

Abstract
We developed a porcine dermal explant model to determine the extent to which Staphylococcus aureus biofilm communities deplete oxygen, change pH, and produce damage in underlying tissue. Microelectrode measurements demonstrated that dissolved oxygen (DO) in biofilm-free dermal tissue was 4.45 ± 1.17 mg/liter, while DO levels for biofilm-infected tissue declined sharply from the surface, with no measurable oxygen detectable in the underlying dermal tissue. Magnetic resonance imaging demonstrated that biofilm-free dermal tissue had a significantly lower relative effective diffusion coefficient (0.26 ± 0.09 to 0.30 ± 0.12) than biofilm-infected dermal tissue (0.40 ± 0.12 to 0.48 ± 0.12; P < 0.0001). Thus, the difference in DO level was attributable to biofilm-induced oxygen demand rather than changes in oxygen diffusivity. Microelectrode measures showed that pH within biofilm-infected explants was more alkaline than in biofilm-free explants (8.0 ± 0.17 versus 7.5 ± 0.15, respectively; P < 0.002). Cellular and nuclear details were lost in the infected explants, consistent with cell death. Quantitative label-free shotgun proteomics demonstrated that both proapoptotic programmed cell death protein 5 and antiapoptotic macrophage migration inhibitory factor accumulated in the infected-explant spent medium, compared with uninfected-explant spent media (1,351-fold and 58-fold, respectively), consistent with the cooccurrence of apoptosis and necrosis in the explants. Biofilm-origin proteins reflected an extracellular matrix-adapted lifestyle of S. aureus. S. aureus biofilms deplete oxygen, increase pH, and induce cell death, all factors that contribute to impede wound healing.
INTRODUCTION
Staphylococcus aureus causes a diversity of infections in both humans and animals, ranging from minor skin infections to life-threatening toxic shock syndrome, endocarditis, and septicemia in humans (1). It is one of the most common bacterial pathogens found in both acute and chronic wounds, and its treatment is complicated by the emergence of drug-resistant strains commonly known as methicillin-resistant S. aureus (MRSA) (2–4). Formation of biofilms in wounds is a major cause of nonhealing wounds (5–7) and treatment failure (8). Oxygen and pH are two critical factors in wound healing (9–12), but at present we have limited knowledge about how the presence of S. aureus biofilms affects oxygen and pH levels in wounds.
Oxygen availability is essential for wound healing (9, 10) and is required for optimal function of leukocytes and fibroblasts (13, 14). Oxygen enhances leukocyte chemotaxis, phagocytosis, and antibacterial activities. Killing of bacteria by leukocytes is compromised when oxygen pressure drops below 40 mm Hg (∼1.81 mg/liter) (15, 16). A minimum oxygen level is required for replication and proper functioning of cells, including fibroblasts, which play a critical role in wound repair. For example, fibroblasts can replicate at oxygen levels below 25 mm Hg (∼1.13 mg/liter), but their capacity for hydroxylation of proline and lysine, which is required for collagen synthesis, is impaired. Cell replication is arrested at oxygen levels below 0.5% (approximately 0.16 mg/liter) (17). Furthermore, angiogenesis in chronic wounds also requires oxygen. Consequently, hypoxia can impair or prevent wound healing through several mechanisms (18). Because biofilm communities consume oxygen, we surmise that S. aureus biofilms negatively impact oxygen content in tissues underlying the wound surface. Similarly, mass transport of essential nutrients in the tissue will influence the healing process. Mass transfer of O2 and nutrients can be characterized by a parameter known as the effective diffusion coefficient. To our knowledge, this parameter has never been measured in tissues underlying biofilms.
Normal wound healing proceeds by tissue degradation and reassembly and therefore requires various enzymes and proteins. Enzymes function optimally in a defined pH range. In chronic wounds, for example, pH increases from 7.5 to 8.9, and this enhances the activity of proteolytic enzymes. As healing ensues, wound pH declines to around 6, where the activity of enzyme inhibitors is optimal (11, 19, 20). Nothing is known about the specific contribution to the pH of chronic wounds by various types of bacterial biofilms in general and S. aureus biofilms in particular, despite S. aureus being an important wound pathogen. In addition to oxygen and pH, successful wound healing requires basic functions from surviving cells. While it has been shown that S. aureus can kill isolated blood cells (erythrocytes and monocytes) and various cultured cells, little is known about how S. aureus biofilms impact host cells within the context of tissue.
We hypothesized that S. aureus biofilms can deplete dissolved oxygen and change the pH of tissue and that these changes together with secretion of bacterial products into the tissue result in cell and underlying tissue damage. To test this hypothesis, we developed a porcine explant dermal model that is conducive to measuring these and related parameters. Explants were 500-μm-thick samples of dermis from pig ears, which we selected in part because porcine skin closely resembles human skin (21) and pigs are generally preferable models for studying human infectious diseases (22). The biofilms were grown on dermal tissue, and microelectrodes were used to measure dissolved oxygen and pH profiles throughout the depth of the biofilms and the tissue. In addition, we estimated relative effective diffusion coefficients using pulsed-field gradient nuclear magnetic resonance imaging (or magnetic resonance microimaging) to understand if S. aureus biofilms negatively impact mass transfer processes. Cytotoxic effects of biofilm were examined by histology, and proteins released as a result of biofilm–dermal-tissue interaction were identified by quantitative label-free shotgun proteomics. The explant model allowed us to accurately determine these parameters, which are either difficult or impossible to determine accurately in an in vivo animal model.
MATERIALS AND METHODS
Explant preparation and infection.
Ears (pinnae) were collected from healthy pigs (Sus scrofa domesticus) within 1 h of slaughter at a local abattoir and transported to the lab on ice (no study-specific animals were purchased or housed for the work reported herein). The ears were rinsed with water, dried with a paper towel, clipped, and shaved. Ear tissue was then lightly wetted with 70% ethanol and then sectioned longitudinally in 2- to 3-cm-wide strips using a sterile surgical blade. A 500-μm-thick slice of skin was removed from each strip using a Padgett's dermatome (Nouvag, Goldach, Switzerland). After removal of the epidermis, one more layer of 500-μm thickness containing mostly dermis was sliced from each strip. These dermal strips were placed in petri dishes containing ice-cold phosphate-buffered saline (PBS; pH = 7.2). Discs 12 mm in diameter were punched out from each dermal strip using surgical biopsy punches. The discs were placed in insert wells (Greiner Bio-one Inc., Monroe, NC, USA) on a membrane (0.4-μm pore size). Care was taken to ensure that the inner surface (in reference to intact skin) of the discs rested on the membrane for proper nutrient acquisition by the explants (Fig. 1). The insert wells were placed in 6-well plates that were prefilled with 2 ml of Dulbecco's modified Eagle medium (DMEM) with high glucose (HyClone Laboratories Inc., Logan, UT, USA) containing the antibiotics streptomycin (100 μg/ml), penicillin (100 units/ml), and chloramphenicol (5 μg/ml) and the antifungal amphotericin B (0.4 μg/ml). The spent explant medium was replaced with fresh medium on the 4th and 7th days after initial explant culture. pH and lactate dehydrogenase (LDH) activity was measured with a CytoTox 96 kit (Promega Corporation, Madison, WI, USA), described below, from a fraction of the spent medium, and the remaining medium was stored at −80°C for other analyses. All equipment was either autoclaved or disinfected before use. The work was primarily carried out in a biological safety cabinet to limit the probability of external contamination of the explants.
FIG 1.
