

Abstract
In this study, the cytotoxicity of the recently described subtilase variant SubAB2-2 of Shiga toxin-producing Escherichia coli was determined and compared to the plasmid-encoded SubAB1 and the chromosome-encoded SubAB2-1 variant. The genes for the respective enzymatic active (A) subunits and binding (B) subunits of the subtilase toxins were amplified and cloned. The recombinant toxin subunits were expressed and purified. Their cytotoxicity on Vero cells was measured for the single A and B subunits, as well as for mixtures of both, to analyze whether hybrids with toxic activity can be identified. The results demonstrated that all three SubAB variants are toxic for Vero cells. However, the values for the 50% cytotoxic dose (CD50) differ for the individual variants. Highest cytotoxicity was shown for SubAB1. Moreover, hybrids of subunits from different subtilase toxins can be obtained which cause substantial cytotoxicity to Vero cells after mixing the A and B subunits prior to application to the cells, which is characteristic for binary toxins. Furthermore, higher concentrations of the enzymatic subunit SubA1 exhibited cytotoxic effects in the absence of the respective B1 subunit. A more detailed investigation in the human HeLa cell line revealed that SubA1 alone induced apoptosis, while the B1 subunit alone did not induce cell death.
INTRODUCTION
Shiga toxin-producing Escherichia coli (STEC) strains are zoonotic bacterial pathogens causing a variety of symptoms in humans, ranging from relatively mild forms, such as diarrhea, to hemorrhagic colitis and the life-threatening hemolytic-uremic syndrome (HUS) (1). Besides Shiga toxin, the best-characterized pathogenicity factor for the development of serious diseases is the locus of enterocyte effacement (LEE), encoding a type III secretion system and associated effector proteins (2, 3). Other pathogenicity factors can be involved in the development of human disease (4, 5). An example is the subtilase cytotoxin (SubAB), which is located on a 163-kb plasmid of STEC E. coli O113:H21 strain 98NK2 (6). This strain was isolated from a case of HUS in the south of Australia. The subtilase cytotoxin was shown to cause HUS-like symptoms in mice and apoptosis in human epithelial cells (7–9). Subtilase cytotoxin genes were detected in a variety of LEE-negative STEC strains of human, ovine, and game origin (10–14). Furthermore, strains from human origin were shown to cause symptoms in humans ranging from watery diarrhea to fully developed HUS (15, 16).
The SubAB toxin is a typical AB5 toxin, composed of an enzymatically active A subunit (SubA) and a B pentamer (SubB), which mediates the uptake of the toxin into target cells by binding to a specific surface sialic acid, namely, N-glycolylneuraminic acid (Neu5Gc) (17). Neu5Gc is present in most mammals but is not synthesized by human cells due to a 92-bp deletion in the cytidine monophospho-N-acetylneuraminic acid hydroxylase (CMAH) gene (18). However, it was shown that human cells are able to incorporate Neu5Gc from external sources (19). Inside the eukaryotic cell, the enzymatic active A subunit acts as a subtilisin-like serine protease. SubA cleaves the cellular endoplasmic reticulum chaperone GRP78/BiP at an L-L motif on position 416 between the N-terminal ATPase domain and the C-terminal protein binding domain (20). This highly specific protease activity is unique within AB5 toxins and leads to the accumulation of unfolded proteins in the endoplasmic reticulum. This accumulation induces the unfolded protein response (UPR), which in the end results in the death of the affected cell (21). Yahiro and coworkers demonstrated that binding of SubAB to one of four different cell surface receptors, namely, NG2, hepatocyte growth factor receptor (Met), L1 cell adhesion molecule (CAM), and ß1 integrin (ITG), induces signals which also result in apoptosis of HeLa cells without internalization of SubAB (22). However, the underlying mechanism is not completely understood so far.
Besides the plasmid-based subAB (syn. subAB1) operon, two new chromosomal variants have been described. The subAB2-1 operon is located on the chromosomal pathogenicity island SEPAI, close to the tia gene encoding an invasin in enterotoxigenic E. coli (ETEC) (23, 24). The subAB2-1 genes are about 90% homologous to the plasmid-carried subAB1 with the catalytic triad being present in subA2-1, but the subB2-1 open reading frame is one triplet shorter than that of subB1. The subAB2-2 operon recently was described by our group (25), and it could be demonstrated that subAB2-2 was located next to a gene encoding an outer membrane efflux protein and associated genes of a type one secretion system. This variant also shows about 90% sequence identity to the plasmid-encoded subAB1 operon and differs by about 2% from subAB2-1. Furthermore, it could be demonstrated that both chromosomal variants can be present as single operons or in parallel (25).
Although all three gene variants have been characterized on the genetic level, analysis of the cytotoxic action of their gene products was not carried out yet. To determine the cytotoxicity of all three toxins, the respective genes were cloned and expressed as recombinant single subunits, each carrying a C-terminal His tag. After purification of the SubA and SubB proteins, they were analyzed as single subunits but also in various combinations as subunit mixtures of the same and different variants for their cytotoxic effects in African Green Monkey kidney (Vero) cell assays. This method offers the ability to operate under safe laboratory conditions with the toxin A and B subunits and analyze their toxic potential after mixing them together directly before monitoring their effects on cell viability. By investigating the cytotoxic effects of hybrids of subunits from different subtilase toxin combinations, we made the observation that higher concentrations of the A subunit exhibited cytotoxic effects even in the absence of the B subunit in Vero as well as HeLa cells. By investigating this effect in more detail, we found that SubA1 alone induced caspase-dependent cell death in the human HeLa epithelial cell line.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The subAB-positive STEC strains TS30/08, LM27564, and LM14603/08 used in this study were described previously (12, 25). Laboratory E. coli strain BL21(DE3) and the BL21 derivative C41(DE3) (26) were used for protein expression. For standard cultivation, LB broth (27) was used with or without 100 μg/ml ampicillin (sodium salt; Carl Roth AG, Karlsruhe, Germany). For cultivation on solid media, LB broth was supplemented with 1.5% (wt/vol) agar (Becton Dickinson, Heidelberg, Germany).
Cloning of subAB variants.
For cloning of subAB1, both subunit genes were amplified with oligonucleotides subAF-His/SubAR-His and SubBF-His/SubBR-His using genomic DNA of STEC strain TS30/08 (12) as the template (Table 1). The oligonucleotides (Eurofins MWG, Ebersberg, Germany) used for PCR are listed in Table 1. The PCR products were purified using a commercial PCR purification kit (Qiagen, Hilden, Germany), digested with restriction enzymes NdeI and XhoI (Thermo Scientific, MA), and ligated in the plasmid vector pET-22b(+) (Merck Millipore, Darmstadt, Germany), which was digested with the same restriction enzymes and dephosphorylated with fast alkaline phosphatase (Thermo Scientific, MA). Subsequently, the plasmids pET-22b(+)-subA1 and pET-22b(+)-subB1 were transformed into electrocompetent E. coli strains BL21(DE3) and C41 (DE3), respectively. For the chromosomal variants subAB2-1 and subAB2-2, STEC strains LM27564 and LM14603/08 (13) were used, and the cloning protocol was modified as follows. According to the sequence similarities in the 5′ and 3′ gene regions of subAB, a nested PCR was carried out prior to the insert amplification using the oligonucleotides subA-L/subAB2-3′out for subAB2-2 and Linkerfor-subAB/subAB3′tia for subAB2-1 (Fig. 1). The PCR products of this amplification were used for a second amplification reaction using the oligonucleotides listed in Table 1. Ligation and transformation of subA and subB genes in electrocompetent strain C41(DE3) were performed according to the cloning protocol for subA1 and subB1 (described above). The success of the cloning procedure was analyzed after plasmid purification by sequence analysis with a CEQ8000 automated sequencer (Beckman Coulter, Germany). Plasmids generated during the cloning experiments are listed in Table 2.
TABLE 1.
