

Abstract
Purpose.
Although hyperglycemia is the main instigator in the development of diabetic retinopathy, dyslipidemia is also considered to play an important role. In the pathogenesis of diabetic retinopathy, cytosolic NADPH oxidase 2 (Nox2) is activated before retinal mitochondria are damaged. Our aim was to investigate the effect of lipids in the development of diabetic retinopathy.
Methods.
Reactive oxygen species (ROS, by 2′,7′-dichlorofluorescein diacetate) and activities of Nox2 (by a lucigenin-based method) and Rac1 (by G-LISA) were quantified in retinal endothelial cells incubated with 50 μM palmitate in 5 mM glucose (lipotoxicity) or 20 mM glucose (glucolipotoxicity) for 6 to 96 hours. Mitochondrial DNA (mtDNA) damage was evaluated by extended-length PCR and its transcription by quantifying cytochrome b transcripts.
Results.
Within 6 hours of exposure of endothelial cells to lipotoxicity, or glucotoxicity (20 mM glucose, without palmitate), significant increase in ROS, Nox2, and Rac1 was observed, which was exacerbated by glucolipotoxic insult. At 48 hours, neither lipotoxicity nor glucotoxicity had any effect on mtDNA and its transcription, but glucolipotoxicity significantly damaged mtDNA and decreased cytochrome b transcripts, and at 96 hours, glucotoxicity and glucolipotoxicity produced similar detrimental effects on mitochondrial damage.
Conclusions.
Although during initial exposure, lipotoxic or glucotoxic insult produces similar increase in ROS, addition of lipotoxicity in a glucotoxic environment further exacerbates ROS production, and also accelerates their damaging effects on mitochondrial homeostasis. Thus, modulation of Nox2 by pharmacological agents in prediabetic patients with dyslipidemia could retard the development of retinopathy before their hyperglycemia is observable.
Keywords: diabetic retinopathy, hyperlipidemia, mitochondria, NADPH oxidase, oxidative stress
Although during initial stages, lipids and glucose produce similar increase in cytosolic reactive oxygen species in the retinal endothelium, addition of lipids in a hyperglycemic environment exacerbates ROS production, and accelerates their damaging effects on mitochondrial homeostasis.
Diabetes, a major systemic chronic disease, is the leading cause of acquired blindness in young adults. Although hyperglycemia is the main instigator of its micro- and macrovascular complications, more than 80% of diabetic patients also present elevated triglycerides, and dyslipidemia also is considered as one of the major contributors associated with the development/progression of diabetic retinopathy.1,2 The Landmark Diabetes Control and Complications Trial has shown that the severity of retinopathy is associated with increasing serum triglycerides.3 Although a recent clinical epidemiological study, examining the long-term relationships between serum cholesterol and high-density lipoproteins and the incidence and prevalence of proliferative diabetic retinopathy, has revealed a modest association between these lipids and proliferative diabetic retinopathy,4 regulation of triglycerides by fenofibrates in type 2 diabetic patients is shown to reduce its laser intervention.5,6 Experimental studies using in vitro and in vivo models have demonstrated that saturated free fatty acids induce apoptosis of retinal microvascular cells, and the administration of a docosahexaenoic acid-rich diet to type 2 diabetes animals prevents retinal inflammation and vascular pathology.7–11
In the pathogenesis of diabetic retinopathy, retinal metabolism is impaired, and oxidative stress is increased.12,13 Recent studies have shown that, in addition to increased production of mitochondrial superoxide by diabetic milieu, cytosolic reactive oxygen species (ROS) are also increased in the retina, and this cytosolic ROS production is mainly mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 (Nox2).14,15 Nox2, a complex enzyme, has both membrane and the cytosolic components, including a small molecular weight GTP-binding protein Rac1, which helps in the membrane stability of its holoenzyme.15–17 Previous studies have shown that lipids also activate Nox218,19; however, the role of lipids in accentuating the effect of Nox2 activation in diabetic retinopathy remains unclear.
Progression of diabetic retinopathy is a slow process, and hyperglycemic insult activates Nox2-Rac1 module during the early stages of the disease, which elevates ROS. These cytosolic ROS serve as a trigger to damage the mitochondria, resulting in mitochondrial dysfunction and accelerating the capillary cell apoptosis, which precedes the development of histopathology characteristic of diabetic retinopathy.15,20–22 The role of increased lipids (lipotoxicity) in exacerbating glucose-induced (glucotoxicity) elevation in ROS and mitochondrial damage in diabetic retinopathy remains unclear.
The aim of this study was to investigate the effect of lipotoxicity in potentiating Rac1-Nox2–mediated ROS generation and mitochondrial damage in the development of diabetic retinopathy. Using retinal endothelial cells, we investigated the effect of a saturated fatty acid, palmitate, on glucotoxicity-induced ROS production and damage of mitochondrial DNA (mtDNA) and its transcription.
Methods
Bovine Retinal Endothelial Cells
Bovine retinal endothelial cells from the fifth to the sixth passage were incubated in the medium containing 2% heat-inactivated fetal bovine serum, 10% replacement serum, 50 μg/mL heparin, 1 μg/mL endothelial growth factor, and antibiotic/antimycotic for 6 hours to 96 hours in 5 or 20 mM glucose. The media were supplemented with or without palmitate or ceramide, each at a concentration of 50 μM.19 A group of cells also was incubated in lipotoxic or glucolipotoxic conditions in the presence of a Rac1 activation inhibitor, N6-[2-[4-(diethylamino)- 1-methylbutyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl- 4,6-quinolinediamine, trihydrochloride (NSC23766, 20 μM; Calbiochem-EMD Millipore, Billerica, MA, USA).15,19 Osmotic control included cells incubated in 20 mM mannitol instead of 20 mM glucose.15
Human Endothelial Cells
To confirm the effect of glucolipotoxicity on Nox2 activation, some of the key parameters were confirmed in the endothelial cells obtained from human retina, as described previously.23 Cells from fourth to sixth passages were incubated for 48 hours in 5 or 20 mM glucose media containing 1% fetal bovine serum, 9% Nu-serum, and 0.5 μg/mL endothelial growth factor in the presence or absence of palmitate (50 μM). As with the bovine retinal endothelial cells, incubation conditions also included supplementation with 20 μM NSC2376.
