

Abstract
Large clostridial toxins (LCTs) are produced by at least four pathogenic clostridial species, and several LCTs are proven pivotal virulence factors for both human and veterinary diseases. TpeL is a recently identified LCT produced by Clostridium perfringens that has received relatively limited study. In response, the current study surveyed carriage of the tpeL gene among different C. perfringens strains, detecting this toxin gene in some type A, B, and C strains but not in any type D or E strains. This study also determined that all tested strains maximally produce, and extracellularly release, TpeL at the late-log or early-stationary growth stage during in vitro culture, which is different from the maximal late-stationary-phase production reported previously for other LCTs and for TpeL production by C. perfringens strain JIR12688. In addition, the present study found that TpeL levels in culture supernatants can be repressed by either glucose or sucrose. It was also shown that, at natural production levels, TpeL is a significant contributor to the cytotoxic activity of supernatants from cultures of tpeL-positive strain CN3685. Lastly, this study identified TpeL, which presumably is produced in the intestines during diseases caused by TpeL-positive type B and C strains, as a toxin whose cytotoxicity decreases after treatment with trypsin; this finding may have pathophysiologic relevance by suggesting that, like beta toxin, TpeL contributes to type B and C infections in hosts with decreased trypsin levels due to disease, diet, or age.
INTRODUCTION
Clostridium perfringens is a Gram-positive, spore-forming, anaerobic, and toxigenic bacterium (1). This microorganism is widely distributed, with a presence in soil, sewage, feces, foods (such as raw meat, fish, and poultry), and surfaces of residential homes. Although C. perfringens is present in the intestines of some healthy humans and animals (1), it is also a pathogen causing a range of diseases, spanning from histotoxic infections, such as gas gangrene, to infections originating in the intestines, e.g., hemorrhagic or nonhemorrhagic diarrhea and often-fatal enterotoxemia. C. perfringens virulence is due largely to its abundant toxin production, which varies by strain (2). Different C. perfringens isolates are assigned to one of five types (A to E) based upon their production of four typing toxins (alpha toxin [CPA], beta toxin [CPB], epsilon toxin [ETX], and iota toxin [ITX]) (3, 4). Besides these four typing toxins, this bacterium can produce at least 13 other toxins, including the enterotoxin (CPE) (1, 5), although no single strain produces all of these toxins.
TpeL, a recently discovered toxin produced by some C. perfringens strains, is a member of the large clostridial toxin (LCT) family (6). Other LCTs include toxins A (TcdA) and B (TcdB) from C. difficile, the hemorrhagic toxin TcsH and the lethal toxin TcsL from C. sordellii, and alpha toxin (TcnA) from C. novyi (7, 8). This toxin group consists of proteins ranging in size from ∼195 to 310 kDa that share primary amino acid sequence identities spanning from 36% to 90% (9). Most LCTs contain four functional domains, including the biologically active domain located in the N terminus, the cysteine protease domain, the putative pore-forming and delivery domain, and a C-terminal receptor binding domain that contains combined repetitive oligopeptides (CROPs) (9–11). Like other LCTs, TpeL possesses the glycosyltransferase activity domain, autocatalytic activity domain, and the transmembrane domain; however, it lacks the CROP sequences present in all other LCTs (12).
Despite the absence of CROP sequences, TpeL exerts cytotoxic effects on Vero cells, HeLa cells, and rat pheochromocytoma PC12 cells (6, 12–15). A recent study identified the low-density lipoprotein receptor-related protein 1 (LRP1) as the TpeL receptor and also demonstrated the existence of a second, CROP-independent receptor-binding domain in LCTs, suggesting a two-receptor model for the cellular uptake of these toxins (15). TpeL is the largest toxin among the ≥17 toxins produced by C. perfringens, with a typical size of ∼205 kDa (12). However, at least one strain produces a truncated, 15-kDa-smaller TpeL variant that is less active (6). Nagahama et al. reported in vitro studies indicating that a recombinant N-terminal TpeL fragment containing the glycosyltransferase domain uses both UDP-GlcNAc and UDP-Glc as donor cosubstrates and mainly modifies Ras subfamily proteins via glycosylation to mediate its cytotoxic effects (13). A later study confirmed that TpeL preferentially glycosylates Ras and, to a lesser extent, Rap1a and R-Ras3, but it only very weakly modifies Rac1 (14). However, Guttenberg et al. found that TpeL preferentially utilizes UDP-GlcNAc as a sugar donor (12). Whether, at natural production levels, TpeL contributes to the cytotoxic or virulence properties of TpeL-positive C. perfringens strains is largely unclear at present.
Some toxins produced by C. perfringens in the intestines are exceptionally sensitive to trypsin, while other toxins made by this bacterium must be activated by trypsin or other proteases (2, 16–18). CPB, produced by C. perfringens type B and type C strains, is very sensitive to endogenous trypsin degradation in the intestines of natural host animals (18, 19). In contrast, a major toxin of both type B and D strains, i.e., ETX, is produced as an inactive prototoxin that must be proteolytically activated by intestinal proteases (such as trypsin, chymotrypsin, and carboxypeptidases) or, perhaps, proteases produced by C. perfringens (20–25). Until now, no information has been available regarding whether TpeL, which can be encoded by strains causing diseases originating in the intestines (26, 27), is trypsin sensitive.
Since TpeL is a recently identified toxin of C. perfringens, many of its features remain poorly understood, including the carriage of the tpeL gene among different C. perfringens types or subtypes, whether natural production levels of TpeL contribute to strain cytotoxicity, the timing of TpeL production, and TpeL sensitivity to trypsin. This study has addressed each of those important topics.
MATERIALS AND METHODS
Bacterial strains and growth media.
Wild-type C. perfringens type A to E isolates used in this study are listed and described in Table 1. The toxin genotypes (A to E) and some phenotypic characteristics of these isolates were determined previously (23, 24, 26–34). All C. perfringens isolates used in this study were maintained as stock cultures in cooked meat medium (Oxoid) and stored at −20°C. All type E isolates were maintained as stock cultures in 15% glycerol and stored at −80°C. FTG medium (fluid thioglycolate medium; Difco Laboratories), TH medium (Bacto Todd Hewitt broth [Becton-Dickinson], with 0.1% sodium thioglycolate [Sigma-Aldrich]), TGY medium (3% tryptic soy broth [Becton-Dickinson] containing 2% glucose [Fisher Scientific], 1% yeast extract [Becton-Dickinson], and 0.1% thioglycolate [Sigma-Aldrich]), or TY medium (3% tryptic soy broth [Becton-Dickinson], 1% yeast extract [Becton-Dickinson], and 0.1% thioglycolate [Sigma-Aldrich]) was used to grow broth cultures of C. perfringens. E. coli strain TOP 10 was grown in Luria-Bertani (LB) broth and used for constructing toxin gene mutagenesis plasmids.
TABLE 1.
Description of wild-type C. perfringens strains used in this study
| Strain type and name | Origin (reference) | tpeL PCR | tpeL Southern blotting |
|---|---|---|---|
| A | |||
| JGS5369 | Chicken necrotic enteritis (24) | + | + |
| JGS4143 | Chicken necrotic enteritis (24) | − | − |
| JGS4140 | Chicken necrotic enteritis (24) | + | + |
| F4396 | Sporadic diarrhea (29) | − | − |
| B38 | Antibiotic associated diarrhea (30) | − | − |
| ATCC3624 | Gas gangrene (30) | − | − |
| F5603 | Sporadic diarrhea (29) | − | − |
| PS16 | Unknown, Centers for Disease Control and Prevention (31) | − | − |
| B2 | Antibiotic associated diarrhea (30) | − | − |
| NCTC10239 | Food poisoning (29) | − | − |
| NCTC8235 | Food poisoning (30) | − | ND |
| NCTC8238 | Food poisoning (30) | − | ND |
| NCTC8239 | Food poisoning (29) | − | ND |
| NCTC8679 | Food poisoning (30) | − | ND |
| NCTC8799 | Food poisoning (30) | − | ND |
| NCTC8798 | Food poisoning (30) | − | ND |
| NCTC8359 | Food poisoning (30) | − | ND |
| FD1041 | Food poisoning (30) | − | ND |
| 042-03 | Intestines of healthy North Americans (29) | − | ND |
| 00-803 | Intestines of healthy North Americans (29) | − | ND |
| 168 | Intestines of healthy North Americans (29) | − | ND |
| 011-03 | Intestines of healthy North Americans (29) | − | ND |
| B | |||
| CN1795 | Veterinary laboratory, toxigenic (27) | + | + |
| CN2416 | Stomach, 5-day-old lamb (27) | + | + |
| CN677 | Acute lamb dysentery (27) | + | + |
| CN684 | Lamb dysentery (39) | + | + |
| CN1301 | Lamb dysenterya | − | − |
| NCTC3110 | Unknown (27) | + | + |
| NCTC8533 | Lamb dysentery (27) | + | + |
| PS49 | Unknown (27) | + | + |
| Bar6 | Lamb dysentery (32) | − | − |
| Bar2 | Sheep (27) | + | + |
| C | |||
| CN882 | Sheep with struck (26) | + | + |
| CN3955 | Ewe peritoneal fluid (39) | + | + |
| CN3763 | Animal (species unknown) (26) | − | − |
| Bar3 | Human pigbel (26) | + | + |
| CN3685 | Sheep with struck (39) | + | + |
| CN2065 | Ewe with strucka | + | + |
| CN885 | Veterinary laboratory (39) | − | − |
| CN5388 | Human pigbel (39) | + | + |
| CN3708 | Calfa | + | + |
| CN3727 | Calfa | + | + |
| D | |||
| CN462 | Goat (28) | − | − |
| CN1634 | Lamb with suspected dysentery (28) | − | − |
| CN3718 | Guinea pig heart blood (23) | − | − |
| CN3842 | Ewe, small intestine (28) | − | − |
| CN2068 | Lamb stomach (28) | − | − |
| JGS1948 | Caprine, enterotoxemia (61) | − | − |
| JGS1945 | Caprine, Diarrhea (33) | − | − |
| JGS1240 | Sheep, bronchopneumonia, and enterotoxemia (33) | − | − |
| NCTC8346 | Sheep (34) | − | − |
| JGS1705 | Ovine (33) | − | − |
| E | |||
| NCIB10748 | Unknown (50) | − | − |
| 51 | Neonatal calves, hemorrhagic enteritis (50) | − | − |
| 294 | Neonatal calves, hemorrhagic enteritis (50) | − | − |
| 572 | Neonatal calves, hemorrhagic enteritis (50) | − | − |
| 576 | Neonatal calves, hemorrhagic enteritis (50) | − | − |
| 853 | Neonatal calves, hemorrhagic enteritis (50) | − | − |
| 1987 | Neonatal calves, hemorrhagic enteritis (50) | − | − |
| B2085 | Neonatal calves, hemorrhagic enteritis (50) | − | − |
The strain was originally from the collection of Russell Wilkinson.
