor feeding and recurrent vomiting were a consistent problem in all the cases, and achieving weight gain was difficult, despite the use of nasogastric tube feeding or total parenteral nutrition. All cases had dry scaly skin requiring regular application of emollients. Proximal renal tubular acidosis required fluid and electrolyte replacement and hypernatraemic dehydration worsened during periods of intercurrent illness. Severe metabolic bone disease was a common feature, and all cases suffered pathological fractures, which raised the need to carefully consider the fragility of these infants when deciding whether to undertake certain investigations or interventions. Similarly, the propensity to bleeding in ARC syndrome secondary to the documented platelet abnormalities was another important consideration in making such decisions. All cases experienced occult or overt bleeding secondary to such procedures as nasogastric tube replacement, with some episodes requiring transfusion. Neurologically, all cases were hypotonic and demonstrated global developmental delay and two of the three cases had severe bilateral sensorineural deafness. All cases had intermittent cholestasis with intermittent derangement of liver function tests but normal Gamma GT results, and normal liver appearance on ultrasound.
Glomerular expression of the ARC proteins, VPS33 and VIPAR
The renal tubular disorder in ARC syndrome has been well described, but the nephrotic syndrome observed in these cases has only been reported in a minority of previous reports in the literature.
In view of the nephrotic syndrome seen in these cases and the albuminuria detected in two of our patients, we were interested to examine the cells of the human glomerulus to determine whether there were any cell type located here that expressed the ARC-associated proteins. We therefore studied the three cells of the glomerulus (podocytes, GEnC and mesangial cells) using conditionally immortalized cells that we have developed in our laboratory [11–13]. We found that the cells that constitute the filtration barrier of the kidney, podocytes and GEnC, both expressed the ARC-associated protein VPS33B and VIPAR but that mesangial cells had greatly reduced expression. (Figures 1 and 2). To confirm these results ex vivo we also performed immuno-fluorescence on normal human glomeruli that had been obtained from discarded renal transplant specimens that were unsuitable for transplantation due to its vasculature. We found that the VIPAR antibody intensely stained the tubular compartment of the kidney as expected, but additionally it also robustly stained the glomerulus (Figure 3). Podocytes were positive for VIPAR as demonstrated by co-localization of signal with nephrin (Figure 3).
Fig. 1.

VPS33 expression in conditionally immortalized cells of the glomerulus—western blots. (A) Western blot of conditionally immortalized cells from the human glomerulus and positive control of proximal tubular cells. (B) Densitometry of VPS33B in human glomerular cells shows increased expression in podocytes and GEnC compared with mesangial cells. N = 3.
Fig. 2.
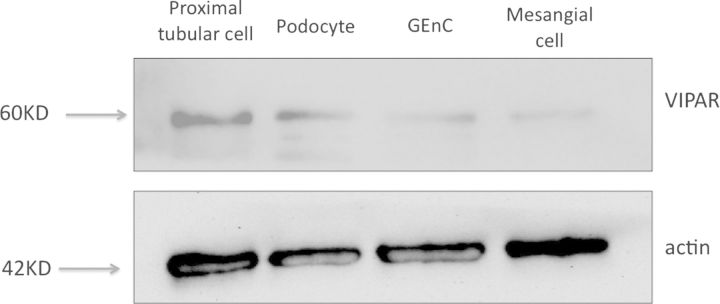
VIPAR expression in human immortalized cells of the glomerulus—western blots. Western blot of conditionally immortalized cells show VIPAR expressed in podocytes and GEnC with less expression in mesangial cells. Positive control of proximal tubular cells.
Fig. 3.