Palmitate and Ceramide
Stock solutions of palmitate (150 mM) and ceramide (150 mM) solutions were prepared by dissolving their sodium salts in ethanol:H2O (1:1 vol/vol) at 50°C. Aliquots of the stock solution were complexed with fatty acid-free BSA by stirring for 1 hour at 37°C. The final molar ratio of palmitate or ceramide to BSA was 5:1.19,24
Reactive Oxygen Species
Total ROS levels were quantified fluorometrically using 2′,7′-dichlorofluorescein diacetate (DCHFDA; Sigma-Aldrich Corp., St. Louis, MO, USA). Briefly, 5 μg protein was incubated with 4 μM DCHFDA for 10 minutes, and the resultant fluorescence was measured at 485 nm and 530 nm as excitation and emission wavelengths, respectively.15,22
Activity of Nox2 and Rac1
Nox2 activity was measured in cell homogenates (10 μg protein) using 20 μM lucigenin as electron acceptor and 100 μM NADPH as a substrate. The specificity of Nox2 activity was evaluated by performing the assay in the presence or absence of 0.2 mM apocynin in the assay medium. In the same cell preparations, Rac1 activation was determined using G-LISA colorimetric assay (Cytoskeleton, Denver, CO, USA), as described by us previously.15,25
Colocalization of Nox2 and Rac1
Cells grown on coverslips were incubated with 5 mM or 20 mM glucose, in the presence or absence of palmitate or ceramide, and/or NSC23766, for 48 hours. They were then fixed with 4% formaldehyde for 15 minutes and rinsed three times with PBS. The cells were permeabilized with 0.2% Triton X-100 for 10 minutes, blocked in 2% BSA for 1 hour, and incubated overnight in a moist chamber with the primary antibodies against Rac1 or Nox2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA, and Abcam, Cambridge, MA, USA, respectively; both at a dilution of 1:500). The cells were then rinsed with PBS, and incubated with their respective secondary antibodies, FITC-conjugated for Nox2 (Molecular Probes, Eugene, OR, USA) and Texas Red–conjugated for Rac1 (Vector Laboratories, Burlingame, CA, USA). Slides were mounted in antifade medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vectashield-DAPI; Vector Laboratories) to counterstain the nuclei, and imaged under a ZEISS ApoTome fluorescence microscope at ×40 magnification (Carl Zeiss, Chicago, IL, USA).26
Mitochondrial DNA Damage
Mitochondrial DNA damage was assessed by isolating total DNA using a DNeasy blood and tissue kit (Qiagen, Valencia, CA, USA), followed by performing extended-length PCR by amplifying long (13.4 kb) and short (210 bp) regions of the mtDNA. The intensity of the PCR amplicons was measured using Carestream digital software (Rochester, NY, USA), and the ratio of the long to the short bands was calculated. A decrease in the ratio of long to short amplicons indicated damage to the mtDNA.22,27
RNA Isolation and Gene Expression
After extracting total RNA by Trizol (Invitrogen, Grand Island, NY, USA), 1 μg RNA was used for cDNA preparation using the High Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). The expression of Cytochrome b (Cytb) and IL-1β was measured by SYBR green–based quantitative real-time PCR (qPCR) using gene-specific primers (Cytb = forward 5′-CGATACATACACGCAAACGG-3′, and reverse 5′- CGATACATACACGCAAACGG-3′ and IL-1β = forward 5′- AGTGACGAGAATGAGCTGTT-3′, and reverse 5′- GATTTTTGCTCTCTGTCCTG-3′), with melting curve analysis on ABI 7500 (Applied Biosystems). β-actin was used as a housekeeping gene, and the transcript quantification was performed using the ΔΔCt method.22,26
Statistical analysis was carried out using Sigma Stat software (Jandel Scientific Corporation, San Rafael, CA, USA). Data are expressed as means ± SD. The Shapiro-Wilk test was used to test for normal distribution of the data, and for variables with normal distribution; Student's t-test was used for comparing two groups and one-way ANOVA followed by Bonferroni's test was applied for multiple groups. For data that did not present normal distribution, Mann-Whitney U or Kruskal-Wallis test, followed by Dunn's test was performed. A P value less than 0.05 was considered statistically significant.
Results
Lipotoxicity Augments Glucotoxicity-Induced Increases in ROS Levels
Hyperlipidemia is considered as one of the systemic factors in the development of diabetic retinopathy, and oxidative stress is shown to play a major role in its development.1–3,13 To investigate the effect of lipotoxicity, ROS levels were quantified in the retinal endothelial cells incubated with a nonesterified fatty acid, palmitate, for 6 to 96 hours. As shown in Figure 1a, exposure of cells to palmitate for 6 hours in normal glucose (5 mM) elevated ROS levels, and the increase in ROS was similar to the one obtained from the cells incubated in high glucose (20 mM) without any palmitate. However, addition of glucose (20 mM glucose) to the palmitate-containing medium further exacerbated the production of ROS, and the values obtained from cells incubated in glucolipotoxic medium were significantly different from cells incubated in lipotoxic or glucotoxic medium (P < 0.05). Similar increases were observed when the duration of lipotoxicity or glucolipotoxicity was extended to 96 hours (Fig. 1b), a duration when glucotoxicity damages mitochondria and accelerates cell apoptosis in these retinal endothelial cells.22,28 Consistent with the increase in ROS by palmitate, addition of ceramide, instead of palmitate, also significantly increased ROS levels, which was exacerbated by the addition of glucose to the medium (Fig. 1b). Incubation of cells with mannitol (20 mM), instead of glucose (20 mM), for 6 to 96 hours did not produce any increase in ROS production.
Figure 1.