DNA extraction.
Genomic DNA from C. perfringens was purified from overnight TGY broth cultures using the MasterPure Gram-positive DNA purification kit (Epicentre Biotechnologies, WI).
PCR analyses of tpeL carriage by C. perfringens strains.
PCR was performed to evaluate tpeL gene carriage among C. perfringens isolates of different toxin types. The primers used for these PCR analyses were described previously (27). Template DNA for these short-range PCRs was obtained from colony lysates or purified genomic DNA from these strains. Each PCR mixture contained 2 μl of template DNA, 10 μl of 2× Taq master mix (New England BioLabs), 1 μl of each primer pair (1 μM final concentration), and 7 μl of H2O. PCR conditions used for these amplifications included 94°C for 3 min and 35 cycles of 94°C for 30 s, 55°C for 30 s, and 68°C for 90 s and a final extension for 10 min at 68°C. PCR products were run on a 1% agarose gel and stained with ethidium bromide for visualization.
PCR analyses to link the cpb and tpeL genes in type C strain CN3685.
To evaluate a linkage between the tpeL and cpb genes in CN3685, overlapping PCR and long-range PCR were performed using previously described primers and conditions (26, 27). PCR products then were electrophoresed on a 1% agarose gel, which was stained with ethidium bromide for product visualization.
Southern blotting.
Purified DNA (3 μg) was digested overnight with EcoRI at 37°C (New England BioLabs) and then separated by electrophoresis on a 0.8% agarose gel. The separated DNA fragments were transferred onto a positively charged nylon membrane (Roche) for hybridization with digoxigenin (DIG)-labeled specific intron or tpeL probes, which were prepared as previously described using primer pairs specific for intron or tpeL genes (27). To detect probe hybridization, CSPD substrate (Roche Applied Science) was utilized in accordance with the manufacturer's instructions.
Construction of CN3685::tpeL and CN3685::αβθtpeL null mutants.
To evaluate TpeL contributions to the cytotoxic properties of C. perfringens supernatants, the tpeL gene of type C isolate CN3685 was inactivated by the targeted insertion of a group II intron using the Clostridium-modified Targetron system (35). Briefly, the tpeL gene sequence was entered into the Sigma-Aldrich Targetron website. Three primers were generated to target insertion of an intron, in the sense orientation, into the tpeL open reading frame (ORF) between nucleotides 1426 and 1427. The primers used for PCR targeting of the intron were TpeL-IBS (5-AAAAAAGCTTATAATTATCCTTATATATCCAGCTGGTGCGCCCAGATAGGGTG-3), TpeL-EBS1d (5-CAGATTGTACAAATGTGGTGATAACAGATAAGTCCAGCTGCTTAACTTACCTTTCTTTGT-3), TpeL-EBS2 (5-TGAACGCAAGTTTCTAATTTCGGTTATATATCGATAGAGGAAAGTGTCT-3), and the EBS universal primer provided in the kit (Sigma-Aldrich). The 350-bp PCR product was inserted into pJIR750ai to construct a tpeL-specific Targetron plasmid (pJIR750tpeLi). Mutant preparation and selection then were performed as described previously (35). Transformants were selected on brain heart infusion (BHI) agar plates containing 15 μg/ml of chloramphenicol, and colonies carrying an intron insertion were screened by PCR using primers tpeL-KOF (5-AATGTTTATCTTCGGCTAT-3) and tpeL-KOR (5-TTTGAAAACTCCTTTACTG-3) for detecting the tpeL null mutation.
Each reaction mixture was subjected to the following PCR amplification conditions: cycle 1, 94°C for 5 min; cycles 2 through 35, 94°C for 30 s, 55°C for 30 s, and 68°C for 90 s; and a final extension for 10 min at 68°C. An aliquot (20 μl) of each PCR sample was electrophoresed on a 1.5% agarose gel and then visualized by staining with ethidium bromide.
The pJIR750tpeLi Targetron plasmid was similarly transformed into a previously constructed CN3685::αβθ mutant named BMC107 (19) to create the CN3685::αβθtpeL null mutant. This mutant was selected and characterized as described above for the CN3685::tpeL mutant.
Reversal of the intron insertion to restore TpeL production by CN3685::tpeL mutant.
A previously described procedure (18, 36) was used to partially restore TpeL production in the CN3685::tpeL strain. Briefly, this mutant was electroporated to receive the Targetron plasmid pJIR750tpeLi and then grown in TH medium (with 15 μg/ml chloramphenicol) for 8 h at 37°C. Those cultures then were transferred to a 30°C water bath for another 16 h of growth to facilitate excision of the intron insertion from some tpeL mRNA.
Preparation of anti-TpeL antibody.
Two predicted antigenic peptides (peptide 1, CPGIKKHIFKDINKPT; peptide 2, CDSIQFDAIPEILKGK) from the translated tpeL DNA ORF were identified by bioinformatics analysis and then synthesized and purified by high-performance liquid chromatography (HPLC). The highly purified peptides then were conjugated to the carrier protein keyhole limpet hemocyanin (KLH). Serum against these TpeL antigenic peptides was raised in rabbits by the Pocono Rabbit Farm and Laboratory using the conjugated peptides as the immunogenic antigen. This immunization followed the standard protocol of Pocono Rabbit Farm and Laboratory under their approved IACUC permit (PRF2A). Briefly, the rabbits were injected subcutaneously with 200 μg of conjugated antigens per rabbit in complete Freund's adjuvant (CFA). After 2, 4, and 8 weeks, 50 to 100 μg of conjugated antigens mixed with incomplete Freund's adjuvant (IFA) were given subcutaneously. At day 91, rabbits were terminally bled from the carotid artery.
Western blotting of C. perfringens culture supernatants or cell lysates.
C. perfringens culture supernatants or boiled lysates of washed C. perfringens cells were mixed with 5× loading buffer and electrophoresed on an SDS-containing 8% polyacrylamide gel for detecting TpeL or an SDS-containing 12% polyacrylamide gel for detecting other toxins. The separated proteins then were transferred onto a nitrocellulose membrane and the blot was blocked for 1 h with washing buffer (20 mM Tris-HCl [pH 8.0], 0.3 M NaCl, 0.5% [vol/vol] Tween 20) containing 5% (wt/vol) nonfat dry milk before incubation with primary TpeL antibody overnight at 4°C. Blots then were rinsed three times with washing buffer and incubated with rabbit anti-mouse IgG-horseradish peroxidase (HRP) or goat anti-rabbit IgG-HRP (Sigma-Aldrich) for 1 h at room temperature. After three more washes, the blots were treated with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) and exposed to X-ray film (Life Science Products) to detect the immunoreactive protein bands.
To quantify TpeL levels after different treatments or under different culture conditions, those Western blots were scanned and analyzed with NIH Image J software.
C. difficile toxin B Western blot analyses.
To analyze the time course of toxin B production and release by C. difficile, cooked meat medium stock cultures of C. difficile strains VPI 10463 (Robert Carman, TechLab) and 630 (ATCC) were inoculated into FTG, and those cultures were grown overnight at 37°C. A 0.2-ml aliquot of each starter culture then was passed into 10 ml of fresh TH medium (containing 0.1% sodium thioglycolate), and those cultures were grown at 37°C for 6, 24, or 48 h. Supernatants from each time point were collected and subjected to Western blot analyses using a rabbit polyclonal antiserum against the C-terminal domain of toxin B that was kindly supplied by Jimmy Ballard. The Western blots then were processed as described earlier for TpeL Western blots.
C. perfringens growth rate measurements.
To measure the culture growth rate of C. perfringens in this study, 0.2-ml aliquots of FTG overnight cultures were inoculated into 10 ml of TH, TY, or TGY and cultured at 37°C. At time intervals of 0, 2, 4, 6, 8, and 16 h, the culture was collected and mixed by gentle vortexing. The optical density at 600 nm (OD600) then was recorded using a Bio-Rad Smartspec.
Preparation of samples for cytotoxicity experiments.
Overnight (16-h) TH cultures of wild-type CN3685, mutants, or reversed mutants were centrifuged, and supernatants were filter sterilized using a 0.45-μm filter (Millipore). Those sterile supernatants then were concentrated 10-fold at 4°C using an Amicon Ultra centrifugal filter unit with an Ultracel-30 membrane (EMD Millipore), with buffer exchange to PBS (pH 7.4). Those concentrated supernatants then were diluted 1:10 using Hanks' balanced salt solution (HBSS; Corning) for cytotoxicity testing.
Trypsin treatment assay.
To test whether TpeL is trypsin sensitive, processed TH culture supernatants of BMC107 containing TpeL were prepared as described above and then either treated at 37°C for 1 h with trypsin (Sigma-Aldrich) at the indicated concentrations (0, 0.04, 0.4, 4.0, 40, or 400 μg/ml) or treated with trypsin at a concentration of 12.5 μg/ml at 37°C for different times (0, 10, 20, or 60 min). After incubation, trypsin then was inactivated by adding an equal volume of trypsin inhibitor (3 to 6 mg of inhibitor ligand per ml gel; MP Biochemicals) at room temperature for 30 min. After centrifugation, the resultant supernatants were diluted 1:10 with HBSS and added to Vero cells to measure cytotoxicity (as described below) or for Western blot analyses (as described earlier).