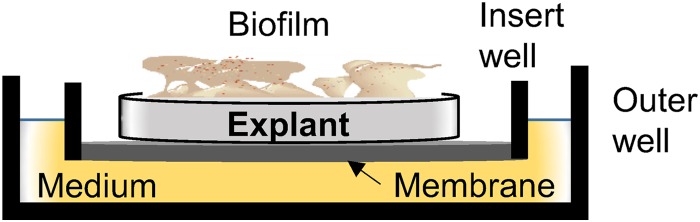
Cross-sectional schematic of a culture well with inner membrane cup, explant tissue, and biofilm.
Explants were infected with a green fluorescent protein (GFP)-expressing S. aureus strain (strain ALC7559) that was derived from S. aureus LAC (USA300), a well-characterized clinical strain (23). ALC7559 carries the plasmid pALC1743 (24), with genes for GFP expression and ampicillin (Ampr) and chloramphenicol (Cmr) resistance. Frozen stocks of bacteria were streaked onto Luria-Bertani (LB; Becton Dickinson and Company, Sparks, MD, USA) agar containing chloramphenicol (5 μg/ml), penicillin (100 μg/ml), and streptomycin (100 μg/ml) and incubated overnight at 37°C. A small portion of the bacterial growth on the agar surface was scraped off and suspended in 1 ml of PBS. A 5-μl volume, containing approximately 500 to 800 CFU, of this suspension was then deposited onto the center of each explant to initiate a local infection and development of a biofilm. CFU were confirmed from each bacterial suspension used to infect the explants.
Growth of S. aureus in DMEM.
To determine if S. aureus grows in the explant culture medium, cells were grown overnight on LB agar, and the resultant cells were detached and retrieved by washing with PBS (1 ml). After adjustment of the optical density at 600 nm (OD600) to 0.7 unit, 50 μl of the suspension was added to phenol red-free DMEM with high glucose (950 μl) and LB broth (950 μl). Aliquots (200 μl) from each of the two inoculated media were separately dispensed into 100-well honeycomb plates (Growth Curves USA, Piscataway, NJ, USA). The plates were loaded into a Bioscreen C plate reader (Growth Curves USA) to record growth (OD600) at 37°C every 30 min.
Assessing explant viability.
Explant viability was assessed using PrestoBlue cell viability reagent (Invitrogen Corporation, Carlsbad, CA, USA), which is a resazurin-based cell viability reagent. Explants were incubated in 300 μl of 10% PrestoBlue (in DMEM with antibiotics) for 2 h at 37°C in a 5% CO2 incubator. After incubation, the absorbance of the medium was measured at 570 and 600 nm. Percent reduction of PrestoBlue was calculated from absorbance and molar extinction coefficients of oxidized and reduced PrestoBlue.
Diffusion coefficient in explants.
Pulsed-field gradient nuclear magnetic resonance (PFG-NMR) was used to measure diffusion coefficients and generate relative effective diffusion coefficient profiles through the Staphylococcus aureus biofilm and explant tissue, similar to the method described by Renslow et al. (25). Specifically, the NMR experiments were conducted at 500.40 MHz for proton (1H) detection using an 89-mm wide-bore 11.7-T magnet with a Bruker Avance III digital NMR spectrometer (Bruker Instruments, Billerica, MA). Bruker ParaVision 5.1 imaging software was used to collect and process the data for the diffusion tensor imaging method (DtiStandard) (26). This method was run with a 750-ms repetition time, an echo time of 15.5 ms, and 128 averages. The pulse gradient width (∂) was 2 ms, and the diffusion time interval (Δ) was 10 ms. Signal intensity was measured for seven independent b factors: 0, 100, 250, 425, 625, 850, and 1050 s/mm2. The field-of-view dimensions were 5 mm in the biofilm/explant depth direction and 5 mm in the perpendicular direction, with a 500-μm-thick slice. A total of 64 complex points were sampled in the depth direction, with 32 phase-encoding steps, for an in-plane resolution of 78 μm by 156 μm. The total experiment time per depth profile was 307 min. Diffusion coefficients were calculated using semilogarithmic regression of the b-factor-dependent intensity value of each measurement pixel in accord with the Block-Torrey differential equation (26, 27). Depth profiles were generated by averaging and performing statistical analysis on the diffusion coefficients in the middle 780 μm of the sample. Therefore, the 78-μm-resolution depth profiles represent the center 780- by 500-μm2 region of the biofilm/explant. PFG-NMR measures diffusion coefficient of water. The measured diffusion coefficient was normalized by dividing the value at a local point by the diffusion coefficient in the bulk. This normalized diffusion coefficient is called the relative effective diffusion coefficient (28).
Histological assessment of explants.
A subset of explants from days 1, 4, and 7 were fixed in 10% neutral buffered formalin, dehydrated by exposure to increasing concentrations of ethyl alcohol, embedded in paraffin, and sectioned to a thickness of 5 μm. The tissue sections were mounted on glass slides, deparaffinized, hydrated, and stained with hematoxylin and eosin according to standard protocols followed by the Washington Animal Disease Diagnostic Laboratory, Washington State University, Pullman, WA. Resulting light micrographs were interpreted by a board-certified veterinary anatomic pathologist.
Biofilm imaging.
Biofilms on 4- and 7-day explants were examined using a Leica SP5 confocal microscope (Leica Microsystems Inc., Deerfield, IL, USA). GFP-labeled bacteria were imaged using 488-nm excitation and 510-nm emission light. Biofilm structure was calculated using Image Structure Analyzer software, developed by our research group (29). We used environmental scanning electron microscopy (ESEM) to image cells on explants at the end of experiments. The samples were first fixed in a solution of glutaraldehyde (2%) and paraformaldehyde (2%) in 0.1 M cacodylate buffer at 4°C. The samples were rinsed with 0.1 M cacodylate buffer twice for 10 min and then treated with osmium tetroxide (2% in 0.1 M cacodylate buffer) overnight at 4°C. Afterwards the samples were rinsed twice in 0.1 M cacodylate buffer for 10 min followed by rinsing with double-distilled water twice for 10 min. The samples were examined using a Quanta 200F (FEI, Hillsboro, OR, USA) scanning electron microscope in environmental mode, in which the samples were frozen to −1°C and the chamber humidity was adjusted to 35% before the electron beam was switched on for scanning the sample surfaces.
Biofilm and explant oxygen and pH depth profiles.
We followed previously described protocols to construct microelectrodes (30). We used dissolved-oxygen microelectrodes with a tip diameter <20 μm in diameter. The dissolved oxygen microelectrodes were polarized to −800 mVAg/AgCl. Prior to use, the microelectrodes were calibrated using two-point calibration: in the air (oxygen saturation) and in Na2SO3 solution (zero oxygen concentration). pH microelectrodes were constructed using a liquid ion-exchange (LIX) membrane tip (28). We used an external Ag/AgCl reference electrode. The pH microelectrodes were calibrated in buffer solutions (pH = 4, 7, and 10).
A Keithley 6517A electrometer/high-resistance meter was used to operate the dissolved-oxygen or pH microelectrode. The surface of the biofilm/explant sample was located using a Zeiss Stemi 2000 stereomicroscope (Carl Zeiss Microscopy, Thornwood, NY, USA). The microelectrode tip was lowered with 10-μm step sizes into the biofilm/explant sample. A stepper motor (PI M-230.10S, part no. M23010SX; Physik Instrumente, Auburn, MA), controlled by custom MicroProfiler software, was used to manipulate the movement of the microelectrodes. Collected data were recorded using an analog/digital converter (ADC; USB-1608FS; Measurement Computing, Norton, MA). The profiles showing the change of oxygen or pH by depth are called “depth profiles.” The pH in spent medium was also measured using a pH electrode (Mettler Toledo, Columbus, OH, USA).
Lactate dehydrogenase activity in spent media.
