

Abstract
Stress and depression are associated with atrophy and loss of neurons in limbic and cortical brain regions that could contribute to the symptoms of depression. Typical monoamine reuptake inhibitor antidepressants have only modest efficacy and require long-term treatment, and are only weakly effective in blocking or reversing these structural changes caused by stress. Recent findings demonstrate that ketamine, an NMDA receptor antagonist, produces rapid antidepressant actions in difficult to treat depressed patients. In addition, preclinical studies demonstrate that ketamine rapidly increases synaptic connections in the prefrontal cortex by increasing glutamate signaling and activation of pathways that control the synthesis of synaptic proteins. Moreover, ketamine rapidly reverses the synaptic deficits caused by exposure to chronic stress in rodent models. Studies of the signaling mechanisms underlying the actions of ketamine have provided novel approaches and targets for new rapid acting antidepressants with decreased side effects, as well as a better understanding of the neurobiology of stress, depression, and treatment response.
Keywords: brain-derived neurotrophic factor, glycogen synthase kinase-3, lithium, mechanistic target of rapamycin, Scopolamine
Introduction
Preclinical and clinical studies of stress and depression have demonstrated a wide range of neurochemical and morphological alterations that could contribute to the pathophysiology of mood disorders. Moreover, the complexity and heterogeneity of depression make it difficult to identify a single underlying abnormality, and suggest that there are multiple causes of depression. However, studies over the past 15 to 20 years have continued to build on evidence that stress and depression are associated with atrophy and loss of neurons and glia, and even reduced volume of key limbic and cortical brain regions implicated in depression. These findings demonstrate alterations at a structural level and studies have focused on identifying the underlying signaling pathways, including regulation of neurotrophic factors that contribute to these morphological changes.
Currently available antidepressants, notably the serotonin selective reuptake inhibitors (SSRIs), require long-term treatment and have limited efficacy, with approximately one-third of patients responding to the first medication prescribed and up to two-thirds after multiple trials, which can take months or even years.[1] This highlights a major unmet need for new antidepressant agents with novel mechanisms that have faster onset of action and greater efficacy. Recent reports indicate that ketamine, an NMDA receptor antagonist, can address these limitations, demonstrating that a single dose of ketamine produces a rapid antidepressant response (within hours) in patients who have failed to respond to two or more typical antidepressants and are considered treatment resistant.[2] In addition, preclinical studies demonstrate that ketamine rapidly increases synaptic connections and thereby reverses the atrophy of neurons caused by chronic stress.
In this review, the studies demonstrating atrophy of limbic and cortical brain regions in stress and depression are discussed, as well as the effects of typical and novel rapid acting antidepressants. In particular, we present evidence on the mechanisms by which rapid acting agents increase spine synapses and cause antidepressant behavioral effects.[3] Together these findings further elucidate the neurobiology of depression and treatment response, and contribute to a synaptic hypothesis of mood disorders
Stress and Depression Cause Loss of Synaptic Connections
Brain imaging studies have provided consistent evidence that depression and other stress related illnesses, including posttraumatic stress disorder (PTSD) are associated with decreased size of brain regions implicated in depression. This includes decreased volume of the hippocampus, a limbic structure with high levels of glucocorticoid receptors that provides negative feedback to the hypothalamic-pituitary-adrenal (HPA) axis.[4] Decreased volume of the hippocampus is associated with the length of depressive illness and inversely related to the time of treatment,[5] and the reduced volume of the hippocampus is reversible with antidepressant treatment.[4] In addition, there is evidence of decreased volume of cortical regions, including the subgenual PFC and cingulate cortex.[6] Decreased volume and function of these brain regions is also associated with altered connectivity.[7]
The underlying cellular changes that account for the decreased hippocampal and cortical volumes have also been examined. Postmortem studies demonstrate that neuronal cell body size is decreased in the PFC and hippocampus with no change in cell number, [8,9] suggesting that neuronal processes and synaptic contacts may also be decreased. Support for the latter possibility is provided by a recent study demonstrating a significant reduction in the number of synapses in the dorsal lateral PFC in a small cohort of depressed subjects.[10] In addition to these neuronal alterations there is strong evidence from postmortem studies that the numbers of glia are decreased in cortical regions, particularly in the dlPFC and cingulate cortex of depressed subjects.[11]
Preclinical studies in rodent models of stress and depression have further examined the cellullar basis of these morphological changes. These reports demonstrate that different types of chronic stress (repeated restraint, unpredictable stress) decrease the number and length of apical dendrites in CA3 pyramidal neurons of the hippocampus and layers II/III and V of the medial PFC.[12,13] Moreover, chronic stress exposure decreases the number of spine synapse connections in both the PFC and hippocampus. This reduction in dendrite complexity and synaptic connections could contribute to the decreased volume of PFC and hippocampus observed in depressed patients. Moreover, loss of synaptic connections could contribute to a functional disconnection and loss of normal control of mood and emotion in depression (Fig. 1). In particular, the medial PFC exerts top down control over other brain regions that regulate emotion and mood, most notably the amygdala, and loss of synaptic connections from PFC to this and other brain regions could thereby result in more labile mood and emotion, as well as cognitive deficits.