C. perfringens supernatant cytotoxicity assay.
Vero cells were used for the C. perfringens supernatant toxin cytotoxicity assay based upon previous studies (6). Vero cells were maintained routinely in M199 (Sigma-Aldrich) supplemented with 5% fetal bovine serum (FBS; Life Technologies), streptomycin (100 U/ml), and penicillin (100 μg/ml). Vero cells normally were harvested with 0.25% trypsin and 2.21 mM EDTA (Gibco), resuspended in cell culture medium, and maintained at 37°C in a 5% CO2 humidified atmosphere.
For assaying C. perfringens supernatant-induced cytotoxicity (morphological damage and quantitative cell death measurement), Vero cells were seeded into 12-well plates and incubated until confluent. For cell cytotoxicity experiments, the culture supernatant samples prepared as described earlier were added to the Vero cells for up to 5 h at 37°C.
The development of morphological changes, indicating cytotoxicity, was checked every hour using a Zeiss Axiovert 25 inverted microscope. After a 5-h treatment, the cytopathic effects caused by the various C. perfringens supernatants were photographed using a Canon Powershot G5 fitted to the Zeiss Axiovert 25 microscope. Images then were processed using Adobe Photoshop CS5.
Lactate dehydrogenase (LDH) release was measured to determine cell death, as described below.
Cytotoxicity neutralization assay.
To help confirm whether the observed damage to Vero cells was caused specifically by the TpeL toxin present at natural concentrations in culture supernatants of CN3685 or derivatives, a serum neutralization approach was applied. Briefly, 50 μl of a neutralizing antiserum (described above) against TpeL toxin or preimmune serum was added to C. perfringens supernatant samples, prepared as described earlier, and those mixtures were incubated at room temperature for 15 min. The mixtures then were applied to Vero cells for 5 h at 37°C.
LDH release assay.
To compare and quantitatively measure Vero cell cytotoxicity caused by different samples, the LDH release assay kit for mammalian cell death (Roche) was used. Vero cell monolayers were treated with different C. perfringens culture supernatant samples, prepared as described earlier, for 5 h at 37°C in a 5% CO2 humidified atmosphere. Supernatants from the treated cell cultures were collected and used for measuring host cell LDH release. The absorbance of each sample then was measured at 490 nm with an iMark microplate reader (Bio-Rad). As described in the kit instructions, Vero cells treated with 1% Triton X-100 were used to determine maximal LDH release. The results are expressed as the percentage of maximal LDH release.
Statistical analysis.
All values are expressed as means ± standard errors of the means (SEM). For statistical evaluations, one-way analysis of variance (ANOVA) with Tukey's post hoc test was performed, and P values of <0.05 were considered significant.
RESULTS
Evaluation of tpeL gene carriage in C. perfringens.
Previous studies (24, 26, 27, 37) had detected the presence of the tpeL gene in some C. perfringens type A, B, and C isolates. To better understand the scope of tpeL gene carriage among C. perfringens isolates, the current study surveyed the presence of the tpeL gene in additional wild-type strains, including representatives of all C. perfringens toxigenic types and major pathogenic subtypes of type A.
PCR assays amplified products of the expected size, indicating carriage of the tpeL gene, using DNA from only two of 22 surveyed type A strains with varied origins (Table 1). Both of those tpeL-positive type A strains were associated with avian necrotic enteritis. All four surveyed type A strains isolated from healthy North Americans, all four plasmid cpe-positive type A strains from non-food-borne human gastrointestinal diseases, and all nine surveyed chromosomal cpe-positive type A food poisoning strains tested tpeL negative. In contrast, eight of 10 surveyed type B or type C strains were tpeL positive (Table 1). Consistent with these PCR results, Southern blot experiments using a tpeL-specific probe confirmed the PCR results for tpeL gene carriage among the surveyed type A, B, and C strains of C. perfringens (Table 1). No tpeL-positive strain was detected by either PCR or Southern blot analyses among the surveyed C. perfringens type D and E strains (Table 1).
Western blot analyses of TpeL production and release from C. perfringens grown in different culture media.
To compare culture medium effects on TpeL production and release into culture supernatants, C. perfringens tpeL-positive type A to C strains were grown in TH, TY, or TGY broth for 16 h. The supernatants of each culture then were collected and subjected to Western blot analyses using TpeL polyclonal antiserum. Results shown in Fig. 1A demonstrate that TH is consistently the best medium for supporting TpeL production and extracellular release, while TpeL production and release is consistently lowest in TGY. In order to determine whether the varied TpeL production and release observed in different culture media was due simply to effects on bacterial growth, growth curve experiments were performed. Measurement of culture OD600 revealed similar growth rates for the strains when cultured in different media (Fig. 1B).
FIG 1.
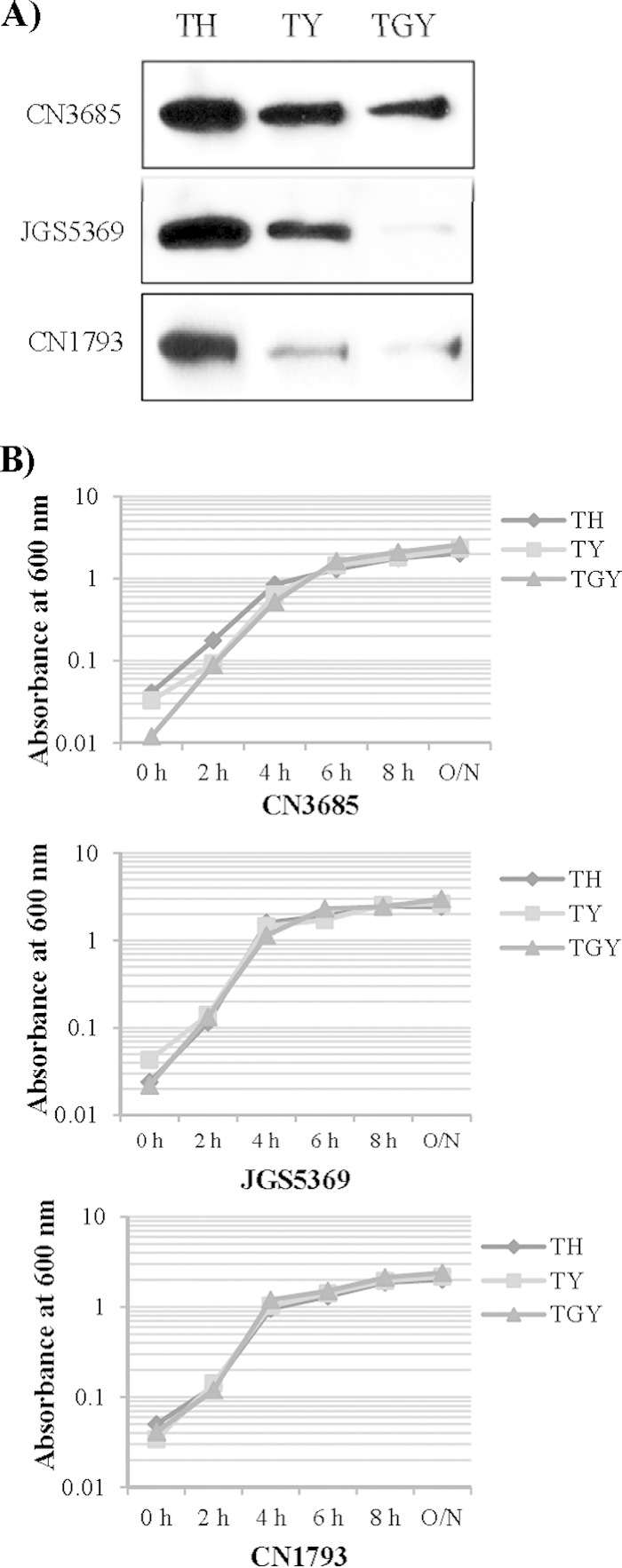
Comparison of TpeL production and extracellular release by C. perfringens growing in different culture media. (A) Western blot analyses of TpeL in culture supernatants from cultures of C. perfringens type A strain JGS5369, type B strain CN1793, and type C strain CN3685 grown for 16 h in TH, TY, or TGY. The immunoreactive protein shown is ∼200 kDa, consistent with the expected mass of TpeL. Results shown are typical of three repetitions. (B) Comparison of growth characteristics of wild-type strains JGS5369, CN1793, and CN3685 at 37°C. The optical density (OD600) of each strain growing in TH, TY, or TGY culture medium was measured using a Bio-Rad Smartspec spectrophotometer. A typical result of three repetitions is shown.
TpeL production begins during early-log-phase growth and peaks during early-stationary-stage growth in TH medium.
Most toxins produced by C. perfringens are maximally expressed within ∼10 h, i.e., during late-log or early-stationary growth phases (1, 2, 4, 38–40). However, other LCTs (such as C. difficile toxin A and toxin B) usually are maximally produced and released extracellularly after 24 h, i.e., at the late stationary stage of growth (11, 41, 42). Figure 2A confirms those previous conclusions for the timing of toxin B production and extracellular release by two C. difficile strains. These C. difficile cultures entered stationary phase after ∼8 h (data not shown).
FIG 2.

Western blot time course analyses of TpeL production and extracellular release. (A) Western blot analyses of toxin B levels in supernatants from cultures of C. difficile strains VPI 10463 and 630 grown at 37°C for 6, 24, or 48 h in TH. (B) Western blot analyses of TpeL levels in supernatants from cultures of C. perfringens strains PS49, CN3690, CN3685, and Bar2 grown for 6, 12, 24, or 48 h in TH. (C) Western blot analyses of TpeL levels in supernatants (Super) or washed and lysed cells (Pellets) of CN3685 cultures grown for 2, 4, 6, 8, or 16 h in TH. (D) Comparison of TpeL levels in 16-h or 72-h supernatants from TH cultures of JGS5369, CN3685, and CN1793. The immunoreactive protein shown is ∼200 kDa, consistent with the expected mass of TpeL. Typical results from three repetitions are shown.