Lactate dehydrogenase activity in the spent medium was assayed using a CytoTox 96 kit (Promega Corporation). Reconstituted substrate mix (50 μl) was added to 50 μl of diluted spent medium, and the solution was incubated at room temperature for 30 min. The reaction was stopped by adding stop solution provided in the kit, and the OD490 was recorded. The LDH activity of the test samples was calculated from standard curve that was prepared using an LDH-positive control (1,600 U of LDH/ml).
Soluble proteins in the spent media.
Soluble proteins in the spent medium were identified by using ultraperformance liquid chromatography/high-bandwidth mass spectrometry (UPLC/MSE). The proteins in the spent medium from S. aureus-infected and S. aureus-free 4-day explants were split and concentrated into two fractions using ultrafiltration devices (Merck Millipore Ltd., Tullagreen, Ireland). One fraction contained proteins of >50 kDa, and the other contained proteins within a range of 3 to 50 kDa. The DMEM in each sample was replaced with PBS by diluting the medium using an excess of PBS and then filtering the diluted suspension through an appropriate ultrafiltration device. A minimum of three washes in PBS were given to each sample. Albumin in the >50-kDa fraction was removed using a Pierce albumin depletion kit (Thermo Scientific, Rockford, IL, USA) according to the instructions of the manufacturer.
The concentrated proteins were quantified using the Qubit protein assay kit (Invitrogen) in accordance with the supplier's protocol for the Qubit 2.0 fluorometer (Invitrogen). Proteins from each sample (100 ng) were digested with trypsin and then analyzed by using a Synapt G2-S HDMS mass spectrometer (Waters Corp., Milford, MA, USA) coupled with a nanoAcquity UPLC system (Waters Corporation). Eluted proteins were identified using a quantitative label-free shotgun proteomic strategy with data-independent scanning (MSE) and ion mobility spectrometry. Data were processed and searched using TransOmics informatics for proteomics (Waters Corporation), and all searches were performed against a protein database of Staphylococcus aureus and Sus scrofa from UniProt 2013_09. Quantities of various proteins in infected- and uninfected-explant media were compared using the TM4 microarray software suite (31).
Statistical analysis.
Data obtained from multiple explants from a minimum of three different animals were used in the statistical analysis. Unless otherwise stated, we used a t test and one- or two-factor analysis of variance (ANOVA) to test the significance among groups, and all the pairwise multiple comparisons were carried out using the Tukey or Holm-Šidák method or with a Bonferroni correction, as applicable. SigmaPlot v. 12.0 (SYSTAT Software Inc.) and the TM4 software suite (31) were used for statistical analyses.
RESULTS
To validate our model, we first verified that S. aureus cannot sustain growth in the culture medium (DMEM) and that therefore any biofilm growth is dependent on essential nutrients from the dermal explants (Fig. 2). In addition, we verified that biofilm-free explants were capable of reducing PrestoBlue, a resazurin-based cell viability testing reagent. Dye reduction was evident for up to 7 days (∼70% reduction by day 1, ∼60% by day 4, and ∼35% by day 7) (see Fig. S1 and Table S1 in the supplemental material). Viable cells reduce PrestoBlue resazurin, a blue compound, to resorufin, a red fluorescent compound. While this assay does not provide an absolute count of viable cells for these tissues, a greater magnitude of chemical reduction is consistent with a greater magnitude of metabolic activity and, presumably, a larger number of viable cells in the explants.
FIG 2.

Comparison of growth curves of S. aureus (strain ALC7559) grown in DMEM and LB broth (15 independent replicates; vertical bars represent standard deviations [SD]).
S. aureus biofilms increase relative effective diffusion coefficient in dermal tissue.
In live uninfected dermal explants, the relative effective diffusion coefficient dropped from 1.00 near the superficial surface to approximately 0.29 near the bottom 100 μm, consistent with a sigmoidal distribution (Fig. 3). In S. aureus-infected explants, these profiles were significantly greater than those in uninfected explants (P < 0.0001), indicating that biofilms actively enhance diffusion in the underlying dermal tissue.
FIG 3.

Relative effective diffusion coefficient depth profiles through uninfected and S. aureus-infected dermal explants. Relative effective diffusion coefficient is the ratio of the diffusion coefficient of water in biological material (explants) to the diffusion coefficient of water in bulk fluid (DMEM). (a) The biofilm or explant superficial surface begins at 0 μm, and the increasing negative numbers indicate increasing depths within the explants. Error bars represent SD for 3 independent NMR measurements. (b) The relative effective diffusion coefficient was significantly higher in S. aureus-infected explants than in uninfected control explants (two-way ANOVA, Holm-Šidák method; P < 0.0001; the model included explant age [4 day and 7 day] plus infection [uninfected and infected] plus interaction; the latter was not significant [P = 0.40]).
S. aureus biofilms kill dermal cells.
Histopathological examination of 1-, 4-, and 7-day uninfected explants showed mild changes in cell and nuclear morphology in the more metabolically active cells. These changes included mild vacuolation of myofibers in the tunica muscularis of arterioles, pyknosis and nuclear fragmentation of endothelial cells, and occasional small clusters of perivascular lymphocytes. In S. aureus-infected explants, the dermal architecture was diffusely intact; however, nearly all cells were hypereosinophilic. Nuclei were nearly completely absent, indicating widespread cell death (Fig. 4). There was no difference in cytotoxicity between 4- and 7-day biofilm-infected explants.
FIG 4.

Histopathological changes in 4-day S. aureus-infected dermal explants. (a) Uninfected explant; (b) S. aureus-infected explant. The arrow in panel a points to a fibroblast nucleus; no equivalent nucleus was present with infected dermal explant. An arrowhead highlights cellular detail in blood vessels in panel a, whereas the basophilic stippling associated with the vascular bundles in panel b appears to be nuclear debris. The double arrowheads in panel b point to biofilm on the explant surface.
S. aureus biofilms deplete oxygen in dermal tissue.
The lowest oxygen concentration (4.45 ± 1.17 mg/liter) was found in high-viability-score, 4-day, biofilm-free explants (Table 1), which corresponds to the approximate partial pressure of oxygen found in most tissues at the capillary level in a resting individual (32). The 7-day biofilm-free explants showed statistically nonsignificant increases in oxygen compared with 4-day explants. The explants with no viable dermal cells had statistically significant (P < 0.05) oxygen content (5.87 ± 0.73 mg/liter) compared with 4- and 7-day live dermal tissue (Table 1). With S. aureus biofilms growing on the top of explants, oxygen was depleted within the first 50 μm of depth, leaving no measurable oxygen in deeper parts of the biofilms or in the explants; a typical example is shown in Fig. 5 (this result was replicated independently three times). We found similar profiles for day 7 biofilms on explant (results not shown).
TABLE 1.
Oxygen content in uninfected control explantsa
| Explant | Oxygen level (mg/liter) | Effective diffusion coefficient (mm2/s) |
|---|---|---|
| 4 day | 4.45 ± 1.17 | 2.32 × 10−3 ± 8.31 × 10−5 |
| 7 day | 4.72 ± 1.12 | 2.41 × 10−4 ± 5.96 × 10−5 |
| Deadb | 5.87 ± 0.73* | 2.32 × 10−3 ± 8.31 × 10−5 |
Values are means ± SD, calculated from multiple data points from oxygen and diffusion coefficient depth profiles of three different explants. Comparisons of oxygen content and diffusion coefficients were carried out using ANOVA (*, P < 0.05).
Explants were killed in 2% paraformaldehyde.
FIG 5.

Typical dissolved-oxygen depth profiles of uninfected and S. aureus-infected explants. The biofilm or explant superficial surface begins at 0 μm, and the increasing negative distance refers to the increasing depth within the explant tissue. No measurable oxygen is present deep inside the tissue explants.