Oligonucleotides used for cloning of the subAB variant genes
| Designation | Sequence (5′–3′) | Tma (°C) | Reference or source |
|---|---|---|---|
| subAF-His | GGG CAT ATG CTT AAG ATT TTA TGG ACG | 61.9 | This study |
| SubA2-2F-His | GGG CAT ATG CTT AAG ATT TTA TGG CC | 61.6 | This study |
| SubA2-1F-His | GGG CAT ATG CTG AAA AAT TTA CGG C | 61.3 | This study |
| SubAR-His | AAA CTC GAG CAG TTC TTC ACT CAT CC | 63.2 | This study |
| SubBF-His | AGG GTG CAT ATG ACG ATT AAG CGT | 61.0 | This study |
| SubBR-His | AAA CTC GAG TGA GTT CTT TTT CCT GTC A | 62.2 | This study |
| SubB2F-His | GGA GGT GCA TAT GAC GAT TAA GAG G | 63.0 | This study |
| SubB2-2R-His | AAA CTC GAG GTT CTT TTT TCT GTC AGG G | 63.7 | This study |
| SubB2-1R-His | AAA CTC GAG GTT CTT TTT CCT GTC GG | 63.2 | This study |
| Linkerfor-subAB | TAT AAG GAG TGC TCC AGG | 53.7 | This study |
| subAB3′tia | ACT GGC TGT TCT AAC CG | 52.8 | 25 |
| subA-L | GGG AGG ATT AAC CAT CG | 52.8 | 25 |
| subAB2-3′out | AGG TCG GCT CAG TGT TC | 55.2 | 25 |
Tm, melting point.
FIG 1.

Cloning strategy for different subAB subunit genes. The genes for the plasmid pO113-encoded subunits SubA1 and SubB1 were cloned into the expression vector pET-22b(+) (A and C) using oligonucleotides subAF-His/SubAR-His and SubBF-His/SubBR-His. For the chromosome-located subAB variants, another cloning approach was chosen. (A) In a first PCR, subAB2-1 and subAB2-2 were amplified, including their flanking DNA regions. For subAB2-2, oligonucleotides subA-L/subAB2-3′out were used, and for subAB2-1, oligonucleotides Linkerfor-subAB/subAB3′tia were used. (B) Amplification of the single-toxin subunits was carried out using the following oligonucleotides: subA2-2, SubA2-2F-His/SubAR-His; subB2-2, SubB2F-His/SubB2-2R/His; subA2-1, SubA2-1F-His/SubAR-His; subB2-1, SubB2-HisF/SubB2-1R-His. (C) PCR products were cloned into pET-22b(+) as shown and as described in the text. All oligonucleotide sequences are listed in Table 1.
TABLE 2.
Plasmids used in this study
| Name | Plasmid characteristic(s) | Source |
|---|---|---|
| pET-22b(+) | T7 promoter, pelB, pBR322 origin, bla+, lacI+, C-terminal His6 | Merck |
| pET-22-subA1-His | pET-22b(+), subA1 | This study |
| pET-22-subB1-His | pET-22b(+), subB1 | This study |
| pET-22-subA2-1-His | pET-22b(+), subA2-1 | This study |
| pET-22-subB2-1-His | pET-22b(+), subB2-1 | This study |
| pET-22-subA2-2-His | pET-22b(+), subA2-2 | This study |
| pET-22-subB2-2-His | pET-22b(+), subB2-2 | This study |
Toxin subunit expression and purification.
For expression of the single-toxin subunits, 400 ml of 2-fold YT medium (28) containing 150 μg/ml ampicillin (sodium salt; Carl Roth Ltd., Karlsruhe, Germany) were inoculated with 8 ml of a bacterial suspension that had been prepared from an overnight culture, which was centrifuged at 2,500 × g at room temperature and resuspended in fresh 2-fold YT medium to remove β-lactamases. The cultures were incubated at 25°C with 180 rpm on a rotary shaker until the optical densities at 600 nm (OD600) reached values of 0.3 to 0.4. Subsequently, the temperature was decreased to 20°C and protein expression was induced with 250 μM isopropyl β-d-1-thiogalactopyranoside after an OD600 of 0.5 was reached. After 20 h of incubation at 20°C, the expression was stopped by incubating the cells for 20 min on ice. Cells were harvested by centrifugation for 20 min at 2,500 × g and 4°C. Following that, cells were washed with precooled resuspension buffer (50 mM NaH2PO4, 100 mM NaCl, pH 7.4) centrifuged for an additional 10 min at 2,500 × g and 4°C and stored at −20°C. Prior to purification, the bacterial cells were resuspended in binding buffer containing 50 mM NaH2PO4, 300 mM NaCl, 5 mM imidazole, 0.1% (vol/vol) Tween 20, 10% (vol/vol) glycerol, and 1 mg/ml lysozyme and then incubated for 1 h on ice. Cell lysis was carried out by ultrasonic disruption using a Sonifier B-12 equipped with a microtip (Branson, Dietzenbach, Germany) and six 10-s ultrasonic steps in 20-s intervals on ice. Insoluble fragments were separated by centrifugation at 25,000 × g and 2°C for 25 min. The supernatant was supplemented with 5 mM Mg-ATP (stock solution contained 1 M Tris, 0.5 M MgCl2, 0.5 M ATP) and 0.75 ml nickel-nitrilotriacetic acid (Ni-NTA) agarose beads and incubated for 2 h on a Dynal sample mixer (Life Technologies, Darmstadt, Germany) at 4°C. The toxin subunits then were purified by gravity chromatography. First the supernatant, including the Ni-NTA beads, was transferred to a disposal column (GE-Healthcare Life Science, Freiburg, Germany) and allowed to flow through after the beads were settled. After that, the beads were washed with 13 ml of buffer 1 (50 mM NaH2PO4, 300 mM NaCl, 5 mM imidazole, 0.1% [vol/vol] Tween 20, 10% [vol/vol] glycerol, 5 mM Mg-ATP, pH 8.0), followed by an additional incubation on the rotary shaker for 20 min with 5 ml buffer 1 and a second washing step also using 13 ml of buffer 1.
Further washing steps were performed using 10 ml buffer 2 (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 10% [vol/vol] glycerol, and 5 mM Mg-ATP, pH 8.0) and 10 ml buffer 2 without glycerol, followed by 4 ml (SubA-His) or 13 ml (SubB-His) buffer 3 (50 mM NaH2PO4, 300 mM NaCl, 40 mM imidazole, 5 mM Mg-ATP, pH 8.0). Elution of the toxin subunits was carried out using 1-ml portions of buffer 4 (50 mM NaH2PO4, 150 mM NaCl, 250 mM imidazole, pH 8.0). Elution fractions one to three were pooled and desalted against Dulbecco's phosphate-buffered saline (PBS) buffer (Merck, formerly Biochrome, Berlin, Germany) using disposal PD10-desalting columns (GE-Healthcare Life Science, Freiburg, Germany). Desalted protein solutions were separated in aliquots and stored at −80°C. Protein concentration was determined using the Roti-Quant Bradford reagent and protease-free bovine serum albumin (Carl Roth Ltd., Karlsruhe, Germany) as a standard reagent. Recombinant His6-C2I was expressed in E. coli BL21 and purified as described earlier (29). The plasmid His6-C2I-pET28 was a kind gift from M. R. Popoff (Institut Pasteur, Paris, France). All recombinant SubA, SubB, and C2I proteins used in this study were His tagged, which is not further mentioned in the text.
SDS-PAGE and Western blotting.
The different purified toxin subunits were analyzed on 15% (wt/vol) SDS polyacrylamide gels according to Laemmli (30) with the following modifications: glycine as tailing ion was replaced in the cathode chamber by tricine for better resolution in the range of 10 to 20 kDa, resulting in a better resolution of the B subunits of the subtilase cytotoxin (14 kDa). SDS-PAGE gels were stained using Roti-Blue colloidal Coomassie solution (Carl Roth Ltd., Karlsruhe, Germany). The purity of the proteins was determined after decoloration and detection using the program ImageJ (version 1.45s; http://imagej.nih.gov/ij) by determination of band intensities over the integral.