To begin evaluating the kinetics of TpeL production and extracellular release, several tpeL-positive C. perfringens strains were grown in TH medium, and supernatants from those cultures then were collected at 6 to 48 h for TpeL Western blot analyses. As shown in Fig. 2B, high supernatant levels of TpeL were detected when these tpeL-positive strains were grown in TH culture medium for 6 to 12 h, which corresponds to late-log-phase or early-stationary-phase growth (Fig. 1B).
To further determine the time course of TpeL production and release, CN3685 was grown in TH medium, and culture supernatants were collected at even earlier time points for Western blot analyses. As shown in Fig. 2C, TpeL was not detectable in 2-h TH culture supernatants but became readily apparent in 4-h TH culture supernatants. For CN3685, peak TpeL levels in culture supernatants were reached within ∼8 h. Note that only limited degradation of extracellular TpeL was detectable in Fig. 2C, as well as in other experiments performed in this study (not shown).
To specifically examine the onset of TpeL production, washed CN3685 cells from TH cultures were lysed and subjected to TpeL Western blotting. This analysis detected a TpeL signal within 2 h (Fig. 2C). Also notable on these blots was the substantial degradation of intracellular TpeL, with this degradation increasing with longer culture time. Considered collectively, the results shown in Fig. 2C indicated that, after production in the cytoplasm of C. perfringens, there is a delayed release of TpeL from the bacterium by a still-unknown mechanism.
As a final evaluation of the time course of TpeL production and extracellular release by C. perfringens, late-stationary-phase (72 h) culture supernatants of three tpeL-positive strains (type A strain JGS5369, type B strain CN1793, and type C strain CN3685) also were tested by TpeL Western blot analyses. Those analyses showed that TpeL levels present in supernatants of 72-h TH cultures were less than those present in 16-h TH culture supernatants (Fig. 2D). Together, the results shown in Fig. 2 indicated that supernatant TpeL levels usually become maximal during the late log or early stationary stages of growth, which is similar to most other toxins produced by C. perfringens, rather than in late stationary phase, as for other LCTs.
Glucose and sucrose repress TpeL supernatant levels.
Previous studies reported that production and release of LCTs often is repressed by glucose (43, 44). Another recent study demonstrated that sucrose, as well as glucose, inhibits the production of C. perfringens PLC (alpha-toxin) and PFO (theta-toxin) (45). Our Fig. 1A results detecting higher TpeL supernatant levels in TY (containing a basal sugar concentration of 0.25%) versus TGY (TY medium with an additional 2% glucose) broth are consistent with a recent report (46) that glucose also represses TpeL production and release by C. perfringens strain JIR12688, despite that strain maximally producing and releasing TpeL during late stationary phase, in contrast to our surveyed strains, where TpeL production starts early in log phase (Fig. 2).
To definitively evaluate if glucose also depresses TpeL supernatant levels for strains that maximally express this toxin during late-log or early-stationary-phase growth, C. perfringens was cultured in TY supplemented with different concentrations of glucose (0, 5, 10, 20, and 40 mg/ml). As shown in Fig. 3A, TpeL supernatant levels in an 8-h culture were significantly decreased by the addition of supplemental glucose. A similar reduction in TpeL supernatant levels also was observed using supplemental sucrose (Fig. 3B).
FIG 3.

Comparison of TpeL supernatant levels for C. perfringens cultures grown in TY medium supplemented with different concentrations of glucose or sucrose. (A) Western blot analyses of TpeL levels in supernatants from 8-h TY cultures, which had been supplemented with glucose at the indicated concentrations of CN3685, CN1793, and PS49. Supernatants from matching TY cultures with no supplemental glucose were used as controls. (B) Western blot analyses of TpeL levels in supernatants from 8-h TY cultures, which had been supplemented with sucrose at the indicated concentrations of CN3685, CN1793, and PS49. Matching TY cultures with no supplemental sucrose were used as controls. The immunoreactive protein shown in panels A and B is ∼200 kDa, consistent with the expected mass of TpeL. (C) Quantitative analysis of TpeL supernatant levels from 8-h TY cultures that had been supplemented with different concentrations of glucose (G) or sucrose (S). An asterisk indicates a statistically significant (P < 0.05) decrease of TpeL production in TY supplemented with glucose or sucrose compared to similar cultures without any carbohydrate supplementation. Shown are mean values ± standard errors of the means (SEM) based on three repetitions.
When quantified from three independent Western blot analyses, the data obtained (Fig. 3C) detected statistically significant different TpeL supernatant levels in the presence or absence of supplemental glucose and sucrose. Measurement of changes in culture OD600 values over time revealed no differences in growth between cultures supplemented with ≤40 mg/ml concentrations of glucose or sucrose for these C. perfringens strains, indicating that the decreased TpeL supernatant levels shown in Fig. 3A and B were not from growth inhibition due to increasing sugar concentrations in the TY culture medium. However, higher supplemental sugar concentrations, i.e., those ranging from 60 to 80 mg/ml, did retard C. perfringens growth (data not shown).
Construction and characterization of a tpeL single null mutant and a cpa, cpb, pfoA, tpeL quadruple null mutant in CN3685.
Recent studies showed that arbitrarily chosen amounts of purified TpeL cause cytotoxicity (6). To specially evaluate if, at natural levels present in culture supernatants, TpeL also can contribute to strain-induced cytotoxicity, the tpeL gene in C. perfringens type C animal disease strain CN3685 was insertionally inactivated with the Clostridium-modified Targetron insertional mutagenesis system (35). Construction of the tpeL isogenic null mutant of CN3685 was verified by PCR analyses using primers specific to tpeL sequences located upstream and downstream of the intron insertion site. These internal tpeL PCR primers specifically amplified a PCR product of ∼600 bp from wild-type CN3685 DNA. However, the same primer pair amplified an ∼1,500-bp PCR product from the DNA of the putative tpeL null mutant due to the insertion of a 900-bp intron into the tpeL ORF (Fig. 4A). Southern blot analyses using an intron-specific probe demonstrated the presence of a single intron insertion in the CN3685::tpeL mutant (Fig. 4B). As expected, no intron signal was detected using wild-type DNA from CN3685 (Fig. 4B).
FIG 4.
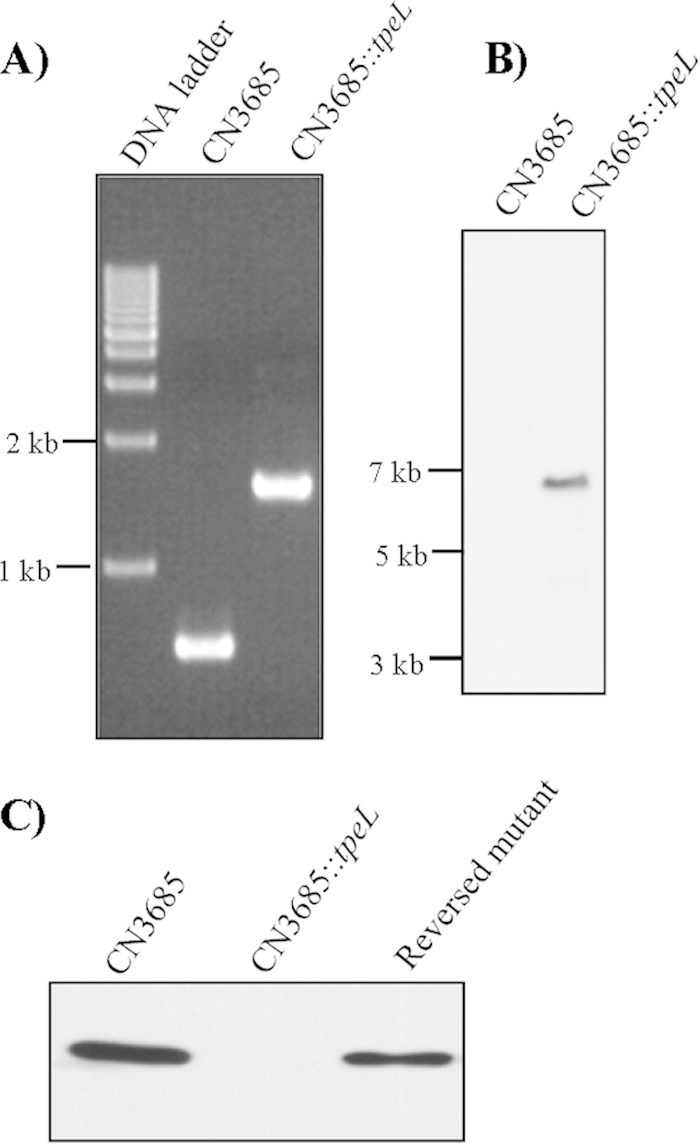
Construction of a CN3685 tpeL null mutant by intron-based insertional mutagenesis. (A) PCR analyses using DNA from wild-type CN3685 or the isogenic tpeL-null mutant (CN3685::tpeL). (B) Southern blot hybridization of wild-type CN3685 and the CN3685::tpeL mutant. DNA from each strain was digested with EcoRI, electrophoresed on a 0.8% agarose gel, and transferred onto a nylon membrane. The separated DNA then was hybridized with a DIG-labeled intron-specific probe. Sizes of DNA fragments in kilobases (kb) are shown on the left. (C) Western blot analyses for TpeL production by wild-type CN3685, the CN3685::tpeL mutant, or reversed mutant strains. The immunoreactive protein shown is ∼200 kDa, consistent with the expected mass of TpeL.