Changes in S. aureus biofilms with age.
Electron microscopy showed that 4-day biofilms were patchy, but these patches coalesced by day 7 (Fig. 6). The bacterial cells in 4-day biofilm were healthy in appearance and rounded, whereas by day 7, the biofilm surface was flat and composed of both lysed and intact bacterial cells that had a flattened appearance (Fig. 6). Confocal laser scanning microscopy (CLSM) examination of dermal biofilms also confirmed bacterial growth interrupted by small spaces in 4-day biofilms, while the growth was dense in 7-day biofilms (see Fig. S2 in the supplemental material). Biofilm structural and volumetric parameters were measured from multiple CLSM images of 4- and 7-day biofilms. The average diffusion distance, defined as the average of all distances between cell clusters and their nearest void area in a biofilm (29), was significantly greater (1.7-fold; P < 0.05) in 7-day biofilm, indicating a progressive coalescence of cell clusters with time (Table 2). The biovolume (total volume of the biofilm biomass), and the average biofilm and biomass thicknesses were also greater (about 2-fold) in day 7 biofilm than day 4 biofilm (Table 2). Biofilm thickness is the volume of the biofilm divided by the total surface area of the substratum, including the areas free of bacterial cell clusters, whereas the average biomass thickness is the volume of the biofilm divided by only that area of the substratum covered by the cell clusters (29). These values nearly doubled in the 7-day biofilms. No significant difference was observed in biovolume-to-surface area ratio between 4- and 7-day biofilms, indicating a uniform bacterial growth in all directions. This is expected in the case of S. aureus, which multiplies in all spatial planes. Despite the volumetric and morphological differences between 4- and 7-day biofilms, biofilms completely depleted oxygen from the top to the bottom of the explants for both time points.
FIG 6.

Environmental scanning electron microscopic images of 4-day (a) and 7-day (b) S. aureus biofilms on dermal explants. The cell clusters in 4-day biofilms appear healthy, with spatially separated cell clusters. The 7-day biofilms show a flat surface with lysed bacteria (arrow in panel b).
TABLE 2.
Differences between 4- and 7-day biofilm structural parameters
| Structural parametera | Value forb: |
|
|---|---|---|
| 4-day biofilm | 7-day biofilm | |
| Biovolume (log μm3) | 7.31 ± 0.06* | 7.58 ± 0.03* |
| Biofilm thickness (μm) | 39.50 ± 5.60* | 86.0 ± 2.50* |
| Biomass thickness (μm) | 46.3 ± 2.90* | 88.2 ± 3.20* |
| Biovolume-to-surface area ratio | 1.7 ± 0.07 | 1.9 ± 0.08 |
| Porosity | 0.71 ± 0.06 | 0.61 ± 0.05 |
| Diffusion distance (μm) | 3.44 ± 0.37* | 5.14 ± 0.31* |
Biovolume is the total volume of the biofilm biomass. Biofilm thickness is the volume of the biofilm divided by the total surface area imaged. Biomass thickness is the volume of the biofilm divided by only that area of the substratum covered by the cells. Porosity is the ratio of total void volume to total biofilm volume. Diffusion distance is the average diffusion distance across all cell clusters in the biofilm.
Values are means ± standard errors from 4 independent experiments. Values that differ significantly (P < 0.05) between 4- and 7-day biofilms are indicated with an asterisk. Comparisons were made using a t test (significance is based on the Bonferroni adjustment; P < 0.008).
In hypoxic tissues, one would expect larger amounts of lactate dehydrogenase and lower pH (33–35). Under hypoxic conditions, LDH converts pyruvate, generated in glycolysis, to lactate, thus decreasing pH (36). The enzyme is also released in larger amounts by damaged cells (37). We measured LDH activity in the spent media from both uninfected and S. aureus-infected explants. As expected, significantly greater LDH activity was detected in spent media from infected explants (1,892 ± 39 mU/ml from 4-day and 1,255 ± 126 mU/ml from 7-day explants; P < 0.001) than in media from uninfected explants (467 ± 68 mU/ml from 4-day and 430 ± 90 mU/ml from 7-day explants) (see Fig. S3 in the supplemental material). Proteomic analysis of the spent media from infected explants revealed the presence of bacterial-origin LDH (see Table S3). This bacterial-origin LDH could contribute to the higher activity of LDH detected in the media of infected explants.
S. aureus biofilms increase pH in dermal tissue.
Because pH can influence wound healing by affecting enzymes and cells involved in the healing process (11, 19), we measured pH depth profiles inside the biofilm and dermal explants. The pH was always significantly more alkaline (P < 0.002) inside the biofilm and the biofilm-infected dermal tissue than in uninfected tissue (8.08 ± 0.03 versus 7.62 ± 0.04 in 4-day explants, and 7.83 ± 0.16 versus 7.39 ± 0.13 in 7-day explants) (Fig. 7). The pH in the spent media was always significantly more alkaline for media from infected dermal explants than biofilm-free dermal explants (P < 0.001) (see Fig. S4 in the supplemental material). These findings were unexpected given that the presence of more LDH with infected biofilms should have made the medium more acidic.
FIG 7.

pH depth profiles in uninfected and S. aureus-infected explants. (a) Representative pH depth profile in uninfected and S. aureus-infected 4-day explants. (b) Histogram for mean pH ± SD in uninfected and S. aureus-infected explants. Values were calculated by pooling measurements from every 10 μm from the top to the bottom of the explant from three different explants. The pH is significantly higher in S. aureus-infected than in uninfected explants (two-way ANOVA, Holm-Šidák method; P < 0.002; the model included explant age [4 day and 7 day] plus infection [uninfected and infected] plus interaction; the latter was not significant [P = 0.97]).
S. aureus-infected dermal tissue releases pro- and antiapoptotic porcine proteins and extracellular matrix-colonizing S. aureus proteins.
S. aureus-infected explants released ∼2.5-fold more protein in the spent medium than uninfected explants. A comparison of soluble proteins from infected and uninfected explants showed a greater diversity of proteins from explants that were infected with S. aureus. A total of 211 porcine-origin and 96 S. aureus-origin proteins were detected in the spent media. Of 211 porcine-origin proteins, 77 accumulated differentially (false discovery rate of <1%) (see Table S2 in the supplemental material). Programmed-cell-death protein 5 (PDCD5) accumulated to levels >1,000-fold higher in biofilm-infected explant media, consistent with explant cells undergoing apoptotic cell death. Accumulation of cytoskeletal proteins was also consistent with dermal cell apoptosis. Compared to uninfected explant media, higher quantities of cofilin-1 (90-fold), tropomyosin-1 (9-fold), and LIM domain and actin-binding protein 1 (LIMA1) (337-fold) and smaller amounts of moesin (11-fold lower) were found in the spent media from infected explants. In addition, the antiapoptotic and proinflammatory cytokines macrophage migration inhibitory factor (58-fold) and interleukin 6 (9-fold) were also found in significantly larger amounts in the spent media from infected explants.
The S. aureus-origin proteins are given in Table S3 in the supplemental material, and the proteins that explain the S. aureus colonization and likely effect of biofilm on dermis explants are summarized in Table S4 in the supplemental material. In addition to known extracellular-matrix adhesins, the amino acid (proline and alanine)- and sugar (glucosamine)-metabolizing enzymes 1-pyrroline-5-carboxylate dehydrogenase (8,192-fold), alanine dehydrogenase (388-fold), and glucosamine-6-phosphate isomerase (1,024-fold) accumulated in high abundance in the spent medium from infected dermal tissue.