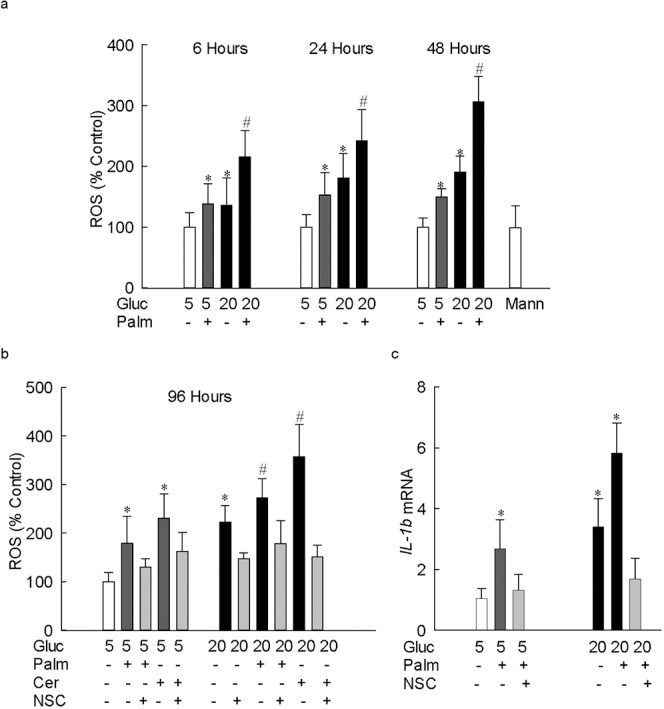
Lipotoxicity augments glucotoxicity-induced increases in ROS and IL-1β levels in retinal endothelial cells. Total ROS levels were quantified in 5 μg protein using DCHFDA, and the resultant fluorescence was measured at 485 nm and 530 nm as excitation and emission wavelengths, respectively, in retinal endothelial cells incubated in 5 mM or 20 mM glucose media (a) for 6 to 48 hours in the presence or absence of 50 μM palmitate, and (b) for 96 hours in the presence palmitate or ceramide (50 μM). (c) The gene transcripts of IL-1β were quantified by real-time PCR in the cells incubated in 5 mM or 20 mM glucose media for 96 hours, with or without 50 μM palmitate; β-actin was used as a housekeeping gene. The results are represented as mean ± SD from three to four cell preparations, with each measurement made in duplicate. *P < 0.05 vs. 5 mM glucose and #P < 0.05 vs. 20 mM glucose; 5 and 20 = 5 mM or 20 mM glucose; Gluc, glucose; Palm, palmitate; Cer, ceramide; NSC, 20 μM NSC23766; Mann, 20 mM mannitol.
Because lipotoxicity is shown to increase proinflammatory mediators, to investigate the effect of glucolipotoxicity, the level of IL-1β was quantified. Although IL-1β was increased by more than 2.5-fold in the cells exposed to either glucose or palmitate compared with the cells exposed to 5 mM glucose, its levels were increased by almost 6-fold when both palmitate and glucose were added together, suggesting that glucotoxicity also potentiates glucose- or lipid- induced increase in inflammatory mediators (Fig. 1c).
Increase in ROS is Mediated via Nox2-Rac1 Signaling
Because Nox2, which is considered as a “professional” ROS producer,29 is activated by high glucose,15 to identify the source of ROS generation, the effect of palmitate on Nox2 activity was investigated. Figure 2a shows that Nox2 was elevated by approximately 50% as early as 6 hours of palmitate exposure, but when the cells were exposed to glucolipotoxic conditions, the increase in Nox2 activity was more than 2.5-fold. As with ROS, similar Nox2 activation was observed when the cells were incubated in lipotoxic or glucolipotoxic conditions for up to 96 hours. Consistent with the increase in ROS, Nox2 activation also was observed when palmitate was replaced by ceramide (Fig. 2b).
Figure 2.
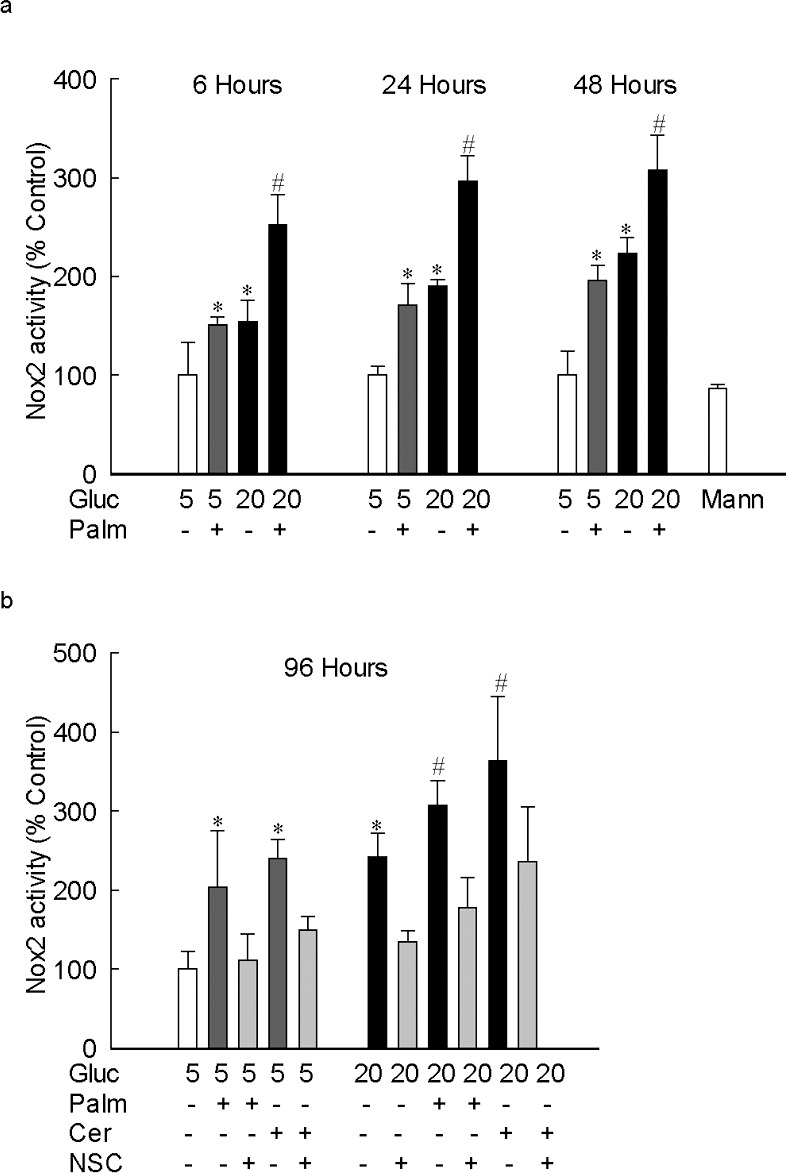
Lipotoxicity-induced increase in ROS is via activation of Nox2. Apocynin-sensitive Nox2 activity was measured in retinal endothelial cells (10 μg protein) using lucigenin as electron acceptor and NADPH as a substrate. Cells incubated (a) with palmitate for 6 to 48 hours, and (b) with palmitate or ceramide (50 μM) for 96 hours in media supplemented with or without NSC23766. The results are represented as mean ± SD from three to four cell preparations, with each measurement made in duplicate. *P < 0.05 vs. 5 mM glucose and #P < 0.05 vs. 20 mM glucose.