The tpeL gene proved difficult to clone into a shuttle plasmid for complementation (data not shown). Instead, TpeL production by the CN3685::tpeL mutant was partially restored via a commonly used, LtrA-mediated trans-splicing approach (18) that removes the inserted intron from some tpeL mRNA, resulting in a partial regain of TpeL translation. For this purpose, the plasmid pJIR750tpeLi-sense, which encodes a functional LtrA protein, was reintroduced into the CN3685::tpeL mutant. The resultant TpeL reversed mutant was grown at 30°C in the presence of antibiotics to maintain selective pressure for producing maximal LtrA protein, as required for splicing-induced intron removal. Western blot results showed that TpeL production was absent from the CN3685::tpeL mutant, while the reversed mutant exhibited partially restored TpeL production (Fig. 4C). This result also demonstrated that the anti-TpeL antibody prepared in this study specifically reacts with the TpeL protein.
C. perfringens type C strain CN3685 produces four known toxins, including CPA, CPB, PFO, and TpeL (18 and this study). To evaluate TpeL-induced cytotoxicity in the absence of any cytotoxic influence from other toxins, a strain producing only TpeL was constructed by using the Targetron procedure described above. For this purpose, the tpeL gene was insertionally inactivated in BMC107, which is a previously constructed CN3685 triple null mutant that does not produce CPA, CPB, or PFO (19). The genotype of this quadruple mutant, named the CN3685::αβθtpeL mutant, was confirmed by PCR (data not shown). The tpeL gene in CN3685 is located 3 kb downstream of its cpb gene (Fig. 5A), which explains why Southern blots only detected three intron signals using DNA from the CN3685::αβθtpeL mutant (Fig. 5B). Long-range PCR confirmed the cpb-tpeL linkage in CN3685 using one primer to internal tpeL ORF sequences and a second primer to internal cpb ORF sequences (Fig. 5C). An overlapping PCR further demonstrated (Fig. 5D) this genetic linkage using a five-pair set of primers described in our previous studies (26, 27). Western blot analyses confirmed that the CN3685::αβθtpeL mutant does not produce PFO, CPA, CPB, or TpeL.
FIG 5.

Construction of the quadruple cpa, cpb, pfoA, tpeL null CN3685::αβθtpeL mutant by intron-based insertional mutagenesis. (A) Arrangement of the cpb and tpeL gene locus in C. perfringens type C strain CN3685 based upon previous studies (26, 27). (B) Southern blot hybridization of wild-type CN3685, the TpeL-producing triple toxin mutant BMC107, and the CN3685::αβθtpeL quadruple null mutant. Sizes of DNA fragments in kilobases are shown to the left. (C) Long-range PCR analyses linking the tpeL and cpb genes in CN3685. (D) Overlapping PCR analyses of the region extending from cpb to tpeL in CN3685 using primers R1 to R5. (E) Western blot analyses of CPA, CPB, PFO, and TpeL levels in 16-h TH culture supernatants of wild-type CN3685, the TpeL-producing triple toxin mutant BMC107, and the CN3685::αβθtpeL quadruple null mutant. The immunoreactive proteins in panel E matched the expected masses, as relevant, of PFO (∼55-kDa), CPA (∼47 kDa), CPB (∼35 kDa), and TpeL (∼200 kDa).
Vero cell cytotoxicity caused by wild-type CN3685 and CN3685::tpeL mutant supernatants.
To evaluate if, at natural production levels, TpeL can contribute to the cytotoxicity of C. perfringens supernatants, the CN3685 tpeL null mutant was tested for its cytotoxic effects on Vero cells. For this purpose, the TH culture supernatants of wild-type CN3685 and CN3685::tpeL and reversed mutants were collected, filter sterilized, concentrated 10-fold with buffer exchange, and then diluted 1:10 into HBSS. To further identify whether the cytotoxic consequences of CN3685 supernatants involved TpeL, an anti-TpeL neutralizing antiserum was added to some supernatant samples prior to the Vero cell cytotoxicity assay.
When the processed culture supernatants from wild-type CN3685, the CN3685 tpeL null mutant, or the reversed mutant were applied to Vero cells for 5 h, the culture supernatant samples from wild-type CN3685 caused significant Vero cell damage, including cell rounding, cell aggregation, and, eventually, cell detachment from the confluent monolayers (Fig. 6A). In contrast, morphological cell damage to Vero cells was visibly weaker, and cell detachment was absent, using similarly prepared culture supernatant from the CN3685 tpeL null mutant strain. Restoring some TpeL production to the tpeL null mutant by partially reversing the intron insertion in tpeL mRNA caused an increase in supernatant cytotoxic activity for Vero cells, which supported the attenuated cell damage caused by supernatants from the tpeL null mutant as being attributable to inactivation of tpeL gene expression (Fig. 6A). Furthermore, preincubating wild-type CN3685 culture supernatants with the TpeL-neutralizing antiserum further confirmed that much of the observed cytotoxicity activity was caused by TpeL toxin. Specifically, preincubation of processed CN3685 supernatant with TpeL-neutralizing antibody attenuated and delayed the development of morphological damage, while a similar preincubation with preimmune serum had little or no protective effects (Fig. 6A).
FIG 6.
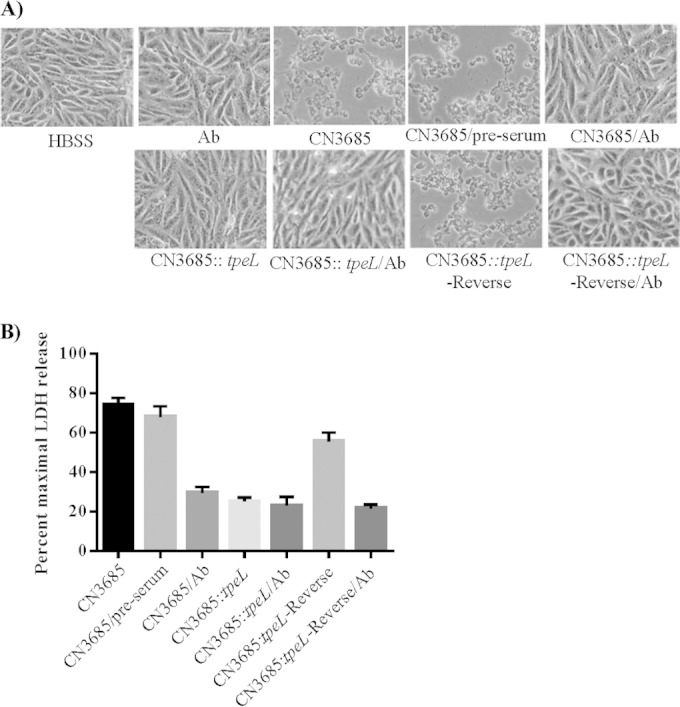
Vero cell cytotoxicity caused by processed supernatants for cultures of wild-type CN3685 and the CN3685::tpeL mutant. (A) Morphological damage to Vero cells. Processed (see Materials and Methods) 16-h TH culture supernatants from wild-type CN3685, CN3685::tpeL, or the reversed mutant were tested for their cytotoxic effects after either preincubation with preimmune serum (preserum) or TpeL neutralizing serum (Ab), as indicated. HBSS shows the effect of buffer alone. (B) Lactate dehydrogenase (LDH) release by Vero cells after treatment with different samples described in panel A. Cytotoxicity caused by processed culture supernatant from the CN3685::tpeL mutant shows a statistically significant (P < 0.05) decrease compared to that of Vero cells treated with similarly processed samples, including wild-type CN3685 culture supernatant, wild-type CN3685 culture supernatant preincubated with anti-TpeL serum, or culture supernatant of the CN3685::tpeL reverse complement strain. The experiment was repeated three times, and results shown are the mean values. Error bars represent SEM.
Lactate dehydrogenase (LDH) release from Vero cells then was measured to quantify the cytotoxicity of these processed culture supernatants (Fig. 6B). Cell death caused by wild-type CN3685 or the reversed mutant strains was significantly higher than that caused by the tpeL null mutant. The lower cell death caused by the reversed mutant versus the wild-type strains is consistent with results shown in Fig. 4C, indicating that reversal of the tpeL mutation was partial and did not completely restore TpeL production, as is typical for this approach (18, 47). When the processed culture supernatants were preincubated with the TpeL-neutralizing antibody, cytotoxicity caused by the wild-type supernatant samples decreased significantly; in contrast, preincubation with preimmune serum did not affect cytotoxicity of this sample, confirming a substantial role for TpeL in the cytotoxic activity of these culture supernatants (Fig. 6B).
Glucose decreases the cytotoxic activity of BMC107 culture supernatants.
As shown in Fig. 3, TpeL production and release by several C. perfringens strains, including CN3685, is repressed by either glucose or sucrose. Therefore, we assessed whether these sugars also modulate the cytotoxic effects of processed culture supernatants obtained from the TY cultures of TpeL-only-producing strain BMC107. After Vero cells were treated for 5 h with culture supernatant samples containing different concentration of glucose, LDH release from dead or damaged cells was measured. As shown in Fig. 7, this analysis detected less cytotoxicity with increasing glucose concentrations in these processed TY culture supernatants. This result further confirmed that glucose and sucrose not only represses TpeL levels in culture supernatants but also can have consequences for cytotoxicity.
FIG 7.