DISCUSSION
S. aureus is a common organism isolated from both chronic and acute wound infections, including surgical and combat-related wounds (38, 39). It has a number of virulence factors suitable for rapid growth and biofilm formation in wounds (40), and treatment is difficult, particularly for antibiotic-resistant strains. Despite its involvement in both acute and chronic wounds (41, 42), little is known about the role of S. aureus biofilms in wound pathogenesis. Three important factors affecting wound healing are oxygen availability, pH, and host cell survival. To examine these parameters in the context of biofilms, we first developed a porcine dermis explant model that resembles a dermal wound. Removal of epidermis exposes the extracellular matrix within the dermis, making it available for binding by S. aureus, which has a greater binding affinity for exposed extracellular matrix proteins (40, 43, 44). In addition, S. aureus is well adapted to survive and form biofilms in the extracellular matrix environment (see Table S3 in the supplemental material). We then demonstrated that biofilm-free explants remain largely viable for a week and that the partial pressure of oxygen toward the deep surface is similar to that found in intact human dermis (45 to 65 mm Hg) (45). The explant model also permits the detection of soluble proteins that are released as a result of biofilm-explant interaction, which is impossible in vivo.
Oxygen is essential in wounds because it is required for ATP generation (46). The rate of diffusion of oxygen in tissue is directly proportional to the rate of diffusion of water molecules (30), where diffusion is a function of random movement of water molecules and is hindered by the compact fibrous nature of tissue, such as can be found in dermal explants. Compared to the diffusion coefficient of water in DMEM, the effective diffusion coefficient of water in explants decreased from 100% to about 29% at the deep surface of the explants (Fig. 3). Dissolved-oxygen content also decreased from top to the bottom in both biofilm-free live and dead explants, but the overall oxygen content was always higher than 4.45 ± 1.17 mg/liter in viable explants (Table 1). Resting partial pressure of oxygen in most of the body tissues falls within the range we found in our explants (32), and therefore, the explant model reflects physiologically meaningful oxygen partial pressure. The oxygen content in 7-day explants was higher than that in the 4-day explants, consistent with reduced consumption of oxygen with decreasing explant viability, although the difference was not statistically significant in this case.
The relative effective diffusion coefficient increased significantly (P < 0.0001) in biofilm-infected explants (Fig. 3). This increase could primarily be due to cell death in the infected explants (Fig. 4). The degradation of extracellular matrix proteins by S. aureus could contribute to an increase in the average relative effective diffusion coefficient, but we did not observe any alterations in connective tissue based on histological examination, nor did we find any significant amounts of connective tissue matrix proteins in medium from S. aureus-infected dermal tissue (see Table S2 in the supplemental material). The increase in protein content in medium from S. aureus-infected explants was mainly of dermal cellular origin (see Table S2).
Despite the increase in relative effective diffusion coefficient, no measurable oxygen could be detected in the biofilm-infected explants. This indicated that changes in oxygen concentration in biofilm and dermal tissue are mainly caused by the oxygen consumption by biofilms rather than a mechanism that otherwise hindered diffusion. The S. aureus biofilms depleted oxygen throughout the full thickness (500 μm) of the explants (Fig. 5). This depletion was irrespective of changes in biofilm structure. We did not determine the minimum biomass of S. aureus biofilm that is needed to cause this degree of oxygen demand throughout an entire dermal explant.
Others have reported that hypoxic cells express more LDH, which results in a subsequent decrease in pH (33, 34). Although we observed more LDH activity in spent media from infected explants, the pH was higher in the biofilm, biofilm-infected explants, and the spent medium than in the uninfected explants. The reason for the higher LDH activity in infected-explant media could partly be explained by substantial amounts of S. aureus-origin l-lactate dehydrogenase, Ldh2 (see Table S3 in the supplemental material). S. aureus can increase LDH expression under anaerobic conditions or in response to nitrosative stress (47, 48), but its role in the absence of lactate production (anaerobic metabolism) and nitrosative stress is not known. In severe cases of infection, S. aureus can cause lactic acidosis (49, 50). It is possible that in these cases, secretion of LDH by S. aureus could contribute to already existing lactic acidosis. Because it is an enzyme, small quantities of LDH can produce relatively large quantities of lactic acid.
S. aureus can increase biofilm and explant pH by consuming explant proteins and amino acids as a source of energy. Protein analysis of the spent medium identified some key S. aureus-origin enzymes involved in amino acid metabolism, including 1-pyrroline-5-carboxylate dehydrogenase (involved in proline catabolism [51]), glucosamine-6-phosphate isomerase (converts d-glucosamine 6-phosphate to d-fructose 6-phosphate and ammonia [52]), and alanine dehydrogenase 1 (which catalyzes conversion of alanine and NAD+ to pyruvate, ammonium, and NADH [53]). The extracellular matrix, which is a binding site of S. aureus (40), contains abundant quantities of collagen, which is rich in proline and alanine, and hyaluronic acid; the latter contains d-glucosamine (54). The essential requirement of proline and alanine for S. aureus growth (55) indicates that S. aureus is well adapted for survival and multiplication in dermal tissue. The increase in pH could also be contributed to by arginine deiminase present in the arginine catabolic mobile element (ACME) of this strain. Arginine deiminase increases pH by producing ammonia and ATP from l-arginine (56). We also observed accumulation of alkaline shock protein in spent media from S. aureus-infected explants; alkaline shock protein expression is increased in S. aureus at elevated pH (57).
Wound pH can have a profound effect on enzyme function and on cells located at the site of injury. Acidic pH favors wound healing by enhancing fibroblast replication, early angiogenesis, and release of oxygen that is bound to hemoglobin (Bohr effect). This is in contrast with chronic wounds, where pH remains alkaline most of the time except during the epithelialization stage (11, 12, 20). S. aureus lives in acidic pH on skin surface (58, 59), but our model indicates that once established in wounds, S. aureus biofilms increase pH, and this is likely to be an impediment to wound healing.
Irrespective of the nature and type of the cells found in the dermis, S. aureus killed all the explant cells by day 4. The diffuse cell death combined with accumulation of large quantities of programmed cell death protein 5 (PDCD5) (more than 1,000-fold) and some apoptosis-associated cytoskeletal proteins indicated that apoptotic cell death may have occurred in the explants. PDCD5 (TFAR19) is a proapoptotic factor (60), and its expression is increased during apoptosis (61–63). It functions in apoptosis either through direct interaction with p53 (63) or via indirect acetylation of p53 (64). P53 plays a key role in cell cycle arrest and apoptosis (65, 66). PDCD5 could also act through impairment of chaperonin CCT (cytosolic chaperonin containing tailless complex polypeptide 1)-mediated beta-tubulin folding (67), thereby inducing apoptosis through an intrinsic pathway.
Previous reports indicate that S. aureus induces apoptosis in osteoblasts (through an extrinsic apoptotic pathway involving a tumor necrosis factor-related apoptosis-inducing ligand) and human umbilical vein and endothelial cell lines only when found intracellularly (68, 69). In our case, histological staining indicated no evidence that bacterial cells invaded the tissue explants, indicating that tissue cell death was driven by a mechanism that does not require intracellular invasion (but see below). S. aureus biofilms may instead induce apoptosis by depleting oxygen. In a hypoxic environment, hypoxia-inducible factor 1 (HIF-1), the key oxygen-sensitive transcription factor mediating cell response in hypoxia, induces high concentrations of proapoptotic genes and stabilizes p53 (70, 71). Under hypoxic conditions, bacterial biofilm products may potentiate the apoptotic effect.