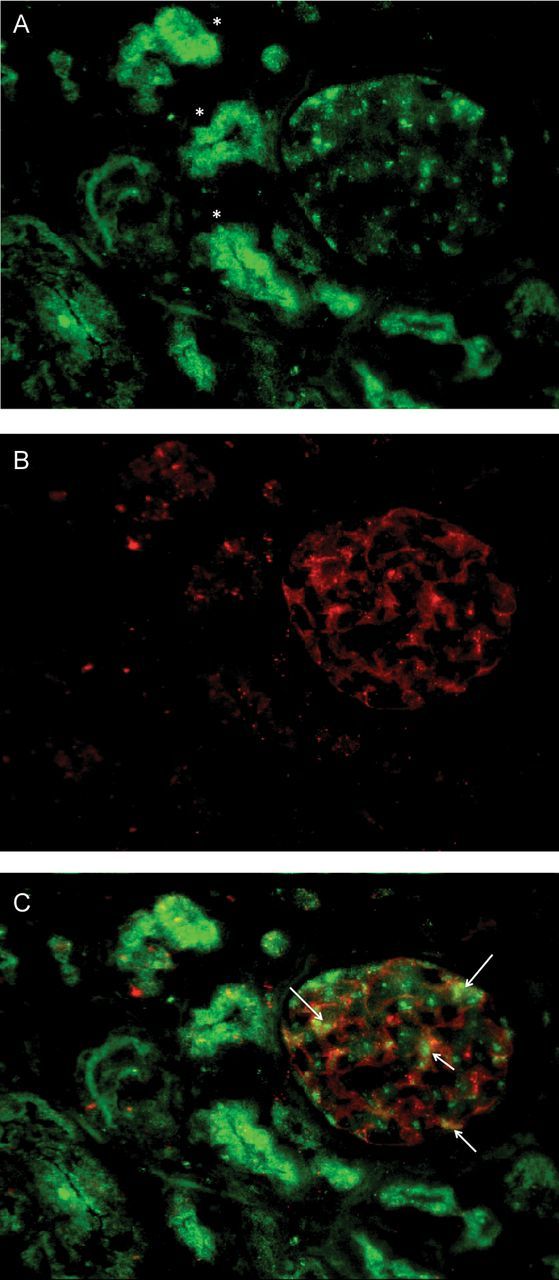
VIPAR immuno-localization in the glomerulus. (A) VIPAR is located in the tubules (asterisk) and glomeruli in human kidneys. (B) Nephrin staining and (C) merge image shows areas of podocyte co-localization (arrows).
Discussion
The three cases described highlight the universality of severe failure to thrive in ARC syndrome and the wide range of associated clinical features, including nephrotic range proteinuria. The three cardinal features of ARC syndrome were not all evident at presentation in any of the cases, only emerging later in the clinical course. This highlights the importance of raising awareness of the constellation of associated features in order to prevent cases being missed, to enable a timely diagnosis in view of the limited prognosis and to ensure optimal management.
Marked proteinuria and albuminuria, together with low serum albumin levels, were present in these infants, and have been mentioned previously in a number of case reports but never explored further [1–7]. We hypothesized that the high degree of urinary protein loss suggested that renal involvement may not be limited to tubular dysfunction and that the glomerulus might also be affected.
Analysis of the cells of the glomerulus showed that VPS33B and VIPAR proteins, whose genes are commonly mutated in ARC syndrome, were expressed here, with greater expression in the two cell types that constitute the filtration barrier of the kidney (podocytes and glomerular endothelial cells).
It is interesting that VPS33B and VIPAR are present and therefore may be functional in both cell types that constitute the filtration barrier of the glomerulus. To date, most genetic causes of albuminuria due to nephrotic syndrome have been shown to map to proteins found exclusively in the podocyte in the glomerulus [17, 18]. However, there is some recent intriguing data showing that mutations exclusively affecting the glomerular endothelium can also result in albuminuric renal disease [19]. It seems likely that in ARC syndrome that both the podocyte and GEnC may be involved in loss of integrity of filtration barrier. However, it remains to be elucidated if this is true, and if the mechanism causing dysfunction in podocytes and GEnC is similar.