Figure 1.
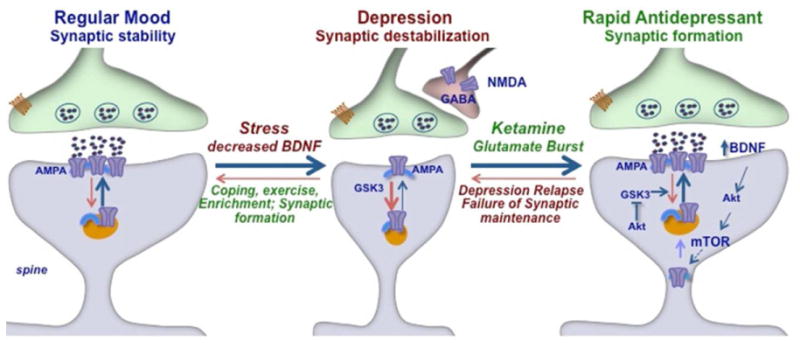
Model for the loss of synapses caused by stress and rapid induction of synaptic connections by ketamine. Synaptic connections in the PFC are maintained by normal circuit activity, but are decreased by chronic stress exposure, which also reduces the expression of BDNF. Ketamine can rapidly reverse the effects of stress by causing a burst of glutamate that causes a fast synaptogenic response. This is thought to occur via blockade of tonic firing GABAergic interneurons that express NMDA receptors. Ketamine induction of synapses requires BDNF and activation of the mTOR signaling pathway, which regulates the synthesis of new synaptic proteins, including glutamate AMPA receptors. The cycling of AMPA receptors to the membrane is also regulated by GSK-3β, a target of lithium that has been implicated in the actions of ketamine. Although relatively stable, the new synapses reverse after 7–10 days, consistent with the time course for relapse of depressed patients after a single dose of ketamine. The effects of stress on synaptic connections can also be influenced by exercise, enriched environment, and coping mechanisms.
In addition to loss of synapses and dendrites, preclinical studies demonstrate that stress decreases the birth of new neurons in the dentate gyrus of adult rodent hippocampus.[14] While there is evidence that antidepressants increase cell birth in hippocampus of depressed subjects (see below), there is currently no evidence that depression is associated with decreased numbers of new cells in this brain region. Finally, rodent studies also demonstrate that exposure to chronic stress decreases the number of glia in the PFC, which could contribute to the decreased volume of this region in depressed patients.[15]
Typical Antidepressant Agents and Regulation of Synaptic Connections
There are currently no reports on the influence of antidepressant treatment on synaptic connections in depressed subjects so this question must await future postmortem studies of spine synapses. However, this issue has been examined in rodent models. Administration of a typical antidepressant such as an SSRI does not influence synapse number in naïve, nonstressed animals, but can block or reverse the synaptic deficits caused by chronic stress exposure.[16] However, this rescue of sympatic loss requires chronic antidepressant administration of several weeks, consistent with the time course for the therapeutic action of these agents. There is also evidence that chronic SSRI administration increases synaptic plasticity in models that require flexibility, including extinction learning.[17]
Chronic antidepressant administration also increases neurogenesis in the adult rat hippocampus and postmortem studies demonstrate that depressed patients on medication at the time of death show elevated levels of cell birth in the hippocampus.[14] Preclinical studies demonstrate that chronic antidepressant administration increases the birth of glia in the PFC.[15] Together these effects on neuronal and glial number could contribute to the actions of antidepressant agents. Moreover, it is possible that altered levels of glia, which play an important role in providing metabolic support for neurons, could contribute to the regulation of dendrite complexity and synapse number in response to stress, depression and antidepressant treatment via direct or indirect mechanisms.[18]
Rapid Acting Antidepressants
The rapid (∼2 hr) antidepressant actions of the NMDA receptor antagonist ketamine are now well established.[19] Moreover, these studies demonstrate that ketamine decreases suicidal ideation and is even effective in depressed patients who have failed to respond to two or more typical antidepressants and are considered treatment resistant. Together the discovery of an agent that acts rapidly and through a completely novel mechanism has had a major impact on the field of depression and how we think about development of new antidepressant medications. Studies of the molecular and cellular-signaling pathways underlying the actions of ketamine have begun to elucidate a novel mechanism of action that may lead to safer rapid acting agents.