To investigate the mechanism of Nox2 activation by lipotoxicity, the effect of palmitate on the activation of Rac1, approximately 21 kDa signaling G-protein (critical in Nox2 activation), was determined. Figure 3a shows that although 6 hours of glucose or palmitate did not activate Rac1, addition of both palmitate and glucose increased the Rac1 activity by approximately 2-fold. However, after 24 hours, and up to 96 hours (total duration of the experiment), of palmitate exposure, Rac1 was increased by 70%. Addition of palmitate to high glucose-containing medium (glucolipotoxicity) further increased Rac1 activity; the values obtained from cells exposed to glucolipotoxic insult were significantly increased compared with the cells incubated in either glucotoxic or lipotoxic conditions. Similar increase in Rac1 activity was observed when ceramide, instead of palmitate, was used as a source of lipotoxicity (Fig. 3b).
Figure 3.
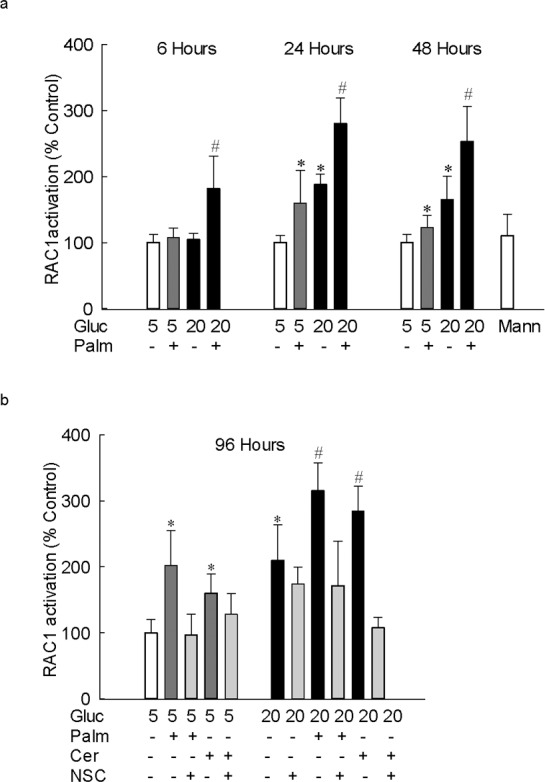
Lipotoxicity-mediated increase in glucotoxicity-induced increase in Nox2 is via Rac1 activation. Rac1 activation was quantified in 20 to 30 μg protein using G-LISA colorimetric assay in cells incubated in 5 mM or 20 mM glucose media supplemented with (a) 50 μM palmitate for 6 to 48 hours, and (b) with 50 μM palmitate or 50 μM ceramide for 96 hours. The results are represented as mean ± SD from three to four cell preparations, and each measurement was made in duplicate. *P < 0.05 vs. 5 mM glucose and #P < 0.05 vs. 20 mM glucose; 5 and 20 = cells in 5 mM or 20 mM glucose.
The role of Nox2-Rac1 in glucolipotoxicity was confirmed by immunofluorescence techniques. Consistent with the results obtained above, the expressions of both Nox2 and Rac1 were significantly increased by lipotoxic insult (palmitate or ceramide), and these increases were further exacerbated when the lipotoxicity was supplemented with glucotoxicity (Fig. 4).
Figure 4.

Glucolipotoxicity increases Nox2 and Rac1, and these two proteins are colocalized. Localization of Nox2 and Rac1 was performed immunohistochemically in cells incubated in 5 mM or 20 mM glucose for 48 hours, in the presence or absence of palmitate or ceramide, supplemented with NSC23766. For Nox2 (green) FITC-conjugated, and for Rac1, Texas Red–conjugated secondary antibodies were used, and the coverslips were mounted using Vectashield with DAPI (blue): 5 mM Glu, 5 mM Glu+Palm, and 5 mM Glu+Cer = cells incubated in 5 mM glucose with no addition or with 50 μM palmitate or ceramide, respectively; 20 mM Glu, 5 mM Glu+Palm, and 20 mM Glu+Palm+NSC = cells incubated in 20 mM glucose with no addition or with palmitate supplemented with or without 20 μM NSC23766; 20 mM Glu+Cer and 20 mM Glu+Cer+NSC = cells incubated in 20 mM glucose and 50 μM ceramide in the absence or presence of NSC23766, respectively.
Glucolipotoxicity Accelerates mtDNA Damage
Our previous work has shown that although glucotoxicity activates Nox2 as early as 3 hours of glucose insult, mitochondrial damage is not observed until the duration of glucose exposure is extended to 96 hours. To investigate the compounding effect of glucolipotoxicity on mtDNA damage, the cells incubated in lipotoxic or glucolipotoxic conditions were analyzed. Figure 5 shows that neither lipotoxicity (palmitate or ceramide) nor glucotoxicity (20 mM glucose) insult alone for 48 hours increased mtDNA damage, but, glucolipotoxic insult significantly increased mtDNA damage. However, at 96 hours, although both glucotoxicity and glucolipotoxicity conditions increased mtDNA damage, lipotoxicity alone failed to damage mtDNA. To confirm the effect of glucolipotoxicity on mtDNA damage, transcripts of mtDNA-encoded Cytb were quantified in the same cell preparations. As shown in Figure 6, exposure of cells to glucolipotoxic conditions, but not to lipotoxic or glucotoxic conditions alone, for 48 hours decreased Cytb transcripts by more than 50%. However, when the exposure was extended to 96 hours, decrease in Cytb transcripts was observed in both glucotoxic and glucolipotoxic conditions, but not in lipotoxic conditions. Furthermore, the extent of mtDNA damage and decrease in Cytb transcripts were similar in the cells exposed to glucotoxic or glucolipotoxic conditions for 96 hours.
Figure 5.