For Western blotting of cell-bound toxins, cells were incubated for 30 min at 4°C with either SubA1 or C2I. Subsequently, cells were washed and equal amounts of lysate protein subjected to SDS-PAGE and blotted onto a nitrocellulose membrane (Whatman, Dassel, Germany). After blocking of the membrane for 60 min with 5% nonfat dry milk in PBS containing 0.2% (vol/vol) Tween 20, the His proteins were detected with a specific antibody against the His tag (Qiagen, Hilden, Germany) and a secondary anti-mouse antibody coupled to horseradish peroxidase (Santa Cruz Biotech Inc., Heidelberg, Germany) by using the enhanced chemiluminescence (ECL) system (Millipore, Schwalbach, Germany) according to the manufacturer's instructions. To analyze the binding of SubB1 and SubB2-1 to Vero and HeLa cells, the SubB proteins were biotinylated and cells were incubated at 4°C for 45 min with the proteins. To analyze the amount of cell-bound biotin-SubB proteins, cells were lysed, proteins separated by SDS-PAGE, and the biotinylated proteins detected by Western blotting with streptavidin-peroxidase (Roche, Mannheim, Germany) and ECL detection as described before. For biotin labeling, recombinant SubB1 and SubB2-1 proteins were incubated for 2 h at 4°C with EZ-Link sulfo-NHS-biotin (Pierce, Bonn, Germany), and the unbound sulfo-NHS-biotin subsequently was removed by using Micro Bio-Spin chromatography columns (Bio-Rad, Munich, Germany) according to the manufacturer's instructions. The PageRuler prestained protein ladder was from Thermo Scientific (Bonn, Germany).
Cytotoxicity of recombinant toxin subunits.
To determine the cytotoxicity of the recombinant proteins, Vero B4 cells (DSMZ no. ACC 33) were cultured in RPMI 1640 cell culture medium with stable glutamine (Merck, formerly Biochrome, Berlin, Germany) supplemented with 10% (vol/vol) fetal calf serum (FCS) and incubation conditions at 37°C with 5% (vol/vol) CO2. For crystal violet (CV) staining assays (31), the cells were seeded in 96-well plates at a total of 1 × 103 cells/well and incubated for an additional 24 h before incubation with the recombinant proteins. Vero cells were incubated with the single-toxin subunits and combinations of A and B subunits in equimolar ratios that were incubated in advance together for 30 min at room temperature. Single- and mixed-toxin subunits were serially diluted in serum-free RPMI medium and applied in 100-μl volumes to the wells to reach final concentrations of 10 μg/ml to 4.9 ng/ml. Dulbecco's PBS (Merck, Berlin, Germany) was used as a negative control. For incubation, cells were washed twice with sterile Dulbecco's PBS, the toxin dilutions were applied, and cells were incubated further for 2 h. Following that, FCS was added to a final concentration of 10%, and cells were incubated for an additional 72 h at 37°C and 5% (vol/vol) CO2. Subsequent to incubation, cells were washed with PBS, fixed with 2% (vol/vol) formalin solution, and stained with crystal violet staining solution consisting of 5% (vol/vol) ethanol, 2% (vol/vol) formalin, and 0.13% (wt/vol) crystal violet for 1 min. Excess color was removed with demineralized H2O, and cells were lysed by adding 70% (vol/vol) ethanol. Staining intensity was measured at a wavelength of 570 nm using the infinite M200 96-well plate reader (Tecan, Crailsheim, Germany). All experiments were performed in triplicate on at least two different days. The 50% cytotoxic dose (CD50) per milliliter was calculated by using a logarithmic trend line after transformation of the values into percent viability according to the value of the PBS control of the respective plate, which was calculated as 100% viability. Data, which differed by more than double the standard deviations from the means in positive or negative values, were excluded from the calculation.
HeLa cells were cultivated at 37°C and 5% (vol/vol) CO2 in minimal essential medium (MEM) containing 10% heat-inactivated FCS, 1.5 g/liter sodium bicarbonate, 1 mM sodium-pyruvate, 2 mM l-glutamine, 0.1 mM nonessential amino acids, and 1% (wt/vol) penicillin G and streptomycin. Cells were detached with trypsin every third day and reseeded at most 20 times. For cytotoxicity assays, cells were seeded in 24-well plates and incubated in MEM plus FCS with the respective proteins. After different incubation periods, pictures from the cells were taken with a Zeiss Axiovert 40CFl microscope (Oberkochen, Germany) with a Jenoptik progress C10 charge-coupled device (CCD) camera (Jena, Germany) to monitor the changes in cell morphology, and the number of cells was determined after trypsin-mediated detachment in a Neubauer chamber.
Cell viability, apoptosis, and caspase-3/7 activity were determined by fluorescence-activated cell sorting analysis (FACScan; BD Bioscience, Heidelberg, Germany). Briefly, HeLa cells were seeded in 24-well plates and incubated in MEM plus FCS and the indicated proteins in the absence or presence of N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD.fmk; Bachem, Weil am Rhein, Germany). Cell viability was assessed by measuring forward scatter/side scatter (FSC/SSC), and apoptotic cell death was determined by DNA fragmentation of propidium iodide (PI)-stained nuclei as described previously (32). To monitor caspase-3/7 activity, HeLa cells were stained with CellEvent caspase-3/7 green detection reagent (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions, incubated for 30 min at 37°C, and then immediately analyzed by flow cytometry.
Reproducibility of the experiments and statistics.
All experiments were performed independently at least twice, and results from representative experiments are given in Results. For statistical analysis, values (n ≥ 3) were calculated as means ± standard deviations (SD) with the software GraphPad Prism, version 4 for Windows (GraphPad Software, San Diego, CA; www.graphpad.com). For the cytotoxicity assays with Vero cells, the averaged values from three technical replicates were used for calculation of the standard errors from three biological replicates derived on three different days.
RESULTS
Preparation of recombinant SubAB toxin variants.
Based on previous results of variant-specific PCRs and sequencing analysis (25), STEC strains TS30/08, LM27564, and LM14603/08 were used as templates for cloning of subAB1, subAB2-1, and subAB2-2, respectively. Whereas subA1 and subB1 could be cloned directly into the expression vector, the subunit genes of both chromosomal variants were cloned after amplification of complete operons and surrounding genetic regions by a nested PCR approach as outlined in Fig. 1. This was necessary because of the high sequence identity at the 5′ and 3′ ends of the two chromosomal variants.
All subunit genes were cloned, including the 5′ signal sequences, according to Paton and coworkers (15). After cloning of the different toxin genes, their sequences were confirmed by DNA sequencing (data not shown) and protein expression was carried out in 2-fold yeast extract-tryptone (YT) medium for 20 h at 20°C, followed by purification using a Ni-NTA resin in a batch approach as described above. In first purification experiments, contamination of the B subunit of SubAB1 with the 58-kDa chaperone GroEL was detected (data not shown). Therefore, the purification protocol of the manufacturer was improved in order to remove chaperone contamination during the purification procedure. In detail, 5 mM Mg-ATP was added to the binding and washing buffers to activate GroEL ATPase activity and separate it from the toxin subunits due to its lower affinity in the ATP-bound state (33). Furthermore, 10% (vol/vol) glycerol was added to reduce unspecific protein binding to the Ni-NTA beads and chaperone binding to the tagged protein, as could be demonstrated for DnaK (34). Due to these changes, a purity of more than 90% for the SubA subunits and more than 70% for the SubB subunits was achieved. The purity of the proteins and the results of the protein purification were determined by 15% (wt/vol) SDS-PAGE, and an example is shown for SubA2-2 and SubB2-2 (Fig. 2A, lane 9, and B, lane 8). The purity of the prepared proteins and the protein amount were determined with the program ImageJ using the protein fraction after buffer exchange against Dulbecco's phosphate-buffered saline. The molecular sizes of the recombinant proteins in the gels of about 36 kDa for SubA1 and 14 kDa for SubB1 corresponded well with the theoretical molecular sizes. As an example, the theoretical molecular mass of SubA1 with the His tag is 36.02 kDa and is 34.96 kDa without that tag. When the signal sequences are included, the molecular masses would be 38.5 kDa with the His tag and 37.38 without that tag. The molecular mass of SubB1 with the His tag is 14.0 kDa and is 12.94 kDa without the His tag. Including the signal sequences, the molecular masses of SubB1 are 16.54 kDa with His tag and 15.39 kDa without that tag, respectively. This is in concordance with data from Paton et al. (15) and indicate that the signal sequences in the recombinant proteins shown here have been cleaved.