Ketamine Rapidly Increases Synaptic Connections: Role of Glutamate and mTORC1
Another discovery that has made an impact on the field is that a single dose of ketamine rapidly increases synaptic connections in the PFC and reverses the deficits caused by chronic unpredictable stress (CUS) exposure[20,21] (Fig. 1). This was surprising as alterations of synaptic connections have been studied primarily in models of learning and memory. The fact that an NMDA receptor antagonist produces this effect is even more puzzling as the induction of synaptic connections requires glutamate and activation, not inhibition of NMDA receptors. One possibility is that ketamine and other NMDA receptor antagonists cause a shift in AMPA to NMDA receptor ratio that contributes to an enhancement of synaptic function (Maeng et al., 2008). Another possibility that has received attention is based on the finding that NMDA receptor blockade increases glutamate transmission.
Despite the blockade of NMDA receptors, administration of ketamine actually increases extracellular levels of glutamate in the PFC.[22] This is thought to occur via blockade of NMDA receptors on GABAergic interneurons that inhibits their tonic firing, thereby causing disinhibition of glutamate transmission[23] (Fig. 1). However, this does not exclude the possibility that ketamine also has direct effects on postsynaptic glutamate synapses that also contribute to the increase in synaptogenesis. For example, the actions of ketamine require phosphorylation and inhibition of glycogen synthase kinase-3β (GSK-3β), which has been shown to regulate the insertion of glutamate receptors and synapse formation.[24] Studies are currently underway to further elucidate the pre- and postsynaptic mechanisms that contribute to the actions of ketamine.
The resulting burst of glutamate caused by ketamine then leads to activation of the signaling machinery that stimulates synapse formation. This includes activation of the mammalian target of rapamycin complex 1 (mTORC1), which controls the translation of synaptic proteins required for new synapse formation and has been implicated in long-term memory induced synaptic plasticity[25] (Fig. 1). Evidence that mTORC1 plays a functional role in the response to ketamine is provided by studies demonstrating that rapamycin, a selective inhibitor of mTORC1, blocks the synaptogenic and behavioral actions of ketamine.[20,21] Further studies have focused on the signaling pathways that stimulate mTORC1, including the role of BDNF and related signaling cascades.
Rapid Antidepressant Actions of Ketamine Require BDNF: Val66Met Allele is a Marker of Treatment Response in Depressed Patients
BDNF is a member of the nerve growth factor family and is one of the most abundant neurotrophic factors in the brain. It plays an important role during development, in the guidance, function and survival of neurons, but is also expressed at high levels in the adult brain and continues to play an important role in neuronal function, plasticity, and survival. BDNF is regulated at multiple levels, including transcriptional regulation of mRNA as well as activity dependent release of protein from nerve terminals, the latter of which is required for new synapse formation. Chronic administration of typical antidepressants increases the expression of BDNF in the PFC and hippocampus, and this effect is required for the behavioral actions of these agents.[26] However, there is no evidence that antidepressants increase the release of BDNF, which may limit the efficacy of typical agents in the regulation of synapse formation.