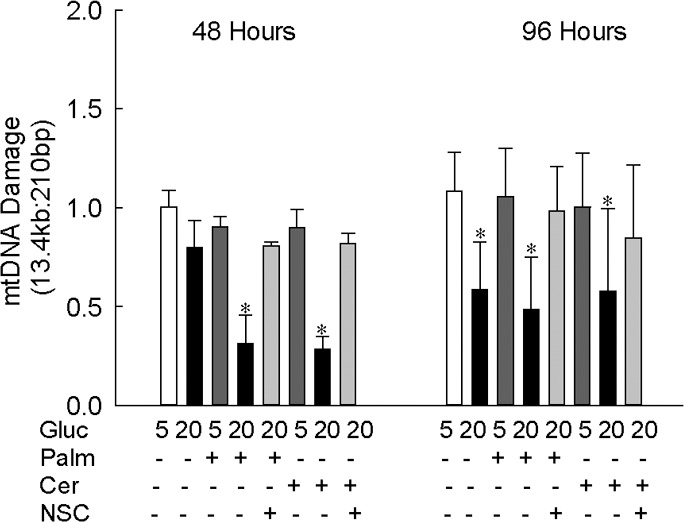
Glucolipotoxicity accelerates mtDNA damage. Damage to mtDNA was assessed in endothelial cells incubated with palmitate or ceramide in the presence of either 5 mM or 20 mM glucose for 48 or 96 hours by extended-length PCR. The ratio of the 13.4 kb to 210 bp amplicons is represented as mean ± SD from two to three cell preparations, each experiment performed in duplicate. *P < 0.05 vs. 5 mM glucose; 5 and 20 = in 5 mM and 20 mM glucose, respectively. NSC, 20 μM NSC23766.
Figure 6.
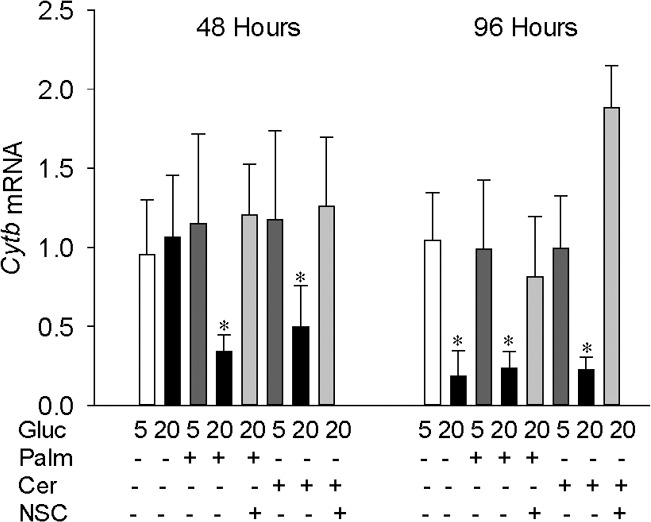
Glucolipotoxicity impairs transcription of mtDNA. The expression of mtDNA-encoded Cytb was quantified by qPCR in the cells incubated in high-glucose medium in the presence of palmitate or ceramide; β-actin was used as the housekeeping gene. The results are represented as mean ± SD from two to three cell preparations. *P < 0.05 vs. 5 mM glucose.
NSC23766 Prevents Glucolipotoxicity-Induced Increase in Nox2-Rac1–Mediated Increase in ROS Levels and Mitochondrial Damage
To confirm the role of Nox2-Rac1 in the ROS production in lipotoxic or glucolipotoxic conditions, the effect of NSC23766, an inhibitor that specifically inhibits Rac1 activity by blocking its interactions with the guanine nucleotide exchange factor T-lymphoma, invasion and metastasis 1 (Tiam1)17,30 were investigated. Addition of NSC23766, in addition to inhibiting glucose-induced Nox2-Rac1–mediated increase in ROS levels, also inhibited lipotoxic (palmitate or ceramide) and glucolipotoxic (20 mM glucose and palmitate or ceramide)-induced increase in ROS levels and activation of Nox2 and Rac1 (Figs. 1b, 2b, 3b, 4). Consistent with the prevention of increase in Nox2-Rac1–mediated ROS production, NSC23766 supplementation also ameliorated damage to the mtDNA and its transcription, induced by glucolipotoxic insult (Figs. 5, 6).
Glucolipotoxicity Also Increases Rac1-Nox2–Mediated ROS Levels in Human Retinal Endothelial Cells
Consistent with the results from bovine retinal endothelial cells, endothelial cells from human retina also showed a significant increase in ROS levels in the cells exposed to palmitate in a 20 mM glucose medium, compared with the cells exposed to palmitate in 5 mM glucose or to just 20 mM glucose alone. Furthermore, this increase in ROS was mediated by Nox2 as evidenced by a more than 3.5-fold increase in Nox2 activity in cells exposed to glucolipotoxicity compared with an approximately 2-fold increase in the cells exposed to gluco- or lipotoxic insults. In the same cells, increase in Rac1 activity also was significantly lower in the gluco- or lipotoxic conditions compared with the values obtained from cells in glucolipotoxic conditions, and addition of NSC23766 prevented glucolipotoxic-induced increase in Rac1-Nox2–mediated ROS production (Fig. 7). These results suggest that the effect of glucolipotoxic insult is not restricted to the bovine retina, and human retinal capillary cells also are sensitive to glucolipotoxic insult.
Figure 7.
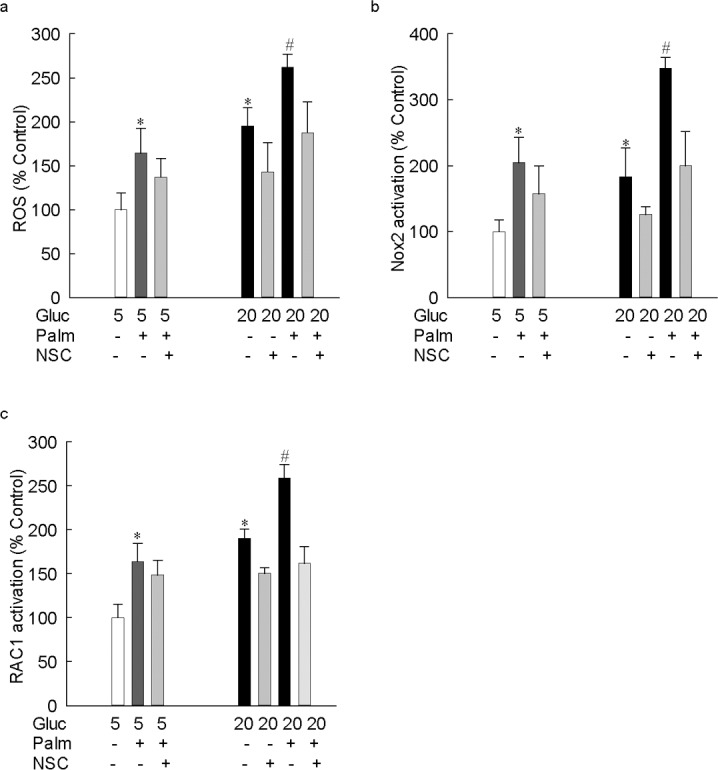
In human retina endothelial cells, glucose-induced increase in ROS is augmented by lipotoxicity. Human retinal endothelial cells, incubated in 5 mM or 20 mM glucose media for 48 hours, in the presence or absence of 50 μM palmitate, were analyzed for (a) ROS levels by using DCHFDA. Activity of (b) Nox2 (apocynin-sensitive) was measured fluorometrically using lucigenin as an electron acceptor and NADPH as a substrate, and (c) Rac1 by G-LISA–based colorimetric assay. The results are presented as mean ± SD from two cell preparations, with each measurement made in duplicate. *P < 0.05 vs. 5 mM glucose and #P < 0.05 vs. 20 mM glucose. 5 and 20 = 5 mM or 20 mM glucose.