Advocates of the ‘albumin retrieval hypothesis’ [20] may argue that the albuminuria observed in ARC syndrome is part of the known renal tubular dysfunction and occurs due to defective tubular re-absorption of filtered protein (as in Fanconi syndrome). However, it is now widely accepted that the glomerulus is the major part of the nephron responsible for the prevention of massive albuminuria [21], which we think was occurring in these cases. The low serum albumin observed in these patients may have been of a renal origin or secondary to their liver dysfunction, as has previously been described. However, in the cases presented, the derangement in liver function tests was intermittent and not temporally consistent with the hypoalbuminaemia observed, which was persistent and severe, suggesting a significant contribution from the kidneys. Furthermore, the levels of albuminuria when measured were up to one hundred times that normally found in the urine (Table 1). This suggests that glomerular protein loss may be contributing, in addition to synthetic hepatic dysfunction, to low circulating albumin levels.
As hypoalbuminaemia is commonly associated with ARC syndrome and there is evidence of glomerular loss of albumin, it may be sensible to consider prophylactic antibiotics, such as penicillin, in this condition as it is highly likely that other immunologically beneficial proteins are also being lost into the urine, as occurs in other nephrotic syndromes. In support of this we found low levels of circulating immunoglobulins in Case 1. It is also common that children with ARC syndrome die very quickly of presumed infective causes and often no precipitating cause is identified. However, this is a severe multi-organ disease with a very poor prognosis so this therapeutic intervention may not make major differences to children with this condition.
In conclusion, we have shown that there is glomerular involvement in the ARC syndrome, and that both podocytes and GEnC express the ARC proteins in the glomerulus. Going forward we would suggest that patients with ARC syndrome have their urinary albumin levels measured and that prophylactic antibiotics may be beneficial in some cases.
Funding
This work was supported by grants from Kidney Research UK and the Medical Research Council (MRC).
Conflict of interest statement
None declared.
Acknowledgements
The authors thank the families of the cases described, the Paediatric Nephrology team at the Bristol Royal Hospital for Children, the University of Bristol Academic Renal Unit and the Medical and Molecular Genetics unit at the University of Birmingham.
References
- 1.Eastham KM, McKiernan PJ, Milford DV, et al. ARC syndrome: an expanding range of phenotypes. Arch Dis Child. 2001;85:415–420. doi: 10.1136/adc.85.5.415. doi:10.1136/adc.85.5.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gissen P, Tee L, Johnson CA, et al. Clinical and molecular genetic features of ARC syndrome. Hum Genet. 2006;120:396–409. doi: 10.1007/s00439-006-0232-z. doi:10.1007/s00439-006-0232-z. [DOI] [PubMed] [Google Scholar]
- 3.Horslen SP, Quarrell OWJ, Tanner MS. Liver histology in the arthrogryposis multiplex congenita, renal dysfunction, and cholestasis (ARC) syndrome—report of 3 new cases and review. J Med Genet. 1994;31:62–64. doi: 10.1136/jmg.31.1.62. doi:10.1136/jmg.31.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jang JY, Kim KM, Kim GH, et al. Clinical characteristics and VPS33B mutations in patients with ARC syndrome. J Pediatr Gastroenterol Nutr. 2009;48:348–354. doi: 10.1097/mpg.0b013e31817fcb3f. [DOI] [PubMed] [Google Scholar]
- 5.Arhan E, Yusufoglu AM, Sayli TR. Arc syndrome without arthrogryposis, with hip dislocation and renal glomerulocystic appearance: a case report. Eur J Pediatr. 2009;168:995–998. doi: 10.1007/s00431-008-0860-5. doi:10.1007/s00431-008-0860-5. [DOI] [PubMed] [Google Scholar]