The behavioral and synaptic actions of ketamine also require BDNF, and the ability of ketamine to stimulate a glutamate burst that causes depolarization suggests that these effects occur via activity dependent release of BDNF. This possibility is supported by studies of a functional BDNF polymorphism, Val66Met, where the Met allele results in decreased processing and activity dependent release of BDNF. The BDNF Met allele is found in approximately 25 percent of the human population, and has been associated with decreased volume of the hippocampus and decreased executive function.[4] In addition, carriers of the Met allele who are exposed to early life stress or trauma are more likely to develop depression.[27]
Recent studies demonstrate that the synaptic and behavioral actions of ketamine are completely blocked in mutant mice with a knockin of the Met allele.[28] Moreover, a follow-up clinical study has found that the rapid actions of ketamine are significantly decreased in depressed patients who are carriers of the Met allele.[29] These findings provide evidence that the Val66Met allele has a functional impact on ketamine responsiveness and can be used as a biomarker of treatment response in future studies.
Scopolamine also Increases Synaptic Connections
Scopolamine, a muscarinic receptor antagonist, has also been reported to produce rapid antidepressant actions based on clinical assessment at 3 days after a single dose and with anecdotal evidence of a therapeutic response after 1 day.[30,31] The ability of scopolamine to produce a rapid antidepressant response raises the possibility that it might also be working through a mechanism similar to ketamine. This turns out to be the case, as we have found that a single dose of scopolamine rapidly increases synapse number and function in the PFC.[32] Moreover, we found that scopolamine increases levels of extracellular glutamate in the PFC, and that the effects of scopolamine are blocked by pretreatment with a glutamate AMPA receptor antagonist. Finally, a single dose of scopolamine also stimulates mTORC1 signaling and the behavioral actions of scopolamine are blocked by pretreatment with rapamycin.
Together, these findings indicate that stimulation of a glutamate burst, activation of mTORC1 signaling, and induction of synapse formation are common features of two different classes of rapid acting antidepressants. Studies are currently underway to determine the muscarinic receptor subtype (M1–M5) that mediates the actions of scopolamine, and to determine if the glutamate burst is due to inhibition of tonic firing GABAergic interneurons.
Lithium Enhances the Synaptogenic and Behavioral Actions of Ketamine
Because ketamine is a drug of abuse with side effects there have been efforts to identify alternative treatment strategies, including combination therapies that are also rapid acting and efficacious. Interestingly, combinations of ketamine with lithium, at doses that have no effects alone, have been reported to produce an antidepressant response in a behavioral model of depression, the forced swim test.[33] This prompted us to ask if a low-dose combination could also increase synaptogenesis and mTORC1 signaling. We found that a combination of ketamine (1 mg/kg) and lithium (10 mg/kg) at doses that have no effect alone significantly increases the number and function of spine synapses in the PFC.[34] The low-dose ketamine and lithium combination also produced significant antidepressant behavioral responses, replicating the previous report, and produced an effect that lasted for 7 days, consistent with the long-lasting synaptogenic actions of this combination treatment. Finally, we also found that the effects of lithium are mediated by inhibition of GSK-3β, as the combination of low-dose ketamine with a selective GSK-3β inhibitor produced the same synaptogenic and behavioral effects.[34]
Together these findings suggest that low-dose ketamine plus lithium could be a new treatment strategy that is safer and produces fewer side effects. It is also possible that this low-dose combination could be administered repeatedly with reduced toxicity. Finally, studies are currently being conducted to determine if lithium administration sustains the synaptogenic and behavioral actions of a single dose of ketamine.
Summary and Future Studies
The studies of rapid acting antidepressants highlight several novel concepts and mechanisms. Ketamine produces a rapid glutamate burst that stimulates the BDNF-mTORC1 cascade, leading to increased synaptic connections in the PFC. Importantly, the acute actions of ketamine on glutamate are transient, which limits excitoxicity, but the synaptogenic response is rapid and long-lasting (∼7 days), consistent with the time course of the therapeutic response. The relevance of this novel mechanism is supported by evidence that another rapid acting antidepressant, scopolamine also causes an induction of mTORC1 and synapse formation, and a behavioral response that is dependent on glutamate, providing markers to test additional rapid acting agents. The focus of current studies is to identify new, safer rapid acting antidepressants using these markers. In addition, work is underway to further elucidate the mechanisms that contribute to synaptic loss and depressive behaviors, to the relapse observed after ketamine treatment, as well as to the resilience to stress (Fig. 1).