FIG 2.

Purification of recombinant SubAB2-2 subunits. Protein fractions stained with colloidal Coomassie brilliant blue were analyzed by 15% SDS-PAGE. (A) purification of SubA2-2: lane 1, crude protein extract after lysis of bacteria; lane 2, unbound proteins after Ni-NTA column; lane 3, 5 mM imidazole washing step; lane 4, 20 mM imidazole washing step; lane 5, 40 mM imidazole washing step; lanes 6 to 8, protein elution fractions; lane 9, protein fraction (4 μg) after buffer exchange; M, PageRuler prestained protein ladder (Thermo Scientific, Germany). MW, molecular weight (in thousands). (B) Purification of SubB2-2: lane 1, crude protein extract after lysis of bacteria; lane 2, unbound proteins after Ni-NTA column; lane 3, 5 mM imidazole washing step; lane 4, 40 mM imidazole washing step; lanes 5 to 7, protein elution fractions; lane 8, protein fraction (3 μg) after buffer exchange; M, PageRuler prestained protein ladder (Thermo Scientific, Germany). Frames in panel A, lane 9, and B, lane 8, label the areas which were used for the determination of protein preparation purity using the software tool ImageJ.
Cytotoxicity of SubAB2-2.
The cytotoxic effects of the single SubA2-2 and SubB2-2 subunits and of their combination were determined in a microtiter-based staining assay. The subunit proteins were applied in a concentration of total protein of 10 μg/ml and subsequently serially diluted to 4.9 ng/ml. For determination of the cytotoxicity after combination of these toxin subunits, 10 μg total protein of SubA2-2 and SubB2-2 (6.6 μg SubA2-2 and 3.4 μg SubB2-2, which corresponds roughly to a molar ratio of 1:1.3) were mixed together. For the cytotoxicity assay, serial dilutions with final concentrations of the mixture of both subunits, ranging from 10 μg/ml to 4.9 ng/ml, were applied. For a control, cells were left untreated. The effects of the individual subunits and the SubAB combination on the viability of Vero B4 cells was determined at 72 h after intoxication using a crystal violet staining assay (described above). In Fig. 3, the percentages of viable cells are shown. Applying the single-toxin subunits SubA2-2 and SubB2-2 alone, only SubA2-2 demonstrated a concentration-dependent cytotoxic effect. When 10 μg/ml of SubA2-2 was applied, there were 7% remaining viable cells, while a final concentration of 1.25 μg/ml had only marginal effects on cell viability. From these results, a CD50 value of 3.45 μg/ml could be calculated for the single SubA2-2 subunit. In contrast, the SubB2-2 subunit did not cause cytotoxic effects under these experimental conditions.
FIG 3.

Cytotoxic effects of SubAB2-2. Vero B4 cells were incubated at 37°C with different concentrations of the single SubA2-2 (squares) and SubB2-2 (triangles) toxin subunits and their combination, SubAB2-2 (diamonds), in the medium. For a control, cells were left untreated. After 72 h, the amount of viable cells was determined. The values of the amount of viable cells were plotted against cell viability of the control cells in percentages. The standard errors are shown in the graph.
The in vitro combined SubA2-2 plus SubB2-2 exhibited strong cytotoxic effects on Vero cells in much lower concentrations than the SubA2-2 subunit alone. Under the conditions applied, a CD50 of 75 ng/ml was calculated for the SubA2-2-SubB2-2 mixture. The concentration of the mixed subunits corresponds to a final concentration of approximately 50 ng/ml of the enzymatically active SubA2-2 subunit; therefore, it demonstrates an effective regain of the toxic potential of the subtilase cytotoxin.
Comparison of the toxicity of different SubAB variants.
To compare the cytotoxic potential of the different variants, the A and B subunits of SubAB1, SubAB2-1, and SubAB2-2 were combined in the same way as that described above and analyzed for cytotoxic effects on Vero B4 cells. To minimize the variations in culture conditions and cell viability that can occur between different experiments, all variants were tested in a parallel approach to be sure that equal conditions were achieved for all toxin variants. Dilutions of the combined toxin subunits were used as described above for SubAB2-2, and the CD50 values (in nanograms per milliliter) were determined mathematically as described above. In this experiment, the CD50 value of SubAB1 was 52 ng/ml after combination of the SubA1 and SubB1 subunits in vitro. This is a factor of about 17 higher than the CD50 of 3 ng/ml, as was calculated by Gerhardt et al. (35) for recombinant SubAB1, which was expressed and copurified as a holotoxin in that study. The CD50 value of the combined SubAB2-2 was calculated to be 75 ng/ml, slightly higher than the calculated CD50 value for SubAB1. These results were in good concordance with the results of the first experiment described above. Finally, we calculated a CD50 value of 320 ng/ml for the combined SubAB2-1, meaning that this variant is 4-fold less toxic than SubAB2-2 under the experimental conditions applied. The differences in the strength of cytotoxicity also are obvious from the cell viability histogram (Fig. 4A), where the application of the combined subunits of SubAB2-1 resulted in considerably higher rates of cell viability, in a range from 2.5 μg to 39 ng/ml of protein, compared to SubAB2-2.
FIG 4.
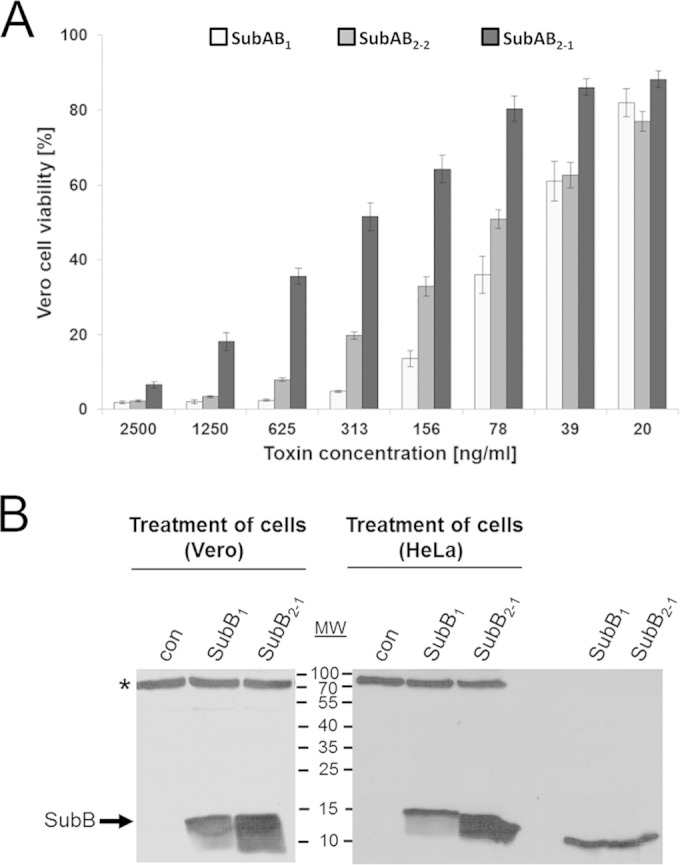
Comparison of the cytotoxic effects caused by different SubAB variants on Vero cells. (A) Cytotoxic effects of the different SubAB variants on Vero B4 cells were measured after a 72-h incubation period using the crystal violet staining assay and were compared to each other. Results obtained by the use of different toxin dilutions between 2,500 ng/ml and 20 ng/ml were plotted against the resulting Vero cell viability of the different variants. The amount of viable Vero B4 cells, which were incubated in the absence of SubAB proteins, were calculated as 100%. The standard errors are shown in the graph. (B) Binding of recombinant SubB1 and SubB2-1 proteins to Vero and HeLa cells. Confluently grown Vero or HeLa cells in 12-well plates were incubated for 45 min at 4°C in serum-free medium with 5 μg/ml of either biotin-labeled SubB1 or biotin-labeled SubB2-1 to enable binding of the SubB proteins to the cells. For a control (con), cells were left untreated. Cells were washed 3 times to remove unbound SubB, lysed in 50 μl of a 2.5-fold SDS sample buffer, and heated for 5 min at 95°C. After SDS-PAGE, the cell-bound biotinylated SubB proteins were detected by Western blotting with streptavidin-peroxidase and the ECL system. The biotin-labeled SubB1 and SubB2-1 proteins (0.5 μg each) were included as running controls (right lanes). A cellular protein between a molecular weight (MW) of 70,000 and 100,000 (marked by an asterisk) also was recognized by streptavidin-peroxidase, which indicates comparable protein loading and blotting of all lysate samples.