Paradoxically, two proinflammatory and antiapoptotic cytokines (macrophage migration inhibitory factor [MIF] and interleukin 6 [IL-6] [72, 73]) accumulated to significantly larger amounts in biofilm-infected-explant media. Both MIF and IL-6 either inhibit or downregulate p53 and consequently should limit apoptosis (74–76). The expression of antiapoptotic cytokines locally at the site of infection, by either some or most cells (most likely during initial infection), may be the host's response to counter biofilm-induced apoptotic death, prolong cell survival, and aid in proliferation of infiltrating phagocytes and lymphocytes. In explants and actual wounds, S. aureus biofilms may induce both apoptosis and oncosis simultaneously, with apoptosis-induced cell death possibly predominating initially during the infection, when abundant ATP is available for energy-dependent apoptotic processes.
Recent work with a mouse model indicates that integrin-linked kinase (ILK) and Rac1 are requisite for S. aureus to invade mouse keratinocytes (77). S. aureus was able to penetrate skin explants (from the epidermis to dermis) of ILK-deficient mice with significantly greater efficiency, and consequently ILK is required for the epidermis to serve as an effective barrier against staphylococcal invasion. Once S. aureus breaches the epidermis, it likely penetrates the dermis through a combination of cell invasion and loss of tight junction integrity from protease activity. Our micrographs showed no evidence that S. aureus penetrated into the porcine dermis, and we hypothesize that cell death was driven by secreted toxins from the biofilm community. Nevertheless, our micrographs may have lacked sufficient analytic sensitivity to detect bacteria that might have been dispersed within the tissue. If they were present, their numbers were not great enough to detect with conventional staining techniques.
We studied the impact of S. aureus biofilms directly on dermal tissue because it contains multiple types of cells and closely resembles the natural wound surface. By using this model, we demonstrated that S. aureus-biofilms deplete oxygen and increase pH in tissues underlying the biofilms. During a host-pathogen interaction, bacterial pathogens may consume oxygen to the extent that host cells are deprived of this vital resource. Here we show that ∼40-μm-thick S. aureus biofilm growing on superficial surface of dermal tissue can increase the diffusion coefficient in underlying tissue while consuming oxygen so rapidly that nearly all oxygen is depleted throughout a full 500-μm-thick dermal tissue explant. Hypoxia and alterations in pH have a global impact on gene expression, enzyme function, and cell behavior that adversely impact wound healing. We report that S. aureus biofilms kill dermal cells, most likely through an intrinsic pathway of apoptosis by inducing proapoptotic protein PDCD5. The intrinsic pathway of apoptosis can be activated by hypoxia, by bacterial products, or by a combination of both. In response to biofilm infection, we found substantial production of an important proinflammatory and antiapoptotic cytokine, MIF, which probably counters apoptosis induced by S. aureus biofilms for at least part of the infectious period. Our findings indicate that S. aureus biofilms can counter the host immune response far below the actual biofilm surface (500 μm). We therefore propose that it is important to consider how bacterial biofilm-host interaction impacts the physical, biochemical, and immunological state of tissues underlying the actual surface of bacterial colonization.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by a grant (DM110308) from the U.S. Department of Defense and by the Agricultural Animal Health Program, Washington State University, and the Washington State Agricultural Research Center, Pullman, WA. Mass spectrometric analysis was performed on an instrument acquired through a Major Research Instrumentation grant (DBI-1229749) from National Science Foundation to DRG. All NMR experiments were performed at the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory. Ryan Renslow was also partially supported by a Linus Pauling Distinguished Postdoctoral Fellowship at Pacific Northwest National Laboratory.
Niles Donegan, the Geisel School of Medicine, Dartmouth, kindly provided GFP-labeled S. aureus. We thank Lisa Orfe for her technical assistance in the lab. Carla Schubiger helped with histopathology. Tom Tevlin and Daniel Broeckel of Garfield Meats (Garfield, WA) and Sam Hunt and Jake Brunton of C&L Locker (Moscow, ID) generously donated pig ears in support of this research. The Franceschi Microscopy Center of Washington State University is gratefully acknowledged for providing the electron and confocal microscopy resources and especially for the help rendered by Valeri Lynch-Holm.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.03075-14.
REFERENCES
- 1.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stryjewski ME, Corey GR. 2014. Methicillin-resistant Staphylococcus aureus: an evolving pathogen. Clin Infect Dis 58(Suppl 1):S10–S19. doi: 10.1093/cid/cit613. [DOI] [PubMed] [Google Scholar]
- 3.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. 2006. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med 355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 4.O'Hara FP, Amrine-Madsen H, Mera RM, Brown ML, Close NM, Suaya JA, Acosta CJ. 2012. Molecular characterization of Staphylococcus aureus in the United States 2004-2008 reveals the rapid expansion of USA300 among inpatients and outpatients. Microb Drug Resist 18:555–561. doi: 10.1089/mdr.2012.0056. [DOI] [PubMed] [Google Scholar]
- 5.James GA, Swogger E, Wolcott R, Pulcini ED, Secor P, Sestrich J, Costerton JW, Stewart PS. 2008. Biofilms in chronic wounds. Wound Repair Regen 16:37–44. doi: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 6.Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, Wolcott RD. 2008. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8:43. doi: 10.1186/1471-2180-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirketerp-Møller K, Zulkowski K, James G. 2011. Chronic wound colonization, infection, and biofilms, p 11–24. In Bjarnsholt T, Jensen P, Moser C, Høiby N (ed), Biofilm infections. Springer, New York, NY. [Google Scholar]
- 8.Bjarnsholt T, Kirketerp-Møller K, Jensen PØ, Madsen KG, Phipps R, Krogfelt K, Høiby N, Givskov M. 2008. Why chronic wounds will not heal: a novel hypothesis. Wound Repair Regen 16:2–10. doi: 10.1111/j.1524-475X.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 9.Gordillo GM, Sen CK. 2003. Revisiting the essential role of oxygen in wound healing. Am J Surg 186:259–263. doi: 10.1016/S0002-9610(03)00211-3. [DOI] [PubMed] [Google Scholar]
- 10.Tandara AA, Mustoe TA. 2004. Oxygen in wound healing—more than a nutrient. World J Surg 28:294–300. doi: 10.1007/s00268-003-7400-2. [DOI] [PubMed] [Google Scholar]
- 11.Schneider LA, Korber A, Grabbe S, Dissemond J. 2007. Influence of pH on wound-healing: a new perspective for wound-therapy? Arch Dermatol Res 298:413–420. doi: 10.1007/s00403-006-0713-x. [DOI] [PubMed] [Google Scholar]
- 12.Sharpe JR, Booth S, Jubin K, Jordan NR, Lawrence-Watt DJ, Dheansa BS. 2013. Progression of wound pH during the course of healing in burns. J Burn Care Res 34:e201–e208. doi: 10.1097/BCR.0b013e31825d5569. [DOI] [PubMed] [Google Scholar]