Discussion
In the development of diabetic retinopathy, although hyperglycemia is the major instigator, lipid dysmetabolism is also considered as an important factor.2,3 Clinical studies have documented that the severity of retinopathy is associated directly with increasing triglycerides, and inversely with high-density lipoprotein cholesterol.1,31–33 Lipid-modifying fenofibrate have beneficial effects in reducing the need for laser treatment in patients with proliferative diabetic retinopathy.6 Dyslipidemia is also a well-established proinflammatory agent,34 and diabetic retinopathy is considered as a low-grade inflammatory disease with a plethora of inflammatory mediators in the retina and vitreous.35 Here, we present exciting results showing that, although lipotoxic and glucotoxic insults independently increase ROS via Nox2-Rac1 signaling pathway, glucolipotoxicity exacerbates ROS production and proinflammatory mediator IL-1β. In addition, sustained exposure to glucolipotoxicity further accelerates the toxic effects of ROS in damaging mtDNA and its transcription. Thus, this study provides the first evidence to indicate that glucolipotoxicity, by exaggerating ROS production, accelerates mitochondria damage, fueling into the vicious cycle of ROS, and potentiating the development of diabetic retinopathy.
Diabetic patients routinely present dyslipidemia, and their saturated lipid levels are elevated,36 and lipid metabolism in the retina also is impaired in diabetes.7–9,37 In the development of diabetic retinopathy, increase in oxidative stress is considered as an early event.22,28 Furthermore, mitochondrial superoxide radicals are associated with a number of interlinking metabolic abnormalities, including the accumulation of advanced glycation end products, activation of protein kinase C, and polyol and hexosamine pathways.12,38,39 Here, we show that the exposure of retinal endothelial cells for just 6 hours in a normal glucose environment to palmitate, one of the most abundant free fatty acids in human serum,40 which can accelerate retinal capillary cell apoptosis,10,41 increases ROS levels. This increase in ROS is similar to the one observed by high glucose alone. However, when lipotoxic and gluco-toxic insults are applied at the same time (glucolipotoxicity), the increase in ROS is further exacerbated, and the process continues, suggesting that lipids may be potentiating the toxic effects of high glucose. Similar increase in ROS levels is also observed in the presence of ceramide, a bioactive lipid whose biosynthesis uses palmitate as a substrate.42 In addition, as with palmitate, the effect of ceramide on ROS production is augmented by the co-supplementation with high glucose, suggesting that the increased ROS production is not specific to palmitate alone, but is rather a general lipotoxic response.
Nox2, one of the major sources of ROS production, is also identified in retinal endothelial cells and in pericytes,41,43 the capillary cells that are the main targets of histopathology associated with diabetic retinopathy.12 It is a highly regulated membrane-associated protein complex, and oxidizes cytosolic NADPH by facilitating the one-electron reduction of oxygen to superoxide.15,16 Our previous work has shown that the initial increase in retinal ROS in diabetes is via the activation of Nox2, and has suggested Nox2-mediated increase in ROS as an early event in the development of diabetic retinopathy.15 Here, we show that palmitate activates Nox2, and the effect of palmitate on Nox2 activation is potentiated by high glucose. In addition to palmitate, ceramide, which, via mediating the fusion of small raft domains to ceramide-enriched membrane platforms and aggregating the subunits,44 also activates Nox2 in retinal endothelial cells. In addition, exposure of endothelial cells to ceramide in glucotoxic conditions, as with palmitate, also potentiated Nox2 activation.
Nox2 has the small G-protein Rac1 as one of its cytosolic regulatory components, and Rac1 activation is considered critical for the assembly of Nox2 holoenzyme.15–17 The Rac1 signaling pathway regulates lipid (palmitate or ceramide)-dependent ROS generation in pancreatic beta cells,19,45 and Rac1 also regulates Nox-2 mediated ROS generation in the retina in diabetes.15 The results presented here clearly demonstrate that, despite increase in ROS levels within 6 hours of palmitate or glucose exposure, Rac1 activation is not observed, but when both are added together, Rac1 activity is significantly increased. In support, in retinal pericyte, palmitate-induced apoptosis is prevented by inactive mutants of p47phox and Rac1.41 However, chronic exposure of the cells to lipotoxic or glucotoxic insults activates Rac1, and this activation is further exacerbated by glucolipotoxic insult. Consistent with our results, a bioactive lipid-mediated Nox2-dependent mechanism has been recently implicated in the development of diabetic retinopathy.46 Although NADPH oxidases are a family of enzymes, among those Nox2 and Nox4 are the main modulators of redox signaling. Both of these members of the Nox family are activated in the retina in diabetes, and their activation regulates growth factors and inflammatory mediators associated with the development of diabetic retinopathy.46–48 The focus of our study was to investigate the role of Nox2; however, we cannot rule out the role of Nox4 in glucolipotoxicity-induced accelerated damage of retinal capillary cells.
In the pathogenesis of diabetic retinopathy, superoxide generated by mitochondria are postulated to act as unifying molecules in the regulation of major pathways implicated in the development of diabetic retinopathy.38 Mitochondria become dysfunctional and enlarged, and leak out cytochrome c in the cytosol, initiating the apoptotic machinery, and apoptosis of retinal capillary cells precedes the development of histopathology, which is the hallmark of diabetic retinopathy.20,22,28,49 In the initial stages of the hyperglycemic insult, mtDNA repair and biogenesis are increased and this helps protect the electron transport chain system, but, with sustained insult, this protection mechanism becomes overwhelmed and the mtDNA is damaged, and the electron transport system is compromised.22 In addition to increased ROS, mitochondrial lipid oxidation is also associated with its dysfunction, as fatty acids can interact with the electron chain components and bind to cytochrome c in complex III, interrupting the transport of electrons.50 Consistent with 48 hours of glucotoxic insult showing no damage to the mtDNA and accelerating apoptosis,22 here our results show that at this duration, the lipotoxic insult also has no effect on mitochondria damage. However, when the cells are exposed to glucolipotoxicity, mitochondria are damaged, as evidenced by significant increase in mtDNA damage and decrease in its transcription. This strongly suggests that the presence of both lipotoxic and glucotoxic insults together, accelerates damage to the mitochondria, and due to the compromised electron transport system, fueling of the vicious cycle of superoxide radicals is expedited. When the duration of the insult(s) is extended to 96 hours, although both glucotoxicity and glucolipotoxicity significantly damage mtDNA and its transcription, but, in contrast, lipotoxicity alone does not produce any significant effect. The reason for this discrepancy is unclear, but the possibility that further extension of lipotoxic insult could have damaged mtDNA cannot be ruled out.