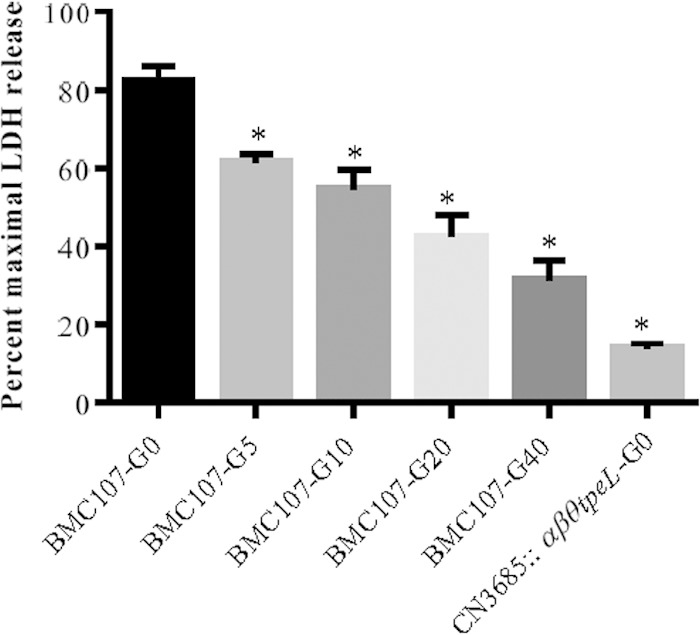
Glucose affects the cytotoxicity caused by supernatants from BMC107 cultures. Lactate dehydrogenase (LDH) release from Vero cells after treatment with processed (see Materials and Methods) supernatants from 8-h cultures of BMC107 grown in TY supplemented with different glucose concentrations. The experiment was repeated three times, and results shown are the mean values. Error bars represent SEM. An asterisk shows a statistically significant difference (P < 0.05) between a sample without glucose supplementation (BMC107-G0) and samples supplemented with different concentrations of glucose. G0, G5, G10, G20, and G40 represent different concentrations of glucose supplementation in TY culture.
TpeL is a trypsin-sensitive toxin.
TpeL-producing type B and C strains, such as CN3685, cause diseases originating in the intestines (1), where secreted TpeL could contact proteases like trypsin. Therefore, the TpeL protein sequence was entered into the Expasy software program (http://web.expasy.org/peptide_cutter/) to predict potential TpeL cleavage sites by proteases. Approximately 200 potential trypsin cut sites were identified by this software in the full-length TpeL protein. To test definitively whether TpeL is a trypsin-sensitive toxin, processed culture supernatants of BMC107 were treated for 1 h at 37°C with different concentrations of purified trypsin. As indicated by Western blotting results shown in Fig. 8A, these trypsin-treated TpeL-containing supernatants contained a decreased amount of TpeL upon Western blot analysis. When similar TpeL-containing supernatants were treated with a single dose of trypsin (12.5 μg/ml) at 37°C for different times, the TpeL Western blot signal intensity decreased with longer trypsin treatment times (data not shown).
FIG 8.
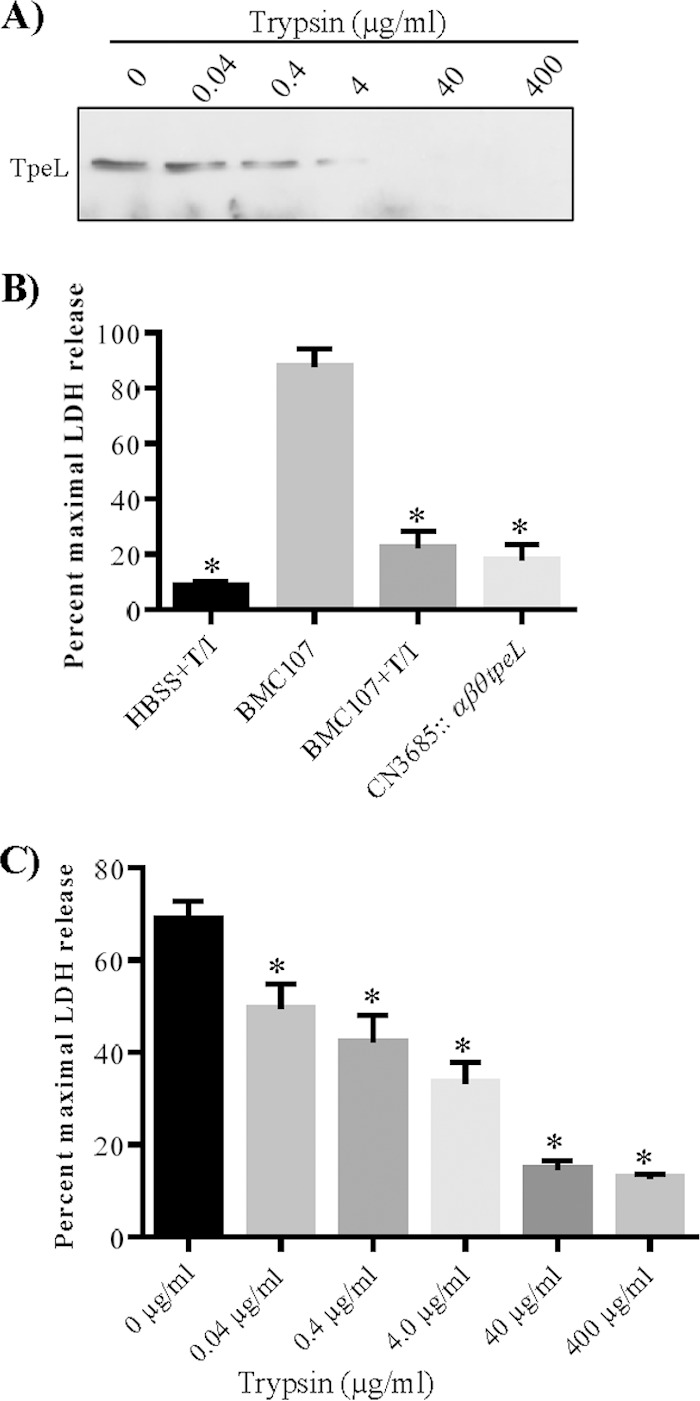
TpeL is sensitive to trypsin. (A) Processed (see Materials and Methods) TpeL-containing culture supernatant from BMC107 was treated with trypsin at the indicated concentration for 1 h at 37°C. Trypsin (T) activity then was removed from the samples by trypsin inhibitor (I) prior to TpeL Western blot analyses. The immunoreactive protein shown is ∼200 kDa, consistent with the expected mass of TpeL. (B) Quantitative Vero cytotoxicity (measured by LDH release) of supernatants from BMC107 treated with trypsin and trypsin inhibitor (T/I). HBSS buffer similarly treated with T/I was used as a trypsin treatment control, and the processed supernatant from the CN3685::αβθtpeL mutant (no T/I treatment) was used as a TpeL-negative control. An asterisk shows a statistically significant (P < 0.05) decrease in LDH cytotoxicity caused by the samples treated with T/I compared to sample without this treatment. (C) Cytotoxic effects were evaluated after treatment of processed supernatants with different trypsin concentrations. LDH release was used to quantitatively evaluate cytotoxicity. An asterisk shows a statistically significant difference (P < 0.05) compared to the value for BMC107. The error bars represent SEM calculated from three independent experiments from panels B and C.
To evaluate the cytotoxic consequences of TpeL cleavage by trypsin, processed culture supernatant from BMC107 was treated with different concentrations of trypsin. After a 1-h treatment, remaining trypsin in these samples was inactivated by the addition of trypsin inhibitor. When these trypsin-treated supernatants were applied to Vero cells, morphological cell damage was much weaker and no cell detachment was observed compared to results for non-trypsin-treated supernatant samples (data not shown). As a control, processed culture supernatants of the CN3685::αβθtpeL quadruple null mutant caused only very weak cell damage in this experiment, even without trypsin treatment (data not shown).
LDH release was quantitatively measured to evaluate the cytotoxic activity of processed supernatants that were or were not trypsin treated (12.5 μg/ml of trypsin). Cell death was much lower in Vero cells challenged with trypsin-treated supernatant. These results confirmed that the reduced cell death caused by the trypsin-treated supernatant samples is attributable to TpeL activity being destroyed by trypsin treatment (Fig. 8B). As a control for this experiment, little cell damage was measured in Vero cells challenged with the processed supernatant from CN3685::αβθtpeL mutant cultures; this result also supports conclusions from Fig. 7 that TpeL contributes to natural CN3685 cytotoxicity.
Lastly, to evaluate the cytotoxic effects of the trypsin-pretreated samples shown in Fig. 8A, various concentrations of the trypsin-treated supernatant samples were applied to Vero cells after removal of residual trypsin activity by the addition of trypsin inhibitor. The percentage of Vero cell death caused by the non-trypsin-treated sample was more than 65%, while Vero cell death resulting from challenge with different concentrations of trypsin-treated supernatants decreased from 49.6% to 12.8% as the trypsin concentration used for supernatant treatment increased (Fig. 8C).
DISCUSSION
This study provides several important insights into tpeL carriage, as well as the production and release, cytotoxic contributions, and trypsin sensitivity of the TpeL protein. With respect to tpeL carriage, the current results are consistent with previous reports indicating that many type C strains, and nearly all type B strains, carry this toxin gene, which is often (but not always) located in these strains near the cpb gene on plasmids of ∼90 kb or ∼60 to 65 kb (26, 27). The current results also detected the tpeL gene in two type A avian necrotic enteritis strains, both netB positive (data not shown); this finding is consistent with previous reports suggesting that TpeL-positive strains often are associated with avian necrotic enteritis, although NetB toxin is considered to play the major role in pathogenesis (24, 48). However, the tpeL gene was not detected in other type A strains, including enteropathogenic type A strains carrying either a chromosomal or plasmid-borne cpe gene. To date, the copresence of both tpeL and cpe genes has never been detected in any single C. perfringens strain. One potential explanation for this situation involves incompatibility between the plasmids carrying tpeL and those carrying the cpe gene. If this is correct, it would still remain unclear why type A chromosomal cpe strains also rarely, if ever, carry the tpeL gene.
When the current study similarly evaluated carriage of the tpeL gene among type D and E strains of C. perfringens, none of the surveyed isolates carried this toxin gene. Again, this pattern could be due to incompatibility issues between tpeL-carrying plasmids and the plasmids carrying etx genes in type D strains or iota toxin genes (itxA and itxB) in type E strains. If, as proposed in the preceding paragraph, tpeL plasmids generally are incompatible with cpe plasmids, this may help to explain the absence of tpeL plasmids from many type E strains, since iota toxin plasmids often are related to cpe plasmids, including their carriage of cpe genes or silent cpe sequences (49–51). While the presence of plasmids carrying tpeL (and cpb) genes in type B strains indicates that not all etx plasmids are incompatible with tpeL plasmids, it is noteworthy that only a single ∼65-kb etx plasmid has been detected in type B strains (27, 52). In contrast, a diversity of etx plasmids exists among type D strains (28), and it has been proposed that many of those plasmids are incompatible with cpb plasmids, since they are never found in type B strains (27, 52). Therefore, it is possible that the tpeL and cpb plasmids found in type B strains are not compatible with the etx plasmids present in many type D strains.