- 13.Behm B, Babilas P, Landthaler M, Schreml S. 2012. Cytokines, chemokines and growth factors in wound healing. J Eur Acad Dermatol Venereol 26:812–820. doi: 10.1111/j.1468-3083.2011.04415.x. [DOI] [PubMed] [Google Scholar]
- 14.Parinello S. 2003. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5:839–839. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaVan FB, Hunt TK. 1990. Oxygen and wound healing. Clin Plast Surg 17:463–472. [PubMed] [Google Scholar]
- 16.McGovern NN, Cowburn AS, Porter L, Walmsley SR, Summers C, Thompson AAR, Anwar S, Willcocks LC, Whyte MKB, Condliffe AM, Chilvers ER. 2011. Hypoxia selectively inhibits respiratory burst activity and killing of Staphylococcus aureus in human neutrophils. J Immunol 186:453–463. doi: 10.4049/jimmunol.1002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardner LB, Li F, Yang XJ, Dang CV. 2003. Anoxic fibroblasts activate a replication checkpoint that is bypassed by E1a. Mol Cell Biol 23:9032–9045. doi: 10.1128/MCB.23.24.9032-9045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen CK. 2009. Wound healing essentials: let there be oxygen. Wound Repair Regen 17:1–18. doi: 10.1111/j.1524-475X.2008.00436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schreml S, Szeimies RM, Karrer S, Heinlin J, Landthaler M, Babilas P. 2010. The impact of the pH value on skin integrity and cutaneous wound healing. J Eur Acad Dermatol Venereol 24:373–378. doi: 10.1111/j.1468-3083.2009.03413.x. [DOI] [PubMed] [Google Scholar]
- 20.Gethin GT, Cowman S, Conroy RM. 2008. The impact of Manuka honey dressings on the surface pH of chronic wounds. Int Wound J 5:185–194. doi: 10.1111/j.1742-481X.2007.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Sullivan TP, Eaglstein WH, Davis SC, Mertz P. 2001. The pig as a model for human wound healing. Wound Repair Regen 9:66–76. doi: 10.1046/j.1524-475x.2001.00066.x. [DOI] [PubMed] [Google Scholar]
- 22.Meurens F, Summerfield A, Nauwynck H, Saif L, Gerdts V. 2012. The pig: a model for human infectious diseases. Trends Microbiol 20:50–57. doi: 10.1016/j.tim.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, Long RD, Dorward DW, Gardner DJ, Lina G, Kreiswirth BN, DeLeo FR. 2006. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis 194:1761–1770. doi: 10.1086/509506. [DOI] [PubMed] [Google Scholar]
- 24.Xiong YQ, Van Wamel W, Nast CC, Yeaman MR, Cheung AL, Bayer AS. 2002. Activation and transcriptional interaction between agr RNAII and RNAIII in Staphylococcus aureus in vitro and in an experimental endocarditis model. J Infect Dis 186:668–677. doi: 10.1086/342046. [DOI] [PubMed] [Google Scholar]
- 25.Renslow R, Babauta J, Majors P, Beyenal H. 2013. Diffusion in biofilms respiring on electrodes. Energy Environ Sci 6:595–607. doi: 10.1039/C2EE23394K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torrey HC. 1956. Bloch equations with diffusion terms. Phys Rev 104:563–565. doi: 10.1103/PhysRev.104.563. [DOI] [Google Scholar]
- 27.Renslow RS, Majors PD, McLean JS, Fredrickson JK, Ahmed B, Beyenal H. 2010. In situ effective diffusion coefficient profiles in live biofilms using pulsed-field gradient nuclear magnetic resonance. Biotechnol Bioeng 106:928–937. doi: 10.1002/bit.22755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewandowski Z, Beyenal H. 2013. Microsensors: construction, instrumentation, and calibration, p 269–351. In Lewandowski Z, Beyenal H (ed), Fundamentals of biofilm research, 2nd ed CRC Press. [Google Scholar]
- 29.Lewandowski Z, Beyenal H. 2013. Quantifying biofilm structure, p 353–436. In Lewandowski Z, Beyenal H (ed), Fundamentals of biofilm reseach, 2nd ed CRC Press, Boca Raton, FL. [Google Scholar]
- 30.Lewandowski Z, Beyenal H. 2003. Use of microsensors to study biofilms, p 375–412. In Lens P, O'Flaherty V, Moran A, Stoodley P, Mahony T (ed), Biofilms in medicine, industry and environmental biotechnology: characteristics, analysis and control. IWA Publishing, London, United Kingdom. [Google Scholar]
- 31.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. 2006. TM4 microarray software suite. Methods Enzymol 411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 32.Carreau A, Hafny-Rahbi BE, Matejuk A, Grillon C, Kieda C. 2011. Why is the partial oxygen pressure of human tissues a crucial parameter?. Small molecules and hypoxia J Cell Mol Med 15:1239–1253. doi: 10.1111/j.1582-4934.2011.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. 1996. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 34.Kay HH, Zhu S, Tsoi S. 2007. Hypoxia and lactate production in trophoblast cells. Placenta 28:854–860. doi: 10.1016/j.placenta.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Marti HH, Jung HH, Pfeilschifter J, Bauer C. 1994. Hypoxia and cobalt stimulate lactate dehydrogenase (LDH) activity in vascular smooth muscle cells. Pflugers Arch 429:216–222. doi: 10.1007/BF00374315. [DOI] [PubMed] [Google Scholar]
- 36.Bender DA. 2009. Glycolysis and the oxidation of pyruvate, p 149–156. In Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA (ed), Harper's illustrated biochemistry, 28th ed McGraw-Hill, New York, NY. [Google Scholar]
- 37.Hoffmann WE, Solter PF. 2008. Diagnostic enzymology of domestic animals, p 351–378. In Kaneko JJ, Harvey JW, Bruss ML (ed), Clinical biochemistry of domestic animals, 6th ed Academic Press, New York, NY. [Google Scholar]
- 38.Co E, Keen EF III, Aldous W. 2011. Prevalence of methicillin-resistant Staphylococcus aureus in a combat support hospital in Iraq. Mil Med 176:89–93. [DOI] [PubMed] [Google Scholar]
- 39.Keen EF III, Robinson BJ, Hospenthal DR, Aldous WK, Wolf SE, Chung KK, Murray CK. 2010. Incidence and bacteriology of burn infections at a military burn center. Burns 36:461–468. doi: 10.1016/j.burns.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Smeltzer M, Beenken KE. 2013. Staphylococcus aureus, p 184–193. In McVey DS, Kennedy M, Chengappa MM (ed), Veterinary microbiology, 3rd ed John Wiley & Sons, New York, NY. [Google Scholar]
- 41.King MD, Humphrey BJ, Wang YF, Kourbatova EV, Ray SM, Blumberg HM. 2006. Emergence of community-acquired methicillin-resistant Staphylococcus aureus USA 300 clone as the predominant cause of skin and soft-tissue infections. Ann Intern Med 144:309–317. doi: 10.7326/0003-4819-144-5-200603070-00005. [DOI] [PubMed] [Google Scholar]
- 42.Demling RH, Waterhouse B. 2007. The increasing problem of wound bacterial burden and infection in acute and chronic soft-tissue wounds caused by methicillin-resistant Staphylococcus aureus. J Burns Wounds 7:e8. [PMC free article] [PubMed] [Google Scholar]
- 43.Hauck CR, Ohlsen K. 2006. Sticky connections: extracellular matrix protein recognition and integrin-mediated cellular invasion by Staphylococcus aureus. Curr Opin Microbiol 9:5–11. doi: 10.1016/j.mib.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 44.McGavin MH, Krajewska-Pietrasik D, Ryden C, Hook M. 1993. Identification of a Staphylococcus aureus extracellular matrix-binding protein with broad specificity. Infect Immun 61:2479–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Howard MA, Asmis R, Evans KK, Mustoe TA. 2013. Oxygen and wound care: a review of current therapeutic modalities and future direction. Wound Repair Regen 21:503–511. doi: 10.1111/wrr.12069. [DOI] [PubMed] [Google Scholar]