In order to investigate whether the low cytotoxicity of SubAB2-1 compared to that of the other variants was due to a reduced binding of the SubB2-1 subunit to the cells, we performed Western blot analysis of cell-bound SubB2-1 in direct comparison to cell-bound SubB1. In this assay, the SubB2-1 subunit bound even more strongly to the cells than SubB1 (Fig. 4B). Taken together, the experiments showed clearly that it is possible to combine subtilase A and B subunits in vitro prior to application to the cells and achieve biologically functional toxins, a characteristic known for binary bacterial toxins.
Based on this principle of simply combining subunits expressed from the same operon, the question arose of whether A and B subunits of different toxin variants can be mixed to create hybrid SubAB cytotoxins with cytotoxic activity. In a new approach, recombinant SubA2-1 was mixed with SubB2-2 and SubA2-2 with SubB2-1, and the mixtures were added to Vero B4 cells as described above. As controls, SubA2-1 was mixed with SubB2-1 and SubA2-2 with SubB2-2. After 72 h, the amounts of viable cells were determined and compared to the amounts of viable cells after incubation of cells with the original SubAB toxins. The results showed that the combination of SubA2-1-SubB2-2 (CD50 = 175 ng/ml) exhibited an effect comparable to that of SubA2-2-SubB2-2 (CD50 = 110 ng/ml), and SubA2-2-SubB2-1 (CD50 = 285 ng/ml) behaved comparably to SubA2-1-SubB2-1 (CD50 = 319 ng/ml) (Fig. 5). Since different A subunits combined with the same B subunit caused similar cytotoxic effects and similar A subunits with different B subunits showed different cytotoxic effects, the B subunits might play a role in determining the level of cytotoxicity of the SubAB complexes.
FIG 5.
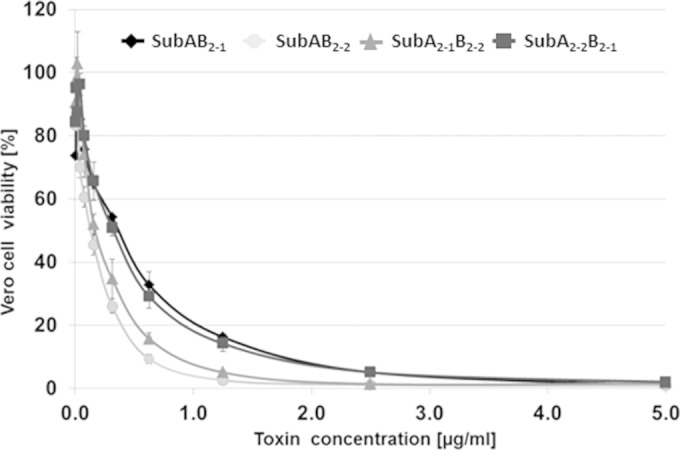
Cytotoxic effects caused by hybrid SubAB toxins. Recombinant SubA2-1 and SubB2-2 (triangles) subunits, as well as SubA2-2 and SubB2-1 (squares) subunits, were mixed in vitro and incubated with Vero B4 cells (for details, see the text). Cells also were incubated with the toxins SubAB2-1 (diamonds) and SubAB2-2 (circles) using the same approach. The standard errors are shown in the graph.
Higher concentrations of SubA1 trigger apoptosis of HeLa cells in the absence of SubB1.
Prompted by the observation that higher concentrations of the SubA subunit alone caused cytotoxic effects on Vero cells (Fig. 3 depicts SubA2-2), we investigated the effects of 10 μg/ml of SubA1 alone, the concentration that exhibited a strong effect on Vero cells, compared to that of SubAB1 on the human epithelial HeLa cell line to exclude a cell line artifact. As shown in Fig. 6, there was a change in cell morphology after 46 h that was more obvious after 72 h of treatment with SubAB1 but also with SubA1 alone. It is worth noting that the concentration of SubA1 alone was much higher than that in combination with SubB1. When SubA1 alone was added to the cells in low concentrations, which were used for experiments where SubA1 was added in combination with the SubB1 subunit, there was no cytotoxic effect on cells. There was a strong decrease in total cell number after treatment with 10 μg/ml SubA1 but only a minor decrease after treatment with 5 μg/ml, which is in agreement with the results obtained for the Vero cells (data not shown). However, no cytotoxic effects were observed when SubA1 was heat inactivated prior to application to the cells, which indicates a specific effect of SubA1 on cells and suggests that its enzyme activity is essential for the cytotoxic mode of action. Moreover, treatment of HeLa cells with 10 μg/ml of His-tagged C2I had no effect on cell morphology or cell number (data not shown). C2I is the enzymatic active subunit of the binary C2 toxin from Clostridium botulinum, which is not taken up into cells in the absence of its separate transport component, C2IIa. This further control experiment let us suggest that the His tag was not responsible for the observed effects caused by His-tagged SubA and confirms the specific mode of action of the SubA1 protein.
FIG 6.

Cytotoxic effects of SubAB1 and SubA1 on HeLa cells. Subconfluently grown HeLa cells were incubated at 37°C with SubA1 (10 nM) plus SubB1 (50 nM) or with SubA1 (10 μg/ml; ∼286 nM) alone. For a control, cells were incubated without any protein. After 24, 46, and 70 h, pictures from the cells were taken to demonstrate the changes in cell morphology.
Moreover, the SubA1 protein was associated with HeLa cells after 30 min of incubation at 4°C, as demonstrated by Western blotting with an anti-His antibody (Fig. 7). Because endocytic processes are blocked at 4°C, this result suggests that SubA1 bound to the surface of the cells. Furthermore, the binding to the cells was mediated via the SubA1 portion and not via the His tag, as His-tagged C2I could not be detected after incubation of HeLa cells with this protein in the same experiment (Fig. 7). Binding of SubA1 to the cells was concentration dependent, and heat-denatured SubA1 showed no binding in the same experiment (data not shown). Taken together, the results show a binding of SubA1 to HeLa cells in the absence of SubB1.
FIG 7.