A recent report (46) indicated that, as for other LCTs, glucose represses TpeL levels in culture supernatants of C. perfringens strain JIR12688, which produces and releases TpeL during late stationary phase. The current study now extends that finding by showing this relationship also holds true for those C. perfringens strains producing TpeL during the log or early stationary phase of growth (see the next paragraph). Furthermore, our results indicated that TpeL production and release by early TpeL-producing strains also is repressed in the presence of sucrose. Thus, tpeL expression, as well as production of CPA and PFO (45), appears to be regulated by both glucose and sucrose catabolite repression in most or all TpeL-positive C. perfringens strains. While the mechanism of this repression in C. perfringens requires further study, it is notable that the C. difficile literature reports that, under nutrient-rich conditions, CodY represses TcdA and TcdB production (53). This may be relevant for understanding the regulation of TpeL production, since C. perfringens also produces a functional CodY protein (54).
There have been discrepancies in the literature regarding the timing of TpeL production by C. perfringens. A recent study reported that, like TcdA and TcdB production by C. difficile, C. perfringens strain JIR12688 produces TpeL during late stationary phase, with maximal production at 72 h (46). However, an earlier study detected tpeL mRNA expression in 6-h cultures of 5 different type C strains (26). In the current study, Western blot kinetic analyses clearly showed that TpeL production by the surveyed C. perfringens strains can begin within 2 h and becomes maximal during the late log or early stationary phase of growth. Therefore, there appears to be heterogeneity in the timing of TpeL production and release among C. perfringens strains, with many or most strains producing and releasing TpeL during the late log or early stationary growth phase (this study), although at least one strain (JIR12688) produces and releases this toxin primarily during late stationary phase (46). The explanation for these timing differences will require further study, but it is notable that JIR12688 appears to be an unusually slow-growing C. perfringens strain (46). It is possible that this growth defect reflects a metabolic difference that impacts the timing of TpeL production and release by this strain.
Previous studies have shown that TpeL-containing supernatants (46), or arbitrarily chosen amounts of purified TpeL (6), are cytotoxic for cultured mammalian cells. By using toxin mutants, the current study now specifically establishes that, at natural production levels, TpeL can contribute to the cytotoxic properties of supernatants from cultures of TpeL-producing C. perfringens. This finding suggests potential TpeL contributions to virulence, but this hypothesis still must be tested in animal models. It is notable that some isogenic toxin mutants of the same CN3685 strain used for constructing mutants in the current study have been tested previously for virulence in animal models (18, 19). Those studies, conducted before the discovery of TpeL, showed that CPB production is required for this strain to cause hemorrhagic necrotic enteritis in rabbit small intestinal loops and is also a major contributor to enterotoxemic lethality in a mouse oral challenge model. In contrast, loss of either CPA or PFO production had no discernible effect on CN3685 pathogenicity in the small intestinal loop model and caused only a slight attenuation in the lethality of this strain in the mouse enterotoxemia model (18, 19). While those previous studies established the importance of CPB for CN3685 virulence, they do not necessarily exclude TpeL contributions to the virulence of this strain. Of note, the earlier studies had challenged animals with TGY cultures of CN3685, which (as now shown in the current study and the study by Carter et al. [46]) is not the most favorable culture medium for TpeL production and release. Alternatively, it is possible that TpeL contributes additively or synergistically to CPB action, similar to the recent discovery of CPB and CPE synergism for the enteric pathogenicity of some TpeL-negative type C strains (47).
Finally, the current study examined whether TpeL is sensitive to trypsin, which is relevant since (i) during intestinal infections, this toxin should be produced by TpeL-positive type B and C strains and (ii) there is considerable variability in the effects of trypsin on C. perfringens toxins, with some (i.e., ETX, CPE, and ITX) toxins being activated by trypsin (41, 55–57), while others (i.e., CPB and beta2 toxin) are inactivated by trypsin (58, 59). Our findings clearly demonstrated that TpeL is trypsin sensitive, like C. difficile toxin B (60), which is another LCT. Demonstrating that TpeL is trypsin sensitive does not diminish the potential importance of this toxin for disease, since TpeL is produced mainly by type B and C strains. Those strains often cause disease in hosts with reduced intestinal trypsin levels due to age, nursing, diet, or other factors (1).
Collectively, these findings provide important new insights into TpeL, an unusual LCT. Future studies will investigate the activity of TpeL in vivo and the regulation of production of this toxin.
ACKNOWLEDGMENTS
These studies were supported by National Institute of Allergy and Infectious Diseases grant RO1 AI056177 (B.A.M.).
We thank James Theoret for help with statistical analyses.
REFERENCES
- 1.McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins TD. 2006. The enterotoxic Clostridia, p 688–752. In Dworkin M, Falkow S, Rosenburg E, Schleifer H, Stackebrandt E (ed), The prokaryotes, 3rd ed Springer Press, New York, NY. [Google Scholar]
- 2.Li J, Adams V, Bannam TL, Miyamoto K, Garcia JP, Uzal FA, Rood JI, McClane BA. 2013. Toxin plasmids of Clostridium perfringens. Microbiol Mol Biol Rev 77:208–233. doi: 10.1128/MMBR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petit L, Gibert M, Popoff MR. 1999. Clostridium perfringens: toxinotype and genotype. Trends Microbiol 7:104–110. doi: 10.1016/S0966-842X(98)01430-9. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Ma M, Uzal FA, McClane BA. 2014. Host cell-induced signaling causes Clostridium perfringens to upregulate production of toxins important for intestinal infections. Gut Microbes 5:96–107. doi: 10.4161/gmic.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClane BA, Robertson SL, Li J. 2013. Clostridium perfringens, p 465–489. In Doyle MP, Buchanan RL (ed), Food microbiology: fundamentals and frontiers, 4th ed ASM Press, Washington, DC. [Google Scholar]
- 6.Amimoto K, Noro T, Oishi E, Shimizu M. 2007. A novel toxin homologous to large clostridial cytotoxins found in culture supernatant of Clostridium perfringens type C. Microbiology 153:1198–1206. doi: 10.1099/mic.0.2006/002287-0. [DOI] [PubMed] [Google Scholar]
- 7.Popoff MR, Bouvet P. 2009. Clostridial toxins. Future Microbiol 4:1021–1064. doi: 10.2217/fmb.09.72. [DOI] [PubMed] [Google Scholar]
- 8.Schirmer J, Aktories K. 2004. Large clostridial cytotoxins: cellular biology of Rho/Ras-glucosylating toxins. Biochim Biophys Acta 1673:66–74. doi: 10.1016/j.bbagen.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Jank T, Aktories K. 2008. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 16:222–229. doi: 10.1016/j.tim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 10.Just I, Hofmann F, Aktories K. 2000. Molecular mode of action of the large clostridial cytotoxins. Curr Top Microbiol Immunol 250:55–83. [DOI] [PubMed] [Google Scholar]
- 11.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guttenberg G, Hornei S, Jank T, Schwan C, Lu W, Einsle O, Papatheodorou P, Aktories K. 2012. Molecular characteristics of Clostridium perfringens TpeL toxin and consequences of mono-O-GlcNAcylation of Ras in living cells. J Biol Chem 287:24929–24940. doi: 10.1074/jbc.M112.347773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagahama M, Ohkubo A, Oda M, Kobayashi K, Amimoto K, Miyamoto K, Sakurai J. 2011. Clostridium perfringens TpeL glycosylates the Rac and Ras subfamily proteins. Infect Immun 79:905–910. doi: 10.1128/IAI.01019-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pauillac S, D'Allayer J, Lenormand P, Rousselle JC, Bouvet P, Popoff MR. 2013. Characterization of the enzymatic activity of Clostridium perfringens TpeL. Toxicon 75:136–143. doi: 10.1016/j.toxicon.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Schorch B, Song S, van Diemen FR, Bock HH, May P, Herz J, Brummelkamp TR, Papatheodorou P, Aktories K. 2014. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc Natl Acad Sci U S A 111:6431–6436. doi: 10.1073/pnas.1323790111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakurai J, Duncan CL. 1978. Some properties of beta-toxin produced by Clostridium perfringens type C. Infect Immun 21:678–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Miyakawa ME, Fisher DJ, Poon R, Sayeed S, Adams V, Rood JI, McClane BA, Uzal FA. 2007. Both epsilon-toxin and beta-toxin are important for the lethal properties of Clostridium perfringens type B isolates in the mouse intravenous injection model. Infect Immun 75:1443–1452. doi: 10.1128/IAI.01672-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sayeed S, Uzal FA, Fisher DJ, Saputo J, Vidal JE, Chen Y, Gupta P, Rood JI, McClane BA. 2008. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol Microbiol 67:15–30. doi: 10.1111/j.1365-2958.2007.06007.x. [DOI] [PubMed] [Google Scholar]