References
- 1.Trivedi M, Rush AJ, Wisniewski SR, et al. STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psych. 2006;163(1):28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 2.Krystal J, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psych. 2013;73(12):1133–1141. doi: 10.1016/j.biopsych.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duman R, Aghajanian GK. Synaptic dysfunction in depression: novel therapeutic targets. Science. 2012;338(6103):68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacQueen G, Frodl T. The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psych. 2011;16(3):252–264. doi: 10.1038/mp.2010.80. [DOI] [PubMed] [Google Scholar]
- 5.Sheline Y, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Amer J Psych. 2003;160(8):1516–1518. doi: 10.1176/appi.ajp.160.8.1516. [DOI] [PubMed] [Google Scholar]
- 6.Drevets W, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213(1–2):93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price J, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35(1):192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajkowska G, Miguel-Hidalgo JJ, Wei J, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 9.Stockmeier C, Mahajan GJ, Konick LC, et al. Cellular changes in the postmortem hippocampus in major depression. Biol Psychiat. 2004;56(9):640–650. doi: 10.1016/j.biopsych.2004.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang H, Voleti B, Hajszan T, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. NatMed. 2012;18(9):1413–1417. doi: 10.1038/nm.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajkowska G, Miguel-Hidalgo JJ. Gliogenesis and glial pathology in depression. CNS Neurol Dis Drug Targets. 2007;6(3):219–233. doi: 10.2174/187152707780619326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McEwen B. The ever-changing brain: cellular and molecular mechanisms for the effects of stressful experiences. DevNeurobiol. 2012;72(6):878–890. doi: 10.1002/dneu.20968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison J, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13(4):240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samuels B, Hen R. Neurogenesis and affective disorders. Eur J Neurosci. 2011;33(6):1460–1468. doi: 10.1111/j.1460-9568.2011.07614.x. [DOI] [PubMed] [Google Scholar]
- 15.Banasr M, Dwyer JM, Duman RS. Cell atrophy and loss in depression: reversal by antidepressant treatment. Curr Opin Cell Biol. 2012;23(6):730–737. doi: 10.1016/j.ceb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bessa J, Ferreira D, Melo I, et al. Hippocampal neurogenesis induced by antidepressant drugs: an epiphenomenon in their mood-improving actions. Mol Psych. 2009;14(8):739. doi: 10.1038/mp.2008.119. [DOI] [PubMed] [Google Scholar]
- 17.Castren E. Neuronal network plasticity and recovery from depression. JAMA Psych. 2013;70(9):983–989. doi: 10.1001/jamapsychiatry.2013.1. [DOI] [PubMed] [Google Scholar]
- 18.Bialas A, Stevens B. Glia: regulating synapatogenesis from multiple directions. Curr Biol. 2013;22(19):R833–5. doi: 10.1016/j.cub.2012.08.036. [DOI] [PubMed] [Google Scholar]
- 19.Aan Het Rot M, Zarate CA, Jr, Charney DS, Mathew SJ. Ketamine for depression: where do we go from here? Biol Psych. 2012;72(7):537–547. doi: 10.1016/j.biopsych.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li N, Lee BY, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li N, Liu RJ, Dwyer J, et al. Glutamate N-methyl-d-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psych. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17(8):2912–2917. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27(43):11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beurel E, Song L, Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psych. 2011;16:1068–1070. doi: 10.1038/mp.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoeffer C, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33(2):67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duman R, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psych. 2006;59(12):1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Gatt J, Nemeroff CB, Dobson-Stone C, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psych. 2009;14:681–695. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- 28.Liu R, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psych. 2012;71(11):996–1005. doi: 10.1016/j.biopsych.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laje G, Lally N, Mathews D, et al. Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol Psych. 2012;72(11):27–28. doi: 10.1016/j.biopsych.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drevets W, Furey ML. Replication of scopolamine's antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psych. 2010;67:432–438. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furey M, Drevets WC. Antidepressant efficacy of the ant-imuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psych. 2006;63:1121–1129. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Voleti B, Navarria A, Liu RJ, et al. Scopolamine rapidly increases mTORC1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psych. 2013;74(10):742–749. doi: 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghasemi M, Raza M, Dehpour AR. NMDA receptor antagonists augment antidepressant-like effects of lithium in the mouse forced swimming test. J Phychopharmacol. 2010;24(4):585–594. doi: 10.1177/0269881109104845. [DOI] [PubMed] [Google Scholar]
- 34.Liu RJ, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38(11):2268–2277. doi: 10.1038/npp.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]