Binding of SubA1 to HeLa cells. Cells were incubated for 30 min at 4°C with increasing concentrations of SubA1 from 1 to 200 μg/ml to enable binding of this protein to the cell surface. Subsequently, the medium was removed and cells were washed to remove any protein, which was not associated with the cells. Cells were lysed and subjected to SDS-PAGE, and the cell-bound His6-labeled SubA1 was detected by Western blotting with an antibody against the His6 tag. For a control, cells were incubated for 30 min at 4°C with His6-labeled C2I (10 μg/ml), which does not bind to cells, to demonstrate the specificity of the SubA1 binding. Purified SubA1 and C2I proteins were used as controls. MW, molecular weight (in thousands).
We next tested whether internalization of cell-bound SubA1 is a prerequisite for the observed cytotoxic effects. If this were the case, then a nonrecurrent pulse incubation period of cells with SubA1 should be sufficient to induce the cytotoxic effects. To test this hypothesis, HeLa cells were incubated for 1 h at 37°C with SubA1, and subsequently the SubA1-containing medium was removed. Subsequently, one portion of the cells was further incubated in the absence and the other portion in the presence of SubA1. As shown in Fig. 8, only the cells which were incubated with SubA1 over the complete incubation period showed the morphological alterations, indicating that a nonrecurring treatment was not sufficient and that SubA1 has to be present over time to induce its cytotoxic effects. This result also implies that an uptake of the cell-bound SubA1 into the cells is not sufficient to induce the observed cytotoxic effects. However, from this result we cannot exclude that SubA1, which was permanently present in the medium, was taken up into the cells over the incubation period.
FIG 8.

Continuous presence of SubA1 in the medium is necessary to induce cytotoxic effects on HeLa cells. Cells were incubated for 1 h at 37°C with SubA1 (10 μg/ml) to enable internalization of this protein. The medium then was removed and cells were washed to remove the protein. Subsequently, one portion of the cells was incubated further at 37°C in the presence of fresh SubA1 (10 μg/ml; middle), and another portion was incubated in fresh medium without SubA1 (lower). For a control, cells were incubated without any protein (upper). Pictures from the cells were taken after 25, 50, and 72 h.
Finally, we performed a series of flow-cytometric analyses to further explore the cytotoxic effect of SubA1 alone. The application of 10 μg/ml SubA1 markedly reduced cell viability in HeLa cells as measured by FSC/SSC (Fig. 9A), and induced DNA fragmentation in the cells (Fig. 9B). To investigate whether caspases are involved in its cell death-inducing effect, we incubated HeLa cells with SubA1 in the presence and absence of the broad-spectrum caspase inhibitor zVAD.fmk. Based on these findings, we performed caspase activity assays and could confirm that SubA1 treatment leads to the activation of caspases 3 and 7 (Fig. 9C). Importantly, inhibition of caspases significantly reduced SubA1-induced cell death (Fig. 9C) as well as SubA1-induced DNA fragmentation (Fig. 9D). Taken together, the results indicate that high concentrations of SubA1 compared to the concentrations used for the SubAB complexes are sufficient to induce caspase-3/7 activation, cell death, and DNA fragmentation in the absence of SubB1, while comparable final concentrations of SubB1 alone did not induce cell death.
FIG 9.

SubA1 induces apoptosis of HeLa cells. HeLa cells were incubated at 37°C with SubA1 (10 μg/ml), SubB1 (50 nM), or the combination of SubA1 (10 nM) plus SubB1 (50 nM). For a negative control, cells were left untreated, and for a positive control, cells were incubated with staurosporine, an established inducer of apoptosis. After 72 h, cell viability was determined by FSC/SSC measurements (A) and apoptosis by analysis of DNA fragmentation of PI-stained nuclei (B). Significance was tested by using Student's t test (**, P < 0.005; ***, P < 0.0005); ns, not significant. Cells were incubated for 72 h with or without SubA1 (10 μg/ml) in the absence (dark gray bars) or presence (light gray bars) of the caspase inhibitor zVAD.fmk (40 μM), and caspase activation was investigated by using the CellEvent caspase-3/7 green detection reagent (C); apoptosis was analyzed by measuring DNA fragmentation of PI-stained nuclei (D).
DISCUSSION
We performed a series of cell-based experiments to characterize the cytotoxic activity of the subtilase toxin variants from STEC strains in more detail. The results of these experiments demonstrate that it is possible to purify the single-toxin subunits of the subtilase AB5 cytotoxin variants and regain their cytotoxic activity after combining the recombinant subunits in vitro. This approach offers the possibility to analyze toxic effects of the single subunits as well as of combinations of both subunits. It was used for the first time for the subtilase cytotoxin but is well known for binary bacterial AB-type toxins, such as the C2 toxin of Clostridium botulinum (36).
Interestingly, we observed residual cytotoxic effects on Vero cells for the A subunits of the subtilase cytotoxin in the absence of the B subunits for all variants analyzed in this study (data not shown for SubAB2-1). This effect of SubA1 alone was about 70-fold less than that of the SubA2-2 subunit in combination with its B subunit in the culture medium, a factor of about 1,150 less than the effect observed for the copurified SubAB1 (35).
We analyzed the cytotoxic effect caused by SubA1 alone in more detail in the human HeLa cell line, and we found that higher concentrations of SubA1 caused apoptosis and caspases 3/7 were activated. This effect also was observed when much lower concentrations of SubA1 were delivered into cells via the SubB1 transport subunit, and comparable results were reported earlier by other groups for the SubAB1 cytotoxin (37–40). This finding explains the observed alterations in cell morphology and the reduced number of viable cells after incubation with high concentrations of SubA1 in the absence of SubB1. Interestingly, we obtained evidence that SubA1 alone was bound to the surface of HeLa cells, but a continuous presence of SubA1 in the culture medium was essential to induce the cytotoxic effects caused by SubA1 in the absence of SubB1. This is in contrast to the mode of action of bacterial AB toxins, including SubAB, where a single binding step via the B subunit is sufficient to trigger the full cytotoxic effects, because the cell-bound toxin is rapidly internalized into the cells by specific endocytotic mechanisms. The toxicity of SubA1 might depend on a nonspecific uptake of this protein under high protein concentrations, as was hypothesized for the Shiga toxin A1 subunit (41). SubA1 does not harbor a classical KDEL motif, such as CTX-A, the catalytic subunit of the cholera toxin (42); however, the intracellular trafficking of internalized SubA1 to the endoplasmic reticulum could be facilitated by a putative C-terminal endoplasmic retrieval signal (SEEL) as predicted by the scanProsite tool (43). On the other hand, we cannot exclude that SubA1 interacts with the recently identified receptors for SubAB on the cell surface, whose signaling contributes to SubAB-mediated apoptosis in HeLa cells (22). However, the mechanism underlying the apoptosis induced by SubA1 alone is not clear, and the pathophysiological relevance for this new observation requires further investigation. Morinaga and coworkers reported earlier that SubB alone showed some effects on the morphology of Vero cells (44). We could also detect a moderate cytotoxic effect of SubB1 alone on Vero cells at a concentration of 10 μg/ml (data not shown). However, we did not detect cytotoxic effects when Vero cells were incubated with the same final concentrations of SubB2-1 or SubB2-2. Moreover, no cell death was detected after incubation of HeLa cells with any of the SubB subunits.
Based on the results obtained for the cytotoxicity of the SubAB2-2 variant, we directly compared its cytotoxicity to that of the chromosomal variant SubAB2-1 and the plasmid-encoded SubAB1 in Vero cell assays. Under the experimental conditions used in this study, the chromosome-encoded variant SubAB2-1 showed less cytotoxicity than the plasmid-encoded SubAB1 and the second chromosomal variant SubAB2-2. These differences might depend on variations in the amino acid sequence between the different variants, which might affect the enzymatic activity of the A subunits, the receptor recognition by the B subunits, or the stability of all of the holotoxins. Based on this hypothesis and the fact that the differences between SubAB1 and SubAB2-2 are less pronounced, variations between the two chromosomal SubAB variants could be responsible for the differences in cytotoxicity.
SubA2-1 and SubA2-2 share 11 identical amino acid variations from the sequence of SubA1. Furthermore, SubA2-1 differs in 5 additional amino acids from SubA1 and SubA2-2, namely, Ser/Pro at position 62, Ile/Leu at position 106, Thr/Ser at position 142, Ile/Val at position 216, and Thr/Ile at position 246 (numbering corresponds to the protein without signal sequence). The variation Ile/Val at position 216 is located inside one of the loops building the active-site cleft of SubA1 (20), and this could affect its substrate specificity. However, it is unlikely that this is the reason for the reduced cytotoxicity, because both amino acids differ in only one methyl group.
SubB2-1 and SubB2-2 share 10 amino acid variations on identical positions with regard to SubB1 and only one additional variation in SubB2-1, namely, Gly/Ser at position 13 (numbering corresponds to the protein without signal sequence).
Considering the crystal structures of the SubA1 and SubB1 subunits (17, 20), as well as that of the SubAB1 holotoxin (45), none of the mutations, shared within the variants or not, can be directly associated with the key amino acids responsible for enzymatic activity of the SubA1 subunit, binding of the SubB1 subunit to the Neu5Gc receptor, or assembly of the SubAB1 holotoxin. Nevertheless, some variation between SubAB2-1 and SubAB2-2 occurs at positions that may affect cytotoxicity; therefore, they should be discussed in more detail.
The exchange of the glycine residue at position 13 to serine in the B subunit could affect cytotoxicity of the SubAB2-1 variant. This sequence variation occurs in the direct vicinity of serine at position 12, which is responsible for the binding of the B subunit to the carboxyl group of Neu5Gc (17). A mutation of this specific amino acid in the B subunit reduced the cytotoxicity of SubAB1 by 99.98% (17). In front of that, the presence of an additional OH group in the direct vicinity to serine 12 might interfere with receptor recognition by the B subunit; thus, it might decrease the cytotoxicity of SubAB2-1. However, in the Western blot analysis performed here, binding of SubB2-1 to Vero and HeLa cells was even stronger than that of SubB1. This let us suggest that other mechanisms are involved in determining the level of cytotoxicity.
Interestingly, the combination of the A and B subunits from different toxins resulted in toxin hybrids with high cytotoxic activity, the level of which was probably determined by the B subunit rather than by different enzymatic activities of the A subunits. This raises the issue of the specificity of the assembly mechanism for the SubAB toxins and whether this plays a role in wild-type strains, which carry more than one toxin variant.
The association of heterologous subunits to form a SubAB hybrid, which is highly cytotoxic, indicates that the responsible regions in the A and B subunits are structurally conserved. Hybrid toxin formation already has been shown for type I and II heat-labile enterotoxins (46), Vero toxins 1 and 2 (47), and heat-labile enterotoxin and cholera toxin (48).
In conclusion, the results of the present study demonstrate that the different variants of SubAB toxins exhibited different intensities of cytotoxicity toward Vero cells and are able to form hybrid toxins which retain cytotoxic activity. Further studies are needed to resolve the molecular mechanisms of assembly of the SubAB toxin, especially in strains encoding two SubAB variants, as well as the gene expression strategy used by SubAB-producing STEC strains.
ACKNOWLEDGMENTS
This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG; grant BA 2087/2-2 to H.B.) and grant 01KI1012C (Food-Borne Zoonotic Infections of Humans) from the German Federal Ministry of Education and Research (BMBF).
We thank Ulrike Binder for excellent technical assistance.
REFERENCES
- 1.Karch H, Tarr PI, Bielaszewska M. 2005. Enterohaemorrhagic Escherichia coli in human medicine. Int J Med Microbiol 295:405–418. doi: 10.1016/j.ijmm.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Jores J, Rumer L, Wieler LH. 2004. Impact of the locus of enterocyte effacement pathogenicity island on the evolution of pathogenic Escherichia coli. Int J Med Microbiol 294:103–113. doi: 10.1016/j.ijmm.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 3.Frankel G, Phillips AD, Rosenshine I, Dougan G, Kaper JB, Knutton S. 1998. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol Microbiol 30:911–921. doi: 10.1046/j.1365-2958.1998.01144.x. [DOI] [PubMed] [Google Scholar]
- 4.Paton AW, Srimanote P, Woodrow MC, Paton JC. 2001. Characterization of Saa, a novel autoagglutinating adhesin produced by locus of enterocyte effacement-negative Shiga-toxigenic Escherichia coli strains that are virulent for humans. Infect Immun 69:6999–7009. doi: 10.1128/IAI.69.11.6999-7009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bielaszewska M, Fell M, Greune L, Prager R, Fruth A, Tschape H, Schmidt MA, Karch H. 2004. Characterization of cytolethal distending toxin genes and expression in Shiga toxin-producing Escherichia coli strains of non-O157 serogroups. Infect Immun 72:1812–1816. doi: 10.1128/IAI.72.3.1812-1816.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paton AW, Woodrow MC, Doyle RM, Lanser JA, Paton JC. 1999. Molecular characterization of a Shiga toxigenic Escherichia coli O113:H21 strain lacking eae responsible for a cluster of cases of hemolytic-uremic syndrome. J Clin Microbiol 37:3357–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Paton JC, Paton AW. 2007. Pathologic changes in mice induced by subtilase cytotoxin, a potent new Escherichia coli AB5 toxin that targets the endoplasmic reticulum. J Infect Dis 196:1093–1101. doi: 10.1086/521364. [DOI] [PubMed] [Google Scholar]
- 8.Amaral MM, Sacerdoti F, Jancic C, Repetto HA, Paton AW, Paton JC, Ibarra C. 2013. Action of shiga toxin type-2 and subtilase cytotoxin on human microvascular endothelial cells. PLoS One 8:e70431. doi: 10.1371/journal.pone.0070431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marquez LB, Velazquez N, Repetto HA, Paton AW, Paton JC, Ibarra C, Silberstein C. 2014. Effects of Escherichia coli subtilase cytotoxin and Shiga toxin 2 on primary cultures of human renal tubular epithelial cells. PLoS One 9:e87022. doi: 10.1371/journal.pone.0087022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michelacci V, Tozzoli R, Caprioli A, Martinez R, Scheutz F, Grande L, Sanchez S, Morabito S. 2013. A new pathogenicity island carrying an allelic variant of the subtilase cytotoxin is common among Shiga toxin producing Escherichia coli of human and ovine origin. Clin Microbiol Infect 19:E149–E156. doi: 10.1111/1469-0691.12122. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez S, Diaz-Sanchez S, Martinez R, Llorente MT, Herrera-Leon S, Vidal D. 2013. The new allelic variant of the subtilase cytotoxin (subAB2) is common among Shiga toxin-producing Escherichia coli strains from large game animals and their meat and meat products. Vet Microbiol 166:645–649. doi: 10.1016/j.vetmic.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 12.Slanec T, Fruth A, Creuzburg K, Schmidt H. 2009. Molecular analysis of virulence profiles and Shiga toxin genes in food-borne Shiga toxin-producing Escherichia coli. Appl Environ Microbiol 75:6187–6197. doi: 10.1128/AEM.00874-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buvens G, Lauwers S, Pierard D. 2010. Prevalence of subtilase cytotoxin in verocytotoxin-producing Escherichia coli isolated from humans and raw meats in Belgium. Eur J Clin Microbiol Infect Dis 29:1395–1399. doi: 10.1007/s10096-010-1014-z. [DOI] [PubMed] [Google Scholar]
- 14.Orden JA, Horcajo P, de la Fuente R, Ruiz-Santa-Quiteria JA, Dominguez-Bernal G, Carrion J. 2011. Subtilase cytotoxin-coding genes in verotoxin-producing Escherichia coli strains from sheep and goats differ from those from cattle. Appl Environ Microbiol 77:8259–8264. doi: 10.1128/AEM.05604-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paton AW, Srimanote P, Talbot UM, Wang H, Paton JC. 2004. A new family of potent AB(5) cytotoxins produced by Shiga toxigenic Escherichia coli. J Exp Med 200:35–46. doi: 10.1084/jem.20040392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karch H, Mellmann A, Bielaszewska M. 2009. Epidemiology and pathogenesis of enterohaemorrhagic Escherichia coli. Berl Munch Tierarztl Wochenschr 122:417–424. [PubMed] [Google Scholar]
- 17.Byres E, Paton AW, Paton JC, Lofling JC, Smith DF, Wilce MC, Talbot UM, Chong DC, Yu H, Huang S, Chen X, Varki NM, Varki A, Rossjohn J, Beddoe T. 2008. Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature 456:648–652. doi: 10.1038/nature07428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irie A, Koyama S, Kozutsumi Y, Kawasaki T, Suzuki A. 1998. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J Biol Chem 273:15866–15871. doi: 10.1074/jbc.273.25.15866. [DOI] [PubMed] [Google Scholar]
- 19.Lofling JC, Paton AW, Varki NM, Paton JC, Varki A. 2009. A dietary non-human sialic acid may facilitate hemolytic-uremic syndrome. Kidney Int 76:140–144. doi: 10.1038/ki.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, Talbot UM, Paton JC. 2006. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443:548–552. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 21.Paton AW, Paton JC. 2010. Escherichia coli subtilase cytotoxin. Toxins 2:215–228. doi: 10.3390/toxins2020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yahiro K, Satoh M, Morinaga N, Tsutsuki H, Ogura K, Nagasawa S, Nomura F, Moss J, Noda M. 2011. Identification of subtilase cytotoxin (SubAB) receptors whose signaling, in association with SubAB-induced BiP cleavage, is responsible for apoptosis in HeLa cells. Infect Immun 79:617–627. doi: 10.1128/IAI.01020-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mammarappallil JG, Elsinghorst EA. 2000. Epithelial cell adherence mediated by the enterotoxigenic Escherichia coli Tia protein. Infect Immun 68:6595–6601. doi: 10.1128/IAI.68.12.6595-6601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tozzoli R, Caprioli A, Cappannella S, Michelacci V, Marziano ML, Morabito S. 2010. Production of the subtilase AB5 cytotoxin by Shiga toxin-negative Escherichia coli. J Clin Microbiol 48:178–183. doi: 10.1128/JCM.01648-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Funk J, Stoeber H, Hauser E, Schmidt H. 2013. Molecular analysis of subtilase cytotoxin genes of food-borne Shiga toxin-producing Escherichia coli reveals a new allelic subAB variant. BMC Microbiol 13:230. doi: 10.1186/1471-2180-13-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 27.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook J, Fritsch E, Maniatis T. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, New York, NY. [Google Scholar]
- 29.Bronnhuber A, Maier E, Riedl Z, Hajos G, Benz R, Barth H. 2014. Inhibitions of the translocation pore of Clostridium botulinum C2 toxin by tailored azolopyridinium salts protects human cells from intoxication. Toxicology 316:25–33. doi: 10.1016/j.tox.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Gentry MK, Dalrymple JM. 1980. Quantitative microtiter cytotoxicity assay for Shigella toxin. J Clin Microbiol 12:361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. 1991. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139:271–279. doi: 10.1016/0022-1759(91)90198-O. [DOI] [PubMed] [Google Scholar]
- 33.Thain A, Gaston K, Jenkins O, Clarke AR. 1996. A method for the separation of GST fusion proteins from co-purifying GroEL. Trends Genet 12:209–210. doi: 10.1016/S0168-9525(96)90022-0. [DOI] [PubMed] [Google Scholar]
- 34.Guo LW, Assadi-Porter FM, Grant JE, Wu H, Markley JL, Ruoho AE. 2007. One-step purification of bacterially expressed recombinant transducin alpha-subunit and isotopically labeled PDE6 gamma-subunit for NMR analysis. Protein Expr Purif 51:187–197. doi: 10.1016/j.pep.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 35.Gerhardt E, Masso M, Paton AW, Paton JC, Zotta E, Ibarra C. 2013. Inhibition of water absorption and selective damage to human colonic mucosa are induced by subtilase cytotoxin produced by Escherichia coli O113:H21. Infect Immun 81:2931–2937. doi: 10.1128/IAI.00287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser E, Haug G, Hliscs M, Aktories K, Barth H. 2006. Formation of a biologically active toxin complex of the binary Clostridium botulinum C2 toxin without cell membrane interaction. Biochemistry 45:13361–13368. doi: 10.1021/bi061459u. [DOI] [PubMed] [Google Scholar]
- 37.Matsuura G, Morinaga N, Yahiro K, Komine R, Moss J, Yoshida H, Noda M. 2009. Novel subtilase cytotoxin produced by Shiga-toxigenic Escherichia coli induces apoptosis in Vero cells via mitochondrial membrane damage. Infect Immun 77:2919–2924. doi: 10.1128/IAI.01510-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yahiro K, Tsutsuki H, Ogura K, Nagasawa S, Moss J, Noda M. 2012. Regulation of subtilase cytotoxin-induced cell death by an RNA-dependent protein kinase-like endoplasmic reticulum kinase-dependent proteasome pathway in HeLa cells. Infect Immun 80:1803–1814. doi: 10.1128/IAI.06164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yahiro K, Tsutsuki H, Ogura K, Nagasawa S, Moss J, Noda M. 2014. DAP1, a negative regulator of autophagy, controls SubAB-mediated apoptosis and autophagy. Infect Immun 82:4899–4908. doi: 10.1128/IAI.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yahiro K, Morinaga N, Moss J, Noda M. 2010. Subtilase cytotoxin induces apoptosis in HeLa cells by mitochondrial permeabilization via activation of Bax/Bak, independent of C/EBF-homologue protein (CHOP), Ire1alpha or JNK signaling. Microb Pathog 49:153–163. doi: 10.1016/j.micpath.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi PL, Binnington B, Sakac D, Katsman Y, Ramkumar S, Gariepy J, Kim M, Branch DR, Lingwood C. 2012. Verotoxin A subunit protects lymphocytes and T cell lines against X4 HIV infection in vitro. Toxins 4:1517–1534. doi: 10.3390/toxins4121517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johannes L, Goud B. 1998. Surfing on a retrograde wave: how does Shiga toxin reach the endoplasmic reticulum? Trends Cell Biol 8:158–162. doi: 10.1016/S0962-8924(97)01209-9. [DOI] [PubMed] [Google Scholar]
- 43.de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. 2006. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morinaga N, Yahiro K, Matsuura G, Watanabe M, Nomura F, Moss J, Noda M. 2007. Two distinct cytotoxic activities of subtilase cytotoxin produced by Shiga-toxigenic Escherichia coli. Infect Immun 75:488–496. doi: 10.1128/IAI.01336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Nours J, Paton AW, Byres E, Troy S, Herdman BP, Johnson MD, Paton JC, Rossjohn J, Beddoe T. 2013. Structural basis of subtilase cytotoxin SubAB assembly. J Biol Chem 288:27505–27516. doi: 10.1074/jbc.M113.462622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Connell TD, Holmes RK. 1992. Characterization of hybrid toxins produced in Escherichia coli by assembly of A and B polypeptides from type I and type II heat-labile enterotoxins. Infect Immun 60:1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito H, Yutsudo T, Hirayama T, Takeda Y. 1988. Isolation and some properties of A and B subunits of Vero toxin 2 and in vitro formation of hybrid toxins between subunits of Vero toxin 1 and Vero toxin 2 from Escherichia coli O157:H7. Microb Pathog 5:189–195. doi: 10.1016/0882-4010(88)90021-6. [DOI] [PubMed] [Google Scholar]
- 48.Takeda Y, Honda T, Taga S, Miwatani T. 1981. In vitro formation of hybrid toxins between subunits of Escherichia coli heat-labile enterotoxin and those of cholera enterotoxin. Infect Immun 34:341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]