- 46.Kivisaari J, Vihersaari T, Renvall S, Niinikoski J. 1975. Energy metabolism of experimental wounds at various oxygen environments. Ann Surg 181:823. doi: 10.1097/00000658-197506000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuchs S, Pane-Farre J, Kohler C, Hecker M, Engelmann S. 2007. Anaerobic gene expression in Staphylococcus aureus. J Bacteriol 189:4275–4289. doi: 10.1128/JB.00081-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richardson AR, Libby SJ, Fang FC. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672–1676. doi: 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 49.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 50.Stewart KD, Brackett DJ, Lerner MR, Archer LT, Wilson MF. 1994. Comparison of Staphylococcus aureus and Escherichia coli infusion in conscious rats. J Surg Res 56:60–66. doi: 10.1006/jsre.1994.1010. [DOI] [PubMed] [Google Scholar]
- 51.Meile L, Leisinger T. 1982. Purification and properties of the bifunctional proline dehydrogenase/1-pyrroline-5-carboxylate dehydrogenase from Pseudomonas aeruginosa. Eur J Biochem 129:67–75. doi: 10.1111/j.1432-1033.1982.tb07021.x. [DOI] [PubMed] [Google Scholar]
- 52.Midelfort CF, Rose IA. 1977. Studies on the mechanism of Escherichia coli glucosamine-6-phosphate isomerase. Biochemistry 16:1590–1596. doi: 10.1021/bi00627a010. [DOI] [PubMed] [Google Scholar]
- 53.Bellion E, Tan F. 1987. An NAD+-dependent alanine dehydrogenase from a methylotrophic bacterium. Biochem J 244:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murray RK, Keeley FW. 2009. The extracellular matrix, p 527–544. In Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA (ed), Harper's illustrated biochemistry, 28th ed McGraw-Hill, New York, NY. [Google Scholar]
- 55.Becker SA, Palsson BO. 2005. Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: an initial draft to the two-dimensional annotation. BMC Microbiol 5:8. doi: 10.1186/1471-2180-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thurlow LR, Joshi GS, Clark JR, Spontak JS, Neely CJ, Maile R, Richardson AR. 2013. Functional modularity of the arginine catabolic mobile element contributes to the success of USA300 methicillin-resistant Staphylococcus aureus. Cell Host Microbe 13:100–107. doi: 10.1016/j.chom.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuroda M, Ohta T, Hayashi H. 1995. Isolation and the gene cloning of an alkaline shock protein in methicillin resistant Staphylococcus aureus. Biochem Biophys Res Commun 207:978–984. doi: 10.1006/bbrc.1995.1281. [DOI] [PubMed] [Google Scholar]
- 58.Kohler T, Weidenmaier C, Peschel A. 2009. Wall teichoic acid protects Staphylococcus aureus against antimicrobial fatty acids from human skin. J Bacteriol 191:4482–4484. doi: 10.1128/JB.00221-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmid-Wendtner M-H, Korting HC. 2006. The pH of the skin surface and its impact on the barrier function. Skin Pharmacol Physiol 19:296–302. doi: 10.1159/000094670. [DOI] [PubMed] [Google Scholar]
- 60.Wang N, Lu HS, Guan ZP, Sun TZ, Chen YY, Ruan GR, Chen ZK, Jiang J, Bai CJ. 2007. Involvement of PDCD5 in the regulation of apoptosis in fibroblast-like synoviocytes of rheumatoid arthritis. Apoptosis 12:1433–1441. doi: 10.1007/s10495-007-0070-z. [DOI] [PubMed] [Google Scholar]
- 61.Liu HT, Wang YG, Zhang YM, Song QS, Di CH, Chen GH, Tang J, Ma DL. 1999. TFAR19, a novel apoptosis-related gene cloned from human leukemia cell line TF-1, could enhance apoptosis of some tumor cells induced by growth factor withdrawal. Biochem Biophys Res Commun 254:203–210. doi: 10.1006/bbrc.1998.9893. [DOI] [PubMed] [Google Scholar]
- 62.Li HS, Zhang X, Song X, Zhu F, Wang Q, Guo C, Liu C, Shi Y, Ma C, Wang X. 2012. PDCD5 promotes cisplatin-induced apoptosis of glioma cells via activating mitochondrial apoptotic pathway. Cancer Biol Ther 13:822–830. doi: 10.4161/cbt.20565. [DOI] [PubMed] [Google Scholar]
- 63.Xu LJ, Hu J, Zhao YB, Hu J, Xiao J, Wang YM, Ma DL, Chen YY. 2012. PDCD5 interacts with p53 and functions as a positive regulator in the p53 pathway. Apoptosis 17:1235–1245. doi: 10.1007/s10495-012-0754-x. [DOI] [PubMed] [Google Scholar]
- 64.Xu LJ, Chen YY, Song QS, Xu D, Wang Y, Ma DL. 2009. PDCD5 interacts with tip60 and functions as a cooperator in acetyltransferase activity and DNA damage-induced apoptosis. Neoplasia 11:345–354. doi: 10.1593/neo.81524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moll UM, Zaika A. 2001. Nuclear and mitochondrial apoptotic pathways of p53. FEBS Lett 493:65–69. doi: 10.1016/S0014-5793(01)02284-0. [DOI] [PubMed] [Google Scholar]
- 66.Vousden KH, Lu X. 2002. Live or let die: the cell's response to p53. Nat Rev Cancer 2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 67.Tracy CM, Gray AJ, Cuéllar J, Shaw TS, Howlett AC, Taylor RM, Prince JT, Ahn NG, Valpuesta JM, Willardson BM. 2014. Programmed cell death protein 5 interacts with the cytosolic chaperonin containing tailless complex polypeptide 1 (CCT) to regulate β-tubulin folding. J Biol Chem 289:4490–4502. doi: 10.1074/jbc.M113.542159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alexander EH, Rivera FA, I M, Anguita J, Bost KL, Hudson MC. 2003. Staphylococcus aureus-induced tumor necrosis factor-related apoptosis-inducing ligand expression mediates apoptosis and caspase-8 activation in infected osteoblasts. BMC Microbiol 3:5. doi: 10.1186/1471-2180-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haslinger-Loffler B, Kahl BC, Grundmeier M, Strangfeld K, Wagner B, Fischer U, Cheung AL, Peters G, Schulze-Osthoff K, Sinha B. 2005. Multiple virulence factors are required for Staphylococcus aureus-induced apoptosis in endothelial cells. Cell Microbiol 7:1087–1097. doi: 10.1111/j.1462-5822.2005.00533.x. [DOI] [PubMed] [Google Scholar]
- 70.Hammond EM, Giaccia AJ. 2005. The role of p53 in hypoxia-induced apoptosis. Biochem Biophys Res Commun 331:718–725. doi: 10.1016/j.bbrc.2005.03.154. [DOI] [PubMed] [Google Scholar]
- 71.Weinmann M, Jendrossek V, Handrick R, Guner D, Goecke B, Belka C. 2004. Molecular ordering of hypoxia-induced apoptosis: critical involvement of the mitochondrial death pathway in a FADD/caspase-8 independent manner. Oncogene 23:3757–3769. doi: 10.1038/sj.onc.1207481. [DOI] [PubMed] [Google Scholar]
- 72.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. 2007. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med 13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 73.Yonish-Rouach E, Resnftzky D, Lotem J, Sachs L, Kimchi A, Oren M. 1991. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 352:345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- 74.Fingerle-Rowson G, Petrenko O, Metz C, Forsthuber T, Mitchell R, Huss R, Moll U, Müller W, Bucala R. 2003. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci U S A 100:9354–9359. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. 2002. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A 99:345–350. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brighenti E, Calabrese C, Liguori G, Giannone FA, Trere D, Montanaro L, Derenzini M. 2014. Interleukin 6 downregulates p53 expression and activity by stimulating ribosome biogenesis: a new pathway connecting inflammation to cancer. Oncogene 33:4396–4406. doi: 10.1038/onc.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sayedyahossein S, Xu SX, Rudkouskaya A, McGavin MJ, McCormick JK, Dagnino L. 2015. Staphylococcus aureus keratinocyte invasion is mediated by integrin-linked kinase and Rac1. FASEB J 29:711–723. doi: 10.1096/fj.14-262774. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.