- 6.Coleman RA, VanHove JLK, Morris CR, et al. Cerebral defects and nephrogenic diabetes insipidus with the ARC syndrome: additional findings or a new syndrome (ARCC-NDI)? Am J Med Genet. 1997;72:335–338. doi:10.1002/(SICI)1096-8628(19971031)72:3<335::AID-AJMG16>3.0.CO;2-U. [PubMed] [Google Scholar]
- 7.Taha D, Khider A, Cullinane AR, et al. A novel VPS33B mutation in an ARC syndrome patient presenting with osteopenia and fractures at birth. Am J Med Genet. 2007;143A:2835–2837. doi: 10.1002/ajmg.a.32051. doi:10.1002/ajmg.a.32051. [DOI] [PubMed] [Google Scholar]
- 8.Cullinane AR, Straatman-Iwanowska A, Seo JK, et al. Molecular investigations to improve diagnostic accuracy in patients with ARC syndrome. Hum Mutat. 2009;30:E330–E337. doi: 10.1002/humu.20900. doi:10.1002/humu.20900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gissen P, Johnson CA, Morgan NV, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400–404. doi: 10.1038/ng1325. doi:10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 10.Cullinane AR, Straatman-Iwanowska A, Zaucker A, et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat Genet. 2010;42:303–312. doi: 10.1038/ng.538. doi:10.1038/ng.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saleem MA, O'Hare MJ, Reiser J, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 12.Satchell SC, Tasman CH, Singh A, et al. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int. 2006;69:1633–1640. doi: 10.1038/sj.ki.5000277. doi:10.1038/sj.ki.5000277. [DOI] [PubMed] [Google Scholar]
- 13.Sarrab RM, Lennon R, Ni L, et al. Establishment of conditionally immortalised human glomerular mesangial cells in culture, with unique migratory properties. Am J Physiol Renal Physiol. 2011;301:F1131–F1138. doi: 10.1152/ajprenal.00589.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilmer MJ, Saleem MA, Masereeuw R, et al. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010;339:449–457. doi: 10.1007/s00441-009-0882-y. doi:10.1007/s00441-009-0882-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gissen P, Johnson CA, Gentle D, et al. Comparative evolutionary analysis of VPS33 homologues: genetic and functional insights. Hum Mol Genet. 2005;14:1261–1270. doi: 10.1093/hmg/ddi137. doi:10.1093/hmg/ddi137. [DOI] [PubMed] [Google Scholar]
- 16.Coward RJ, Welsh GI, Yang J, et al. The human glomerular podocyte is a novel target for insulin action. Diabetes. 2005;54:3095–3102. doi: 10.2337/diabetes.54.11.3095. doi:10.2337/diabetes.54.11.3095. [DOI] [PubMed] [Google Scholar]
- 17.Brown EJ, Schlondorff JS, Becker DJ, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010;42:72–76. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lavin PJ, Gbadegesin R, Damodaran TV, et al. Therapeutic targets in focal and segmental glomerulosclerosis. Curr Opin Nephrol Hypertens. 2008;17:386–392. doi: 10.1097/MNH.0b013e32830464f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George M, Rainey MA, Naramura M, et al. Renal thrombotic microangiopathy in mice with combined deletion of endocytic recycling regulators EHD3 and EHD4. PLoS One. 2011;6:e17838. doi: 10.1371/journal.pone.0017838. doi:10.1371/journal.pone.0017838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russo LM, Sandoval RM, McKee M, et al. The normal kidney filters nephrotic levels of albumin retrieved by proximal tubule cells: Retrieval is disrupted in nephrotic states. Kidney Int. 2007;71:504–513. doi: 10.1038/sj.ki.5002041. doi:10.1038/sj.ki.5002041. [DOI] [PubMed] [Google Scholar]
- 21.Haraldsson B, Nyström J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. doi:10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- 22.Justesen T, Petersen J, Ekbom P, et al. Albumin-to-creatinine ratio in random urine samples might replace 24-h urine collections in screening for micro- and macroalbuminuria in pregnant woman with type 1 diabetes. Diabetes Care. 2006;29:924–925. doi: 10.2337/diacare.29.04.06.dc06-1555. doi:10.2337/diacare.29.04.06.dc06-1555. [DOI] [PubMed] [Google Scholar]