The active (GTP-bound) and inactive conformations of Rac1 are regulated by Tiam1, which acts upstream of Rac1.17,30,51 We have shown that Tiam1 has a regulatory role in hyperglycemia-induced Rac1-Nox2–mediated ROS generation and mitochondrial damage in the development of diabetic retinopathy.15 Rac1 inhibition by its highly soluble and membrane-permeable small molecular weight antagonist NSC23766, which acts as a specific inhibitor of its guanine nucleotide exchange factor, Tiam1,15,30 also inhibits glucolipotoxicity-induced increase in Rac1-Nox2-ROS signaling. Furthermore, NSC23766 also ameliorates mtDNA damage and its transcription, suggesting that glucolipotoxic conditions activate Tiam-Rac1 signaling. In support, NSC23766 is shown to attenuate palmitate-induced, Rac1-Nox2–mediated, mitochondrial damage in pancreatic beta cells.19
In summary, we demonstrate that although both lipotoxic and glucotoxic insults, via Rac1-Nox2 signaling, increase cytosolic ROS in the early stages, addition of lipotoxic insult in a glucotoxic environment exacerbates ROS production, and accelerates the damaging effects of ROS on the mitochondria and its DNA. Dysfunctional mitochondria initiate the apoptotic machinery, and damaged mtDNA further compromise the electron transport system, propagating the vicious cycle of superoxide radicals. Thus, early modulation of Nox2 by pharmacological agents in hyperlipidemic prediabetic patients has potential to prevent/retard the development of diabetic retinopathy before their hyperglycemia becomes overt, and this blinding disease reaches a point of no return.
Acknowledgments
The authors thank Mangayarkarasi Thandampallayam Ajjeya for technical help.
Supported in part by the National Institutes of Health and the Juvenile Diabetes Research Foundation (RAK and AK), the Thomas Foundation (RAK), the Department of Veterans Affairs (AK), and an unrestricted grant to the Department of Ophthalmology from Research to Prevent Blindness.
Disclosure: B. Kumar, None; A. Kowluru, None; R.A. Kowluru, None
References
- 1.Chew EY,, Klein ML,, Ferris FL, III,, et al. Association of elevated serum lipid levels with retinal hard exudate in diabetic retinopathy. Early treatment diabetic retinopathy study (ETDRS) report 22. Arch Ophthalmol. 1996; 114: 1079–1084. [DOI] [PubMed] [Google Scholar]
- 2.Ucgun NI,, Yildirim Z,, Kilic N,, Gursel E.The importance of serum lipids in exudative diabetic macular edema in type 2 diabetic patients. Ann N Y Acad Sci. 2007; 1100: 213–217. [DOI] [PubMed] [Google Scholar]
- 3.Lyons TJ,, Jenkins AJ,, Zheng D, et al. Diabetic retinopathy and serum lipoprotein subclasses in the DCCT/EDIC cohort. Diabetes. 2004; 45: 910–918. [DOI] [PubMed] [Google Scholar]
- 4.Klein BE,, Myers CE,, Howard KP,, Klein R.Serum lipids and proliferative diabetic retinopathy and macular edema in persons with long-term type 1 diabetes mellitus: The Wisconsin Epidemiologic Study of Diabetic Retinopathy [published online ahead of print December 112014]. JAMA Ophthalmol. doi: http://dx.doi.org/10.1001/jamaophthalmol.2014.5108. [DOI] [PMC free article] [PubMed]
- 5.Chew EY,, Davis MD,, Danis RP,, et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) eye study. Ophthalmology. 2014; 121: 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keech AC,, Mitchell P,, Summanen PA, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007; 370: 1687–1697. [DOI] [PubMed] [Google Scholar]
- 7.Chen W,, Esselman WJ,, Jump DB,, Busik JV.Anti-inflammatory effect of docosahexaenoic acid on cytokine-induced adhesion molecule expression in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2005; 46: 4342–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu D,, Yu JY,, Wu M, et al. Immune complex formation in human diabetic retina enhances toxicity of oxidized LDL towards retinal capillary pericytes. J Lipid Res. 2014; 55: 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gustavsson C,, Agardh CD,, Hagert P,, Agardh E.Inflammatory markers in nondiabetic and diabetic rat retinas exposed to ischemia followed by reperfusion. Retina. 2008; 28: 645–652. [DOI] [PubMed] [Google Scholar]
- 10.Ido Y,, Carling D,, Ruderman N.Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the amp-activated protein kinase activation. Diabetes. 2002; 51: 159–167. [DOI] [PubMed] [Google Scholar]
- 11.Yamagishi S,, Okamoto T,, Amano S, et al. Palmitate-induced apoptosis of microvascular endothelial cells and pericytes. Mol Med. 2002; 8: 179–184. [PMC free article] [PubMed] [Google Scholar]
- 12.Frank RN.Diabetic retinopathy. N Engl J Med. 2004; 350: 48–58. [DOI] [PubMed] [Google Scholar]
- 13.Madsen-Bouterse SA,, Kowluru RA.Oxidative stress and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Rev Endocr Metab Disord. 2008; 9: 315–327. [DOI] [PubMed] [Google Scholar]
- 14.Kanwar M,, Chan PS,, Kern TS,, Kowluru RA.Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007; 48: 3805–3811. [DOI] [PubMed] [Google Scholar]
- 15.Kowluru RA,, Kowluru A,, Veluthakal R, et al. TIAM1-Rac1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia. 2014; 57: 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bokoch GM,, Zhao T.Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid Redox Signal. 2006; 8: 1533–1548. [DOI] [PubMed] [Google Scholar]
- 17.Kowluru A,, Kowluru RA.Phagocyte-like NADPH oxidase [NOX2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem Pharmacol. 2014; 88: 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chinen I,, Shimabukuro M,, Yamakawa K, et al. Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese zucker diabetic fatty rats. Endocrinology. 2007; 148: 160–165. [DOI] [PubMed] [Google Scholar]
- 19.Syed I,, Jayaram B,, Subasinghe W,, Kowluru A.Tiam1/rac1 signaling pathway mediates palmitate-induced ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol. 2010; 80: 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kern TS,, Tang J,, Mizutani M,, Kowluru R,, Nagraj R,, Lorenzi M.Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. 2000; 41: 3972–3978. [PubMed] [Google Scholar]