- 19.Uzal FA, Saputo J, Sayeed S, Vidal JE, Fisher DJ, Poon R, Adams V, Fernandez-Miyakawa ME, Rood JI, McClane BA. 2009. Development and application of new mouse models to study the pathogenesis of Clostridium perfringens type C enterotoxemias. Infect Immun 77:5291–5299. doi: 10.1128/IAI.00825-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freedman JC, Li J, Uzal FA, McClane BA. 2014. Proteolytic processing and activation of Clostridium perfringens epsilon toxin by caprine small intestinal contents. mBio 5:e01994–14. doi: 10.1128/mBio.01994-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, McClane BA. 2012. Role of the Agr-like quorum-sensing system in regulating toxin production by Clostridium perfringens type B strains CN1793 and CN1795. Infect Immun 80:3008–3017. doi: 10.1128/IAI.00438-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minami J, Katayama S, Matsushita O, Matsushita C, Okabe A. 1997. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol Immunol 41:527–535. doi: 10.1111/j.1348-0421.1997.tb01888.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Rood JI, McClane BA. 2011. Epsilon-toxin production by Clostridium perfringens type D strain CN3718 is dependent upon the agr operon but not the VirS/VirR two-component regulatory system. mBio 2:e00275–11. doi: 10.1128/mBio.00275-11.. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Coursodon CF, Glock RD, Moore KL, Cooper KK, Songer JG. 2012. TpeL-producing strains of Clostridium perfringens type A are highly virulent for broiler chicks. Anaerobe 18:117–121. doi: 10.1016/j.anaerobe.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Harkness JM, Li J, McClane BA. 2012. Identification of a lambda toxin-negative Clostridium perfringens strain that processes and activates epsilon prototoxin intracellularly. Anaerobe 18:546–552. doi: 10.1016/j.anaerobe.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurjar A, Li J, McClane BA. 2010. Characterization of toxin plasmids in Clostridium perfringens type C isolates. Infect Immun 78:4860–4869. doi: 10.1128/IAI.00715-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sayeed S, Li J, McClane BA. 2010. Characterization of virulence plasmid diversity among Clostridium perfringens type B isolates. Infect Immun 78:495–504. doi: 10.1128/IAI.00838-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sayeed S, Li J, McClane BA. 2007. Virulence plasmid diversity in Clostridium perfringens type D isolates. Infect Immun 75:2391–2398. doi: 10.1128/IAI.02014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, McClane BA. 2014. Contributions of NanI sialidase to Caco-2 Cell adherence by Clostridium perfringens type A and C strains causing human intestinal disease. Infect Immun 82:4620–4630. doi: 10.1128/IAI.02322-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher DJ, Miyamoto K, Harrison B, Akimoto S, Sarker MR, McClane BA. 2005. Association of beta2 toxin production with Clostridium perfringens type A human gastrointestinal disease isolates carrying a plasmid enterotoxin gene. Mol Microbiol 56:747–762. doi: 10.1111/j.1365-2958.2005.04573.x. [DOI] [PubMed] [Google Scholar]
- 31.Craven SE, Blankenship LC, McDonel JL. 1981. Relationship of sporulation, enterotoxin formation, and spoilage during growth of Clostridium perfringens type A in cooked chicken. Appl Environ Microbiol 41:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hunter SE, Clarke IN, Kelly DC, Titball RW. 1992. Cloning and nucleotide sequencing of the Clostridium perfringens epsilon-toxin gene and its expression in Escherichia coli. Infect Immun 60:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sawires YS, Songer JG. 2005. Multiple-locus variable-number tandem repeat analysis for strain typing of Clostridium perfringens. Anaerobe 11:262–272. doi: 10.1016/j.anaerobe.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Robertson SL, Li J, Uzal FA, McClane BA. 2011. Evidence for a prepore stage in the action of Clostridium perfringens epsilon toxin. PLoS One 6:e22053. doi: 10.1371/journal.pone.0022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, McClane BA, Fisher DJ, Rood JI, Gupta P. 2005. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl Environ Microbiol 71:7542–7547. doi: 10.1128/AEM.71.11.7542-7547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao J, Zhong J, Fang Y, Geisinger E, Novick RP, Lambowitz AM. 2006. Use of targetrons to disrupt essential and nonessential genes in Staphylococcus aureus reveals temperature sensitivity of Ll.LtrB group II intron splicing. RNA 12:1271–1281. doi: 10.1261/rna.68706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nowell VJ, Poppe C, Parreira VR, Jiang YF, Reid-Smith R, Prescott JF. 2010. Clostridium perfringens in retail chicken. Anaerobe 16:314–315. doi: 10.1016/j.anaerobe.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 38.Rood JI. 1998. Virulence genes of Clostridium perfringens. Annu Rev Microbiol 52:333–360. doi: 10.1146/annurev.micro.52.1.333. [DOI] [PubMed] [Google Scholar]
- 39.Fisher DJ, Fernandez-Miyakawa ME, Sayeed S, Poon R, Adams V, Rood JI, Uzal FA, McClane BA. 2006. Dissecting the contributions of Clostridium perfringens type C toxins to lethality in the mouse intravenous injection model. Infect Immun 74:5200–5210. doi: 10.1128/IAI.00534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sayeed S, Fernandez-Miyakawa ME, Fisher DJ, Adams V, Poon R, Rood JI, Uzal FA, McClane BA. 2005. Epsilon-toxin is required for most Clostridium perfringens type D vegetative culture supernatants to cause lethality in the mouse intravenous injection model. Infect Immun 73:7413–7421. doi: 10.1128/IAI.73.11.7413-7421.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geric B, Carman RJ, Rupnik M, Genheimer CW, Sambol SP, Lyerly DM, Gerding DN, Johnson S. 2006. Binary toxin-producing, large clostridial toxin-negative Clostridium difficile strains are enterotoxic but do not cause disease in hamsters. J Infect Dis 193:1143–1150. doi: 10.1086/501368. [DOI] [PubMed] [Google Scholar]
- 42.Carter GP, Rood JI, Lyras D. 2012. The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol 20:21–29. doi: 10.1016/j.tim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Antunes A, Martin-Verstraete I, Dupuy B. 2011. CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79:882–899. doi: 10.1111/j.1365-2958.2010.07495.x. [DOI] [PubMed] [Google Scholar]
- 44.Dupuy B, Sonenshein AL. 1998. Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 27:107–120. doi: 10.1046/j.1365-2958.1998.00663.x. [DOI] [PubMed] [Google Scholar]
- 45.Mendez MB, Goni A, Ramirez W, Grau RR. 2012. Sugar inhibits the production of the toxins that trigger clostridial gas gangrene. Microb Pathog 52:85–91. doi: 10.1016/j.micpath.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 46.Carter GP, Larcombe S, Li L, Jayawardena D, Awad MM, Songer JG, Lyras D. 2014. Expression of the large clostridial toxins is controlled by conserved regulatory mechanisms. Int J Med Microbiol 304:1147–1159. doi: 10.1016/j.ijmm.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Ma M, Gurjar A, Theoret JR, Garcia JP, Beingesser J, Freedman JC, Fisher DJ, McClane BA, Uzal FA. 2014. Synergistic effects of Clostridium perfringens enterotoxin and beta toxin in rabbit small intestinal loops. Infect Immun 82:2958–2970. doi: 10.1128/IAI.01848-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keyburn AL, Bannam TL, Moore RJ, Rood JI. 2010. NetB, a pore-forming toxin from necrotic enteritis strains of Clostridium perfringens. Toxins 2:1913–1927. doi: 10.3390/toxins2071913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyamoto K, Yumine N, Mimura K, Nagahama M, Li J, McClane BA, Akimoto S. 2011. Identification of novel Clostridium perfringens type E strains that carry an iota toxin plasmid with a functional enterotoxin gene. PLoS One 6:e20376. doi: 10.1371/journal.pone.0020376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Miyamoto K, McClane BA. 2007. Comparison of virulence plasmids among Clostridium perfringens type E isolates. Infect Immun 75:1811–1819. doi: 10.1128/IAI.01981-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Billington SJ, Wieckowski EU, Sarker MR, Bueschel D, Songer JG, McClane BA. 1998. Clostridium perfringens type E animal enteritis isolates with highly conserved, silent enterotoxin gene sequences. Infect Immun 66:4531–4536. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Miyamoto K, Li J, Sayeed S, Akimoto S, McClane BA. 2008. Sequencing and diversity analyses reveal extensive similarities between some epsilon-toxin-encoding plasmids and the pCPF5603 Clostridium perfringens enterotoxin plasmid. J Bacteriol 190:7178–7188. doi: 10.1128/JB.00939-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. 2007. Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66:206–219. doi: 10.1111/j.1365-2958.2007.05906.x. [DOI] [PubMed] [Google Scholar]
- 54.Li J, Ma M, Sarker MR, McClane BA. 2013. CodY is a global regulator of virulence-associated properties for Clostridium perfringens type D strain CN3718. mBio 4:e00770–13. doi: 10.1128/mBio.00770-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibert M, Petit L, Raffestin S, Okabe A, Popoff MR. 2000. Clostridium perfringens iota-toxin requires activation of both binding and enzymatic components for cytopathic activity. Infect Immun 68:3848–3853. doi: 10.1128/IAI.68.7.3848-3853.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richardson M, Granum PE. 1983. Sequence of the amino-terminal part of enterotoxin from Clostridium perfringens type A: identification of points of trypsin activation. Infect Immun 40:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bokori-Brown M, Savva CG, Fernandes da Costa SP, Naylor CE, Basak AK, Titball RW. 2011. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J 278:4589–4601. doi: 10.1111/j.1742-4658.2011.08140.x. [DOI] [PubMed] [Google Scholar]
- 58.Vidal JE, McClane BA, Saputo J, Parker J, Uzal FA. 2008. Effects of Clostridium perfringens beta-toxin on the rabbit small intestine and colon. Infect Immun 76:4396–4404. doi: 10.1128/IAI.00547-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gibert M, Jolivet-Reynaud C, Popoff MR. 1997. Beta2 toxin, a novel toxin produced by Clostridium perfringens. Gene 203:65–73. doi: 10.1016/S0378-1119(97)00493-9. [DOI] [PubMed] [Google Scholar]
- 60.Katoh T, Honda T, Miwatani T. 1988. Purification and some properties of cytotoxin produced by Clostridium difficile. Microbiol Immunol 32:551–564. doi: 10.1111/j.1348-0421.1988.tb01417.x. [DOI] [PubMed] [Google Scholar]
- 61.Sawires YS, Songer JG. 2006. Clostridium perfringens: insight into virulence evolution and population structure. Anaerobe 12:23–43. doi: 10.1016/j.anaerobe.2005.10.002. [DOI] [PubMed] [Google Scholar]