

Abstract
Human multidrug efflux transporters are known for their ability to extrude antibiotics and toxic compounds out of cells, yet accumulating data indicate they have additional functions in diverse physiological processes not related to drug efflux. Here, we show that the human multidrug transporter P-glycoprotein (P-gp) (also named MDR1 and ABCB1) is transcriptionally induced in the monocytic cell line THP-1 upon infection with the human intracellular bacterial pathogen Listeria monocytogenes. Notably, we found that P-gp is important for full activation of the type I interferon response elicited against L. monocytogenes bacteria. Both inhibition of P-gp function by verapamil and inhibition of its transcription using mRNA silencing led to a reduction in the magnitude of the type I response in infected cells. This function of P-gp was specific to type I interferon cytokines elicited against cytosolic replicating bacteria and was not observed in response to cyclic di-AMP (c-di-AMP), a molecule that was shown to be secreted by L. monocytogenes during infection and to trigger type I interferons. Moreover, P-gp was not involved in activation of other proinflammatory cytokines, such as those triggered by vacuolar-restricted L. monocytogenes or lipopolysaccharide (LPS). Taken together, these findings demonstrate a role for P-gp in proper development of an innate immune response against intracellular pathogens, highlighting the complexity in employing therapeutic strategies that involve inhibition of multidrug resistance (MDR) efflux pumps.
INTRODUCTION
Multidrug transporters mediate the active efflux of a wide range of drugs and xenobiotics, including antibiotics and chemotherapeutics (1). This permissive efflux ability engenders multidrug resistance (MDR)—a phenomenon that largely underlies the failure of various chemotherapeutic treatments (2–4). Human MDR transporters harbor an ATP-binding cassette (ABC), which defines the ABC-type superfamily, comprising more than 45 proteins in the human genome (5). Among these, several transporters have been extensively studied, such as the P-glycoprotein (P-gp) (also named MDR1 and ABCB1) (6), BCRP (ABCG2) (7), and MRP1 (ABCC1) (8), which were all shown to exhibit clinically relevant MDR functions (9). P-gp, encoded by the MDR1 gene, is the most prominent and best-characterized member of the ABC-type superfamily, first isolated in clinical cancers (6, 10). Aside from its well-documented multidrug resistance function in cancer cells, P-gp is naturally expressed in a variety of normal tissues and cells, including immune cells, such as macrophages, dendritic cells, T and B lymphocytes, and natural killer (NK) cells, and was shown to possess physiological functions beyond detoxification (11–15). Several studies have indicated roles for P-gp in lipid transport, intracellular trafficking of cholesterol, cell death, cell differentiation, and immune responses (16, 17). Regarding the last, P-gp was shown to exhibit immunomodulatory activity and to influence the secretion of various inflammatory mediators, such as steroids, prostaglandins, platelet-activating factor, and cytokines (13, 18–21). Specifically, it was demonstrated that P-gp mediates the secretion of interleukin 2 (IL-2), IL-4, tumor necrosis factor alpha (TNF-α), and gamma interferon (IFN-γ) in T lymphocytes (19, 22, 23) and of cytotoxic compounds in NK cells (24). Furthermore, certain cytokines were shown to induce MDR1 transcription during inflammation (25, 26). P-gp's function in immune cells appears to impact distinct immune processes, such as activation of inflammatory cells and maturation of antigen-presenting cells (13, 15, 23, 27). Taken together, these findings indicate an important role for P-gp in the development and function of immune cells and in the progression of inflammatory responses (15).