- 21.Piconi L,, Quagliaro L,, Assaloni R, et al. Constant and intermittent high glucose enhances endothelial cell apoptosis through mitochondrial superoxide overproduction. Diabetes Metab Res Rev. 2006; 22: 198–203. [DOI] [PubMed] [Google Scholar]
- 22.Santos JM,, Tewari S,, Kowluru RA.A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Rad Biol Med. 2012; 53: 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kowluru RA,, Kanwar M.Oxidative stress and the development of diabetic retinopathy: contributory role of matrix metalloproteinase-2. Free Rad Biol Med. 2009; 46: 1677–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briaud I,, Harmon JS,, Kelpe CL,, Segu VB,, Poitout V.Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001; 50: 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammed AM,, Kowluru A.Activation of apocynin-sensitive NADPH oxidase (NOX2) activity in ins-1 832/13 cells under glucotoxic conditions. Islets. 2013; 5: 129–131. [DOI] [PubMed] [Google Scholar]
- 26.Santos JM,, Tewari S,, Goldberg AFX,, Kowluru RA.Mitochondria biogenesis and the development of diabetic retinopathy. Free Rad Biol Med. 2011; 51: 1849–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madsen-Bouterse SA,, Mohammad G,, Kanwar M,, Kowluru RA.Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal. 2010; 13: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowluru RA,, Abbas SN.Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003; 44: 5327–5334. [DOI] [PubMed] [Google Scholar]
- 29.Lambeth JD.Nox enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004; 4: 181–189. [DOI] [PubMed] [Google Scholar]
- 30.Nassar N,, Cancelas J,, Zheng J,, Williams DA,, Zheng Y.Structure-function based design of small molecule inhibitors targeting rho family GTPases. Curr Top Med Chem. 2006; 6: 1109–1116. [DOI] [PubMed] [Google Scholar]
- 31.Davis MD,, Fisher MR,, Gangnon RE, et al. Risk factors for high-risk proliferative diabetic retinopathy and severe visual loss: Early Treatment Diabetic Retinopathy study report #18. Invest Ophthalmol Vis Sci. 1998; 39: 233–252. [PubMed] [Google Scholar]
- 32.DCCT/EDIC Research Group. Effect of intensive diabetes therapy on the progression of diabetic retinopathy in patients with type 1 diabetes: 18 years of follow-up in the DCCT/EDIC. Diabetes. 2015; 64: 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Leiden HA,, Dekker JM,, Moll AC,, et al. Blood pressure, lipids, and obesity are associated with retinopathy: the HOORN study. Diabetes Care. 2002; 25: 1320–1325. [DOI] [PubMed] [Google Scholar]
- 34.Unger RH.Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes. 1995; 44: 863–870. [DOI] [PubMed] [Google Scholar]
- 35.Tang J,, Kern TS.Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011; 30: 343–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mooradian AD.Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009; 5: 150–159. [DOI] [PubMed] [Google Scholar]
- 37.Fox TE,, Han X,, Kelly S, et al. Diabetes alters sphingolipid metabolism in the retina: a potential mechanism of cell death in diabetic retinopathy. Diabetes. 2006; 55: 3573–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brownlee M.Biochemistry and molecular cell biology of diabetic complications. Nature. 2001; 414: 813–820. [DOI] [PubMed] [Google Scholar]
- 39.Kowluru RA,, Santos JM,, Mishra M.Epigenetic modifications and diabetic retinopathy. Biomed Res Int. 2013; 2013: 635284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Symons JD,, Abel ED.Lipotoxicity contributes to endothelial dysfunction: a focus on the contribution from ceramide. Rev Endocr Metab Disord. 2013; 14: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cacicedo JM,, Benjachareowong S,, Chou E,, Ruderman NB,, Ido Y.Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase oxidant stress, and ceramide. Diabetes. 2005; 54: 1838–1845. [DOI] [PubMed] [Google Scholar]
- 42.Deevska GM,, Nikolova-Karakashian MN.The twists and turns of sphingolipid pathway in glucose regulation. Biochimie. 2011; 93: 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Shabrawey M,, Rojas M,, Sanders T, et al. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci. 2008; 49: 3239–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ushio-Fukai M,, Alexander RW.Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol Cell Biochem. 2004; 264: 85–97. [DOI] [PubMed] [Google Scholar]
- 45.Veluthakal R,, Madathilparambil SV,, McDonald P,, Olson LK,, Kowluru A.Regulatory roles for TIAM1 a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic beta-cells. Biochem Pharmacol. 2009; 77: 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ibrahim AS,, Elshafey S,, Sellak H,, et al. A lipidomic screen of hyperglycemia-treated HRECs links 12/15-lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase. J Lipid Res. 2015; 56: 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J,, Wang JJ,, Yu Q,, Chen K,, Mahadev K,, Zhang SX.Inhibition of reactive oxygen species by lovastatin down-regulates VEGF expression and ameliorates blood-retinal barrier breakdown in db/db mice: role of NADPH oxidase 4. Diabetes. 2010; 59: 1528–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilkinson-Berka JL,, Rana I,, Armani R,, Agrotis A.Reactive oxygen species, Nox and angiotensin II in angiogenesis: implications for retinopathy. Clin Sci (Lond). 2013; 124: 597–615. [DOI] [PubMed] [Google Scholar]
- 49.Kowluru RA,, Mohammad G,, dos Santos JM,, Zhong Q.Abrogation of MMP-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes. 2011; 60: 3023–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bugger H,, Abel ED.Mitochondria in the diabetic heart. Cardiovasc Res. 2010; 88: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamauchi J,, Miyamoto Y,, Tanoue A,, Shooter EM,, Chan JR.Ras activation of a Rac1 exchange factor, Tiam1, mediates neurotrophin-3-induced Schwann cell migration. Proc Natl Acad Sci U S A. 2005; 102: 14889–14894. [DOI] [PMC free article] [PubMed] [Google Scholar]