Listeria monocytogenes is a Gram-positive, foodborne facultative intracellular pathogen that has been extensively studied due to its interactions with the human innate immune system (28–32). L. monocytogenes enters mammalian cells either by phagocytosis or by active invasion. The bacterium evades phagosomal killing by escaping the vacuole into the host cell cytosol. This action involves several bacterial virulence factors, primarily the pore-forming hemolysin listeriolysin O (LLO) (encoded by the hly gene); two phospholipases, PlcA and PlcB; and some components of the competence system (33–35). Following phagosomal escape, L. monocytogenes replicates in the cytosol and spreads from cell to cell using actin-based motility without causing cell lysis (36, 37). The presence of replicating bacteria within host cells is rapidly sensed by cytosolic receptors of the innate immune system, leading to robust induction of a type I interferon response, which is manifested by expression and secretion of IFN-β (28, 31, 38–40). This response was shown to be independent of Toll-like receptors (TLRs) but dependent on various cytosolic innate immune receptors and adaptor molecules (e.g., IRF3, TBK1, RIG-I, MDA5, STING, and DDX41 helicase) (41–46). In contrast to wild-type cytosolic replicating bacteria, L. monocytogenes mutants that fail to access the cytosol (i.e., hly mutants) do not activate the type I interferon response but rather induce a TLR-dependent vacuolar-specific response (42, 47).
We have previously shown that activation of the type I interferon response by L. monocytogenes relies on the expression of a set of bacterial multidrug-resistant transporters—MdrM, MdrT, MdrA, and MdrC—that together are responsible for most of the response in murine macrophages (48, 49). Among these transporters, MdrM was found to be most critical, as deletion of its gene alone led to ∼70% reduction in IFN-β induction compared to that induced by wild-type bacteria. Further studies have identified cyclic di-AMP (c-di-AMP) as a substrate of MdrM and as the ligand that triggers the innate immune system to express type I interferons (46, 50). While immune cells rapidly sense this cyclic dinucleotide as a signal for bacterial invasion, within the bacteria it was shown to play a regulatory role in cell wall stress responses and homeostasis (49, 51).
We were prompted by our findings that bacterial MDR transporters play a role in activation of innate immune responses to ask whether host MDR transporters might also be involved. Here, we show that P-gp, the mammalian MDR transporter, is transcriptionally induced upon L. monocytogenes infection of THP-1 monocytes. Notably, this response correlated with the magnitude of the type I interferon response that was elicited during Listeria infection and was shown to be specific to cytosolic replicating bacteria and not to occur upon exposure to bacterium-derived ligands, such as c-di-AMP. Inhibition of P-gp function or transcription led to significant reduction in IFN-β expression in infected cells. These observations suggest an amplification role for P-gp in development of the type I interferon response during L. monocytogenes infection.
MATERIALS AND METHODS
Materials.
RPMI 1640 and β-mercaptoethanol were purchased from Gibco. Fetal bovine serum (FBS), glutamine, pyruvate, gentamicin, and minimal essential medium (MEM)-Eagle-nonessential amino acids were purchased from Biological Industries, Beit-ha-Emek, Israel. Verapamil, Escherichia coli lipopolysaccharide (LPS), and TRIzol were purchased from Sigma and fumitremorgen C from Enzo. Purified c-di-AMP and c-di-GMP were purchased from Biolog. FVB mdr1a/mdr1b double-knockout mice and their parental strain were purchased from the Taconic knockout repository.
The use of animals and the experimental protocols were approved by the Tel Aviv University Animal Care and Use Committee (L-10-040 and L-13-039) according to the Israel Welfare Law (1994) and the National Research Council guide (Guide for the Care and Use of Laboratory Animals, 2010).
Bacterial strains.
L. monocytogenes 10403S was used as the wild-type (WT) strain and served as the parental strain to generate in-frame deletions of the indicated genes (48). Single colonies were inoculated into 2 ml of brain heart infusion (BHI) broth (BD237500; Merck) and incubated overnight at 30°C without shaking for infection assays or at 37°C with shaking (at 250 rpm) for growth in laboratory media.
Cell cultures.
The human monocyte THP-1 cell line was a kind gift from Isaac P. Witz (Tel Aviv University, Tel Aviv, Israel). The murine RAW264.7 cell line was obtained from the Tsaffrir Zor laboratory (Tel Aviv University, Tel Aviv, Israel), and human ovarian carcinoma Ovcar-8 cells were obtained from the Yehuda G. Assaraf laboratory (Technion, Israel). THP-1 cells were propagated in suspension in RPMI 1640 medium (Gibco) supplemented with 20% inactivated FBS, glutamine (2 mM), pyruvate (1 mM), β-mercaptoethanol (0.05 mM), and MEM-Eagle-nonessential amino acids (Biological Industries, Beit-ha-Emek, Israel). RAW264.7 cells were propagated in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% inactivated FBS and glutamine (2 mM) and Ovcar-8 cells in RPMI 1640 medium (Gibco) supplemented with 10% inactivated FBS and glutamine (2 mM).
Reverse transcription (RT)-qPCR analysis.
RNA was harvested from cells using TRIzol reagent (Sigma), followed by DNase treatment, phenol-chloroform extraction, and ethanol precipitation. cDNA was synthesized from 1 μg of total RNA using a high-capacity reverse transcription kit (Applied Biosystems). For regular PCR analysis, 10 ng of cDNA was used with specific primers. SYBR green-based quantitative-PCR (qPCR) amplification was performed in 96-well plates using SYBR green PCR master mix and the StepOnePlus real-time PCR system (Applied Biosystems). For each indicated gene, ΔΔCT was calculated using a standard relative-quantity (RQ) algorithm applied using the StepOnePlus program, with the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene as a reference gene. Data analysis of at least three biological repeats was performed using the StepOnePlus V2.3 study algorithm. Error bars represent 95% confidence intervals, i.e., the values fall within the bar range in 95% of repeat experiments. When the error bars of two samples do not overlap, the significance of the difference (P value) is ≪0.01.
Bacterial growth analysis and drug resistance assays.
Growth of bacteria in the rich BHI medium at 37°C was assayed by following the optical density at 600 nm (OD600) every 15 min utilizing the Synergy HT Biotek plate reader with continuous shaking. Overnight cultures were diluted to an OD600 of 0.05 and supplemented with the indicated compounds: rhodamine 6G (R6G) (2 μM), ciprofloxacin (4.5 μg/ml), and verapamil (20 μM). The OD600 was recorded for 16 h of incubation, using Breathe-Easy (USA Scientific; 9123-6100) gas-permeable sealing for the plates.
Mammalian-cell infection.
WT L. monocytogenes (1 × 107 CFU; 0.15 ml of overnight culture grown at 30°C) was used to infect 3 × 106 mammalian cells in 3 ml of medium. One hour postinfection (p.i.), the cells were washed with phosphate-buffered saline (PBS) and resuspended in medium with 50 μg/ml gentamicin. For RNA analysis, infections were terminated at 6 h p.i. To determine intracellular bacterial growth in THP-1 cells, 0.5-ml aliquots of infected THP-1 cell suspension were withdrawn, centrifuged, and lysed with 0.5 ml of water. Bacteria released by lysis were plated on BHI-agar plates in serial dilutions. CFU were counted after 24 h of growth at 37°C. Bone marrow-derived macrophages (BMDMs) were isolated and infected as described previously (52).
Stimulation of THP-1 cells with bacterium-derived ligands.
For activation of cytokines by bacterial ligands, 2 × 106 THP-1 cells in 2 ml of permeabilization buffer (50 mM HEPES, pH 7.0, 100 mM KCl, 3 mM MgCl2, 0.1 mM dithiothreitol [DTT], 85 mM sucrose, 0.2% bovine serum albumin [BSA], 1 mM ATP, 0.1 mM GTP, 10 μg/ml digitonin [53]) were incubated for 30 min in the presence of 2 μg/ml listerial DNA, 2 μg/ml listerial RNA, 1.8 μg/ml c-di-AMP, or 3.5 μg/ml c-di-GMP with or without 20 μM verapamil. Both listerial RNA and DNA were extracted from exponentially grown bacteria using standard procedures; 150 ng/ml LPS was added directly to cells without permeabilization buffer. Following incubation, the buffer/medium was removed and the cells were resuspended in fresh medium. After 3.5 h, RNA from the THP-1 cells was harvested and analyzed for cytokine induction by RT-qPCR (as described above).
RNA interference.
THP-1 cells (2.5 × 105) in 2 ml of medium were transfected with 25 pmol of small interfering RNA (siRNA) targeted against the MDR1 gene (sense strand sequence, UCGAGUCACUGCCUAAUAA, for siP-gp) or against an irrelevant sequence of the luciferase gene (sense strand sequence, CUUACGCUGAGUACUUCGA, for siLuc). Lipofectamine 2000 (5 μl; Life Technologies) was used for transfection according to the manufacturer's instructions. Cells were washed 6 h posttransfection and resuspended in medium supplemented with penicillin and streptomycin. The cells were incubated for 72 h and then infected with L. monocytogenes as described above.
Flow cytometry analysis of P-gp expression.
Flow cytometry analysis of P-gp was performed as reported previously (54, 55). Briefly, 72 h posttransfection with siLuc siRNA or siP-gp siRNA, 0.5 × 106 cells from each sample were incubated with either mouse monoclonal anti-human P-gp antibody (clone 4E3), which recognizes an external epitope of P-gp (ABCAM, Cambridge, MA, USA) or mouse IGg2a antibody isotype as a control (BioLegend, San Diego, CA, USA) on ice for 30 min. After being washed, the samples were incubated with a phycoerythrin (PE)-labeled F(ab′)2 anti-mouse antibody (ABCAM, Cambridge, MA, USA) for 30 min at 4°C in the dark. The samples were centrifuged and resuspended in PBS containing 1% fetal calf serum (FCS) for analysis on a Becton Dickinson FACSCalibur flow cytometer with CellQuest software (Becton Dickinson, Franklin Lakes, NJ). Ten thousand events were determined for each test sample. Excitation was done with a single 15-mW argon ion laser beam (488 nm). Emission was collected through a 530-nm band-pass filter. Data analysis was performed using FlowJo software (Tree Star, Inc., OR, USA).
RESULTS
P-gp is transcriptionally induced upon L. monocytogenes infection and correlates with IFN-β response.
To examine if P-gp plays a role during L. monocytogenes infection, we first analyzed its transcription level using RT-qPCR analysis of human monocyte THP-1 cells infected with L. monocytogenes strain 10403S. MDR1 mRNA levels were observed to be upregulated ∼2-fold in L. monocytogenes-infected cells (P < 0.01) relative to noninfected cells at 6 h p.i. (Fig. 1A). This is a relatively modest transcriptional induction but is typical of membrane transporters. The trend of induction was corroborated by our discovery that MDR1 mRNA levels further increased if cells were infected with L. monocytogenes bacteria overexpressing MdrM (∼4-fold, using a mutant with the MdrM repressor, MarR, gene deleted: the marR mutant [48]), whereas the levels did not change in cells infected with an L. monocytogenes mutant that becomes trapped within the phagosome (the hly mutant). The toxic dye R6G, which is a known P-gp substrate and induces its transcriptional expression (56), was exploited as a positive control and induced MDR1 levels (∼4-fold) comparably to the marR mutant (Fig. 1A).
FIG 1.
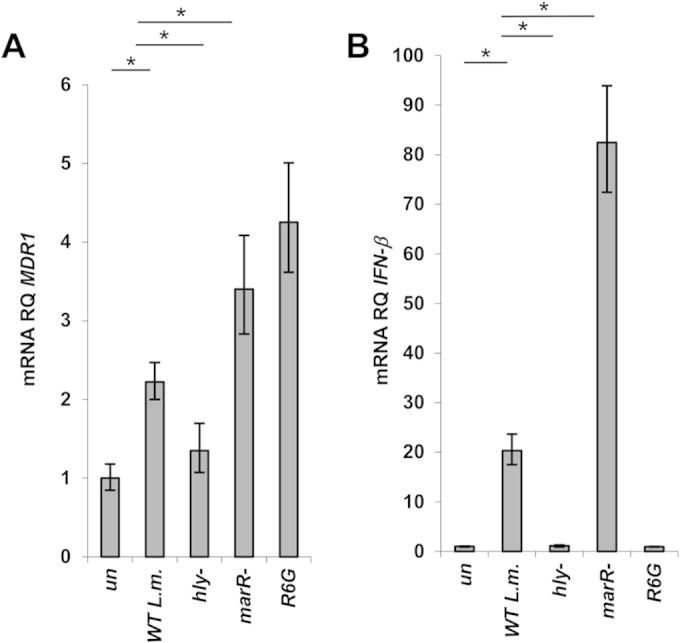
MDR1 transcription is specifically upregulated in response to L. monocytogenes infection of THP-1 cells. (A) RT-qPCR analysis of P-gp transcription levels in THP-1 cells infected with wild-type L. monocytogenes (WT L.m.) and hly and marR deletion mutants of L. monocytogenes or treated with R6G. (B) RT-qPCR analysis of IFN-β transcription levels in THP-1 cells infected with WT L. monocytogenes and hly and marR deletion mutants of L. monocytogenes or treated with R6G. Transcription levels are represented as RQs relative to uninfected/untreated cells (un). The data represent at least 3 biological repeats. The error bars indicate 95% confidence intervals. *, P < 0.01.
We noted that MDR1 transcription in infected cells directly correlated with IFN-β levels elicited by the different listerial strains (Fig. 1B). While the marR mutant resulted in enhanced induction of both P-gp and IFN-β in comparison to WT bacteria, the hly mutant failed to induce either P-gp or IFN-β. These findings demonstrate upregulation of MDR1 transcription in response to L. monocytogenes intracellular infection but also raise the possibility that P-gp may be associated with the type I interferon response.
P-gp contributes to induction of IFN-β in response to L. monocytogenes infection.
We first examined whether P-gp plays any role in IFN-β activation. To this end, THP-1 cells treated and not treated with the P-gp preferential inhibitor verapamil (57) were infected with WT bacteria, and IFN-β induction was analyzed at 6 h p.i. Two concentrations of verapamil were used, 20 and 50 μM, which were shown previously to effectively block P-gp function (references 56 and 57 and FDA). As shown in Fig. 2A, infected THP-1 cells treated with verapamil transcribed reduced levels of IFN-β in comparison to nontreated infected cells (40% and 20%, respectively).
FIG 2.

Verapamil inhibits P-gp and leads to a reduced IFN-β response in infected THP-1 cells. (A) RT-qPCR analysis of IFN-β transcription levels in THP-1 cells infected with wild-type L. monocytogenes supplemented with 20 μM or 50 μM verapamil (Ver 20 and Ver 50, respectively). Transcription levels are represented as RQs relative to uninfected cells. An experiment representative of 3 independent repeats is shown. The error bars indicate 95% confidence intervals. (B) Intracellular growth curves of WT L. monocytogenes in THP-1 cells supplemented with 20 or 50 μM verapamil. Representative growth curves from 3 independent experiments are shown. The error bars represent standard deviations of triplicates. (C) Intracellular bacterial counts of THP-1 cells infected with wild-type L. monocytogenes and supplemented as indicated with 4.5 μM ciprofloxacin (Cipro) and 20 μM verapamil. An experiment representative of 3 independent repeats is shown. The error bars represent standard deviations of triplicates. *, P < 0.05.
To exclude the possibility that verapamil inhibits L. monocytogenes intracellular growth, which by itself can lead to reduced IFN-β induction, the intracellular growth of WT bacteria was analyzed in THP-1 cells treated with 0, 20, and 50 μM verapamil. As demonstrated in Fig. 2B, verapamil had no effect on L. monocytogenes intracellular growth in THP-1 cells or on growth in the rich laboratory medium BHI (see Fig. S2 in the supplemental material). Next, to confirm that verapamil inhibits P-gp function during L. monocytogenes infection, the ability of P-gp to export ciprofloxacin, a P-gp substrate and an antibacterial drug (58, 59), was assessed. Ciprofloxacin has been shown to inhibit L. monocytogenes growth in laboratory media (also shown in Fig. S2 in the supplemental material) and, likewise, intracellularly if added to THP-1 cells (58, 60). Indeed, treating infected THP-1 cells with ciprofloxacin (4.5 μM) strongly inhibited intracellular growth of L. monocytogenes (by 2.5 orders of magnitude) (Fig. 2C). This growth restriction was further enhanced (up to 3 orders of magnitude) when verapamil was added to the cells (20 μM) (Fig. 2C), demonstrating its ability to inhibit ciprofloxacin efflux. Of note, the antibacterial effect of ciprofloxacin on L. monocytogenes was not enhanced by verapamil when the bacteria were grown in broth culture (BHI medium) (see Fig. S1 in the supplemental material). Taken together, the results corroborate the fact that verapamil indeed inhibits P-gp during L. monocytogenes infection and, moreover, support the premise that P-gp is involved in stimulation of the IFN-β response. Of note, in addition to P-gp, we examined another clinically relevant human MDR transporter, BCRP (also named ABCG2). While BCRP was found to also be induced during L. monocytogenes infection (∼2-fold), inhibition of its function, using the specific inhibitor fumitremorgen C, did not affect IFN-β transcription (see Fig. S1 in the supplemental material). We therefore continued to focus our study on P-gp.
P-gp specifically influences activation of type I interferon-related cytokines in response to intracytosolic replicating L. monocytogenes.
We next asked if P-gp influences the expression of cytokines other than IFN-β. To this end, the transcriptional levels of additional cytokines, such as IL-8, IL-1α, MCP-1, IP-10, and TNF-α, were measured upon L. monocytogenes infection of verapamil-treated and untreated cells using RT-qPCR analysis (at 6 h p.i.) (Fig. 3A). Of note, all of these cytokines, except TNF-α, were shown to be part of the type I interferon response to L. monocytogenes infection or associated with its activation (61). We found that P-gp does indeed contribute to induction of each tested cytokine, except TNF-α, as their transcription levels were reduced in verapamil-treated infected cells (by 40 to 70%). Notably, induction of P-gp itself in L. monocytogenes-infected cells was independent of verapamil treatment (Fig. 3A). These results suggest that during L. monocytogenes infection, P-gp specifically enhances activation of the type I interferon response and not of other proinflammatory responses, such as those associated with TNF-α.
FIG 3.

P-gp contributes to elicitation of cytokines in response to L. monocytogenes infection. RT-qPCR analysis of cytokine transcription levels in THP-1 cells infected with L. monocytogenes and treated with the indicated reagents in the presence or absence of 20 μM verapamil. Transcription levels are represented as RQs relative to uninfected/untreated cells. The error bars indicate 95% confidence intervals. *, P < 0.01. The data represent at least 3 biological repeats. (A) THP-1 cells infected with WT L. monocytogenes. (B) THP-1 cells infected with the hly mutant of L. monocytogenes. (C) THP-1 cells stimulated with 150 ng/ml LPS. (D) THP-1 cells supplemented with 2 μM R6G.
To corroborate this model, the role of P-gp in cytokine induction was evaluated upon infection with the L. monocytogenes hly mutant (a phagosomally restricted mutant) and in response to stimulation with LPS. As mentioned above, the hly mutant does not induce a type I interferon response but rather induces many proinflammatory cytokines in a TLR/Myd88-dependent manner (some of these cytokines overlap cytokines induced by the type I interferon response) (61). As shown in Fig. 3B, the hly mutant induced neither IFN-β nor P-gp levels but induced IL-8, IL-1α, MCP-1, IP-10, and TNF-α, all independently of P-gp (Fig. 3B). Similar results were observed with LPS stimulation (150 ng/ml), supplemented extracellularly, which did not induce P-gp or a strong IFN-β response yet triggered robust induction of various proinflammatory cytokines, such as IL-8, IL-1α, and TNF-α, in a P-gp-independent manner (Fig. 3C). To exclude the possibility that P-gp induction by itself leads to enhanced cytokine response, THP-1 cells were treated with R6G (a toxic dye that triggers induction of P-gp transcription) with and without verapamil treatment, and cytokine levels were analyzed. We observed that while R6G indeed activates MDR1 transcription (∼3- to 4-fold), this by itself does not elicit a cytokine response (Fig. 3D). Overall, these findings support the premise that P-gp is specifically involved in elicitation of the type I interferon response against intracytosolic replicating L. monocytogenes bacteria. Specifically, verapamil does not generally inhibit cytokine transcription but instead inhibits P-gp under conditions where P-gp is expressed and thereby impacts the type I interferon response.
P-gp neither is induced by nor mediates IFN-β induction upon stimulation with L. monocytogenes-derived ligands.
The activation of type I interferons by cytosolic replicating L. monocytogenes bacteria has been extensively studied. So far, several Listeria-derived ligands, such as c-di-AMP, double-stranded DNA (dsDNA), and RNA, have been suggested to stimulate type I interferons (44, 50, 62). Although c-di-AMP is the most prominent ligand candidate (studied mainly in murine cells), a recent report indicated that sensing of L. monocytogenes by THP-1 cells treated with phorbol 12-myristate 13-acetate (PMA) relies on recognition of Listeria-derived dsDNA rather than c-di-AMP (63). In light of these observations, we examined whether any of these ligands can recapitulate the P-gp-dependent IFN-β stimulation we observed upon L. monocytogenes infection. To this end, commercially available c-di-AMP and c-di-GMP, as well as listerial RNA and dsDNA extracted from BHI-grown bacteria, were employed to stimulate THP-1 cells, and the transcription levels of MDR1 and IFN-β were measured. As shown in Fig. 4A, c-di-AMP and c-di-GMP triggered high levels of IFN-β, while listerial RNA and dsDNA had no stimulatory effect. The discrepancy with the aforementioned study likely arises from the use of PMA-treated versus nontreated THP-1 cells (the latter were used in the present study). Next, induction levels of IFN-β by the cyclic dinucleotides with and without verapamil treatment were compared and found to be similar (Fig. 4A). Remarkably, we observed that MDR1 was not induced by the cyclic dinucleotides (it was even downregulated by c-di-AMP) or by any of the tested bacterial ligands (Fig. 4B), suggesting that P-gp induction by L. monocytogenes bacteria is part of the general host response to infection and is not mediated by a specific ligand. In addition, the data indicate that P-gp is not essentially part of the type I interferon response but, rather, can enhance it independently.
FIG 4.

P-gp is not involved in IFN-β activation upon stimulation with L. monocytogenes-derived ligands. Shown is RT-qPCR analysis of IFN-β transcription levels (A) and P-gp transcription levels (B) in THP-1 cells treated with the indicated ligands in the presence or absence of 20 μM verapamil. Transcription levels are represented as RQs relative to untreated cells. The error bars indicate 95% confidence intervals. *, P < 0.01. The data represent at least 3 biological repeats.
Specific silencing of P-gp transcription leads to a reduced type I interferon response upon L. monocytogenes infection.
Although the effect of verapamil on P-gp is well documented (references 56 and 57 and FDA), we wanted to validate our observations by directly inhibiting MDR1 transcription using the siRNA approach. To this end, THP-1 cells were transfected with siRNA sequences targeting the MDR1 gene (siP-gp) or the luciferase gene (siLuc), with the latter as a control. Following transfection, MDR1 transcription was measured using RT-qPCR analysis, and P-gp abundance on THP-1 cells was determined by fluorescence-activated cell sorter (FACS) analysis using a P-gp-specific antibody. An ∼30 to 40% reduction in P-gp transcription was observed in cells treated with siP-gp (Fig. 5A and B), with ∼60% of the cells exhibiting an ∼60% reduction in P-gp abundance (Fig. 5B). Next, the impact of P-gp transcriptional inhibition on the induction of several cytokines was examined during L. monocytogenes infection. siRNA-transfected cells were infected with WT L. monocytogenes bacteria, and the P-gp, IFN-β, IP-10, and TNF-α mRNA levels were analyzed using RT-qPCR analysis at 6 h p.i. As shown in Fig. 5C, cells treated with siP-gp exhibited an ∼30% decrease in MDR1 transcription and a similar reduction in the transcription levels of IFN-β and IP-10, but not in TNF-α levels, similar to what was observed with verapamil treatment (Fig. 3A). Collectively these data support the hypothesis that P-gp plays a role in full activation of the type I interferon response during L. monocytogenes infection.
FIG 5.

P-gp expression is required for full activation of the type I interferon response to L. monocytogenes infection. (A) RT-qPCR analysis of P-gp transcription levels upon siRNA treatment (siP-gp). siRNA targeted to the luciferase gene was employed as a control (siLuc). (B) FACS analysis of cells expressing P-gp on their surfaces, using mouse monoclonal anti-human P-gp antibody (clone 4E3), and probed with PE-labeled F(ab′)2 anti-mouse antibody. (C to F) RT-qPCR analyses of various host genes in uninfected cells and cells infected with WT L. monocytogenes bacteria, treated or not treated with siRNA. (C) MDR1. (D) IFN-β. (E) IP-10. (F) TNF-α. Mock, untransfected cells; siP-gp, cells transfected with siRNA against the MDR1 gene; siLuc, cells transfected with siRNA against the luciferase gene (control). Transcription levels are presented as RQs relative to levels in uninfected cells. Data analysis of at least two independent biological repeats was performed using the StepOnePlus study algorithm. The error bars indicate 95% confidence intervals. *, P < 0.05.
P-gp enhances type I interferon response during L. monocytogenes infection only in human cells.
Finally, to address the question of whether the contribution of P-gp to the activation of the type I interferon response to L. monocytogenes infection is a general feature of mammalian cells or specific to human cells, several human and murine cell types were examined. First, the activation of the murine mdr1a and mdr1b genes (homologues of the human MDR1 gene) in both murine BMDMs (prepared from FVB mice) and RAW264.7 monocytes were examined. As shown in Fig. 6A and C, both murine-derived cells induced mdr1a transcription in response to L. monocytogenes infection (up to 5-fold in BMDMs and 2-fold in RAW264.7 cells), whereas mdr1b was induced 2-fold only in the BMDMs. To address the role of the murine P-gp transporters in the activation of the type I interferon response to L. monocytogenes, BMDMs were generated from isogenic mice with both mdr1a and mdr1b genes deleted (FVB mdr1a/mdr1b−/−; Taconic), while RAW264.7 cells were supplemented with verapamil (20 μM). Contrary to what we observed with THP-1 cells, deletion of the mdr genes or their inhibition with verapamil did not lead to a decrease in IFN-β transcription. In BMDMs, an increase in IFN-β levels was observed, while in RAW264.7 cells, no effect was apparent (Fig. 6B and D). Next, we analyzed additional human cell lines frequently used in L. monocytogenes research, such as embryonic kidney HEK293, epithelial Caco-2, and HeLa cells. Surprisingly, we found that none of these cells express P-gp in response to L. monocytogenes infection (data not shown). However, another human cell line commonly used in P-gp studies, the ovarian tumor Ovcar-8 cell line, demonstrated 2-fold induction of P-gp upon L. monocytogenes infection, similar to THP-1 cells (Fig. 6E). Furthermore, inhibition of P-gp by verapamil in these cells resulted in a decrease in IFN-β transcription by ∼50% (Fig. 6F), essentially recapitulating the observations in THP-1 cells. Taken together, these results indicate a role for P-gp in activation of the type I interferon response to L. monocytogenes infection in human cells that express P-gp.
FIG 6.

P-gp contribution to elicitation of cytokines by host cells in response to L. monocytogenes infection is specific to human cells. Shown is RT-qPCR analysis of the transcription levels of the indicated genes in different cell types infected with WT L. monocytogenes. (A) BMDMs derived from FVB mice. (B) BMDMs derived from FVB mice and FVB mdr1a/mdr1b−/− mice (Taconic). (C and D) RAW264.7 cells supplemented or not with 20 μM verapamil. (E and F) Ovcar-8 cells supplemented or not with 20 μM verapamil. Transcription levels are represented as RQs relative to uninfected cells. The error bars indicate 95% confidence intervals. The data represent at least 3 biological repeats. *, P < 0.01.
DISCUSSION
In this study, we analyzed the role of the human MDR transporter P-gp during L. monocytogenes infection. Here, we show that P-gp is induced upon L. monocytogenes intracellular invasion and that its expression levels directly correlate with the type I interferon response elicited by the bacteria. We further found that P-gp is required for full activation of this response, while it has no effect on other proinflammatory responses. P-gp activation was specific to cytosolic replicating bacteria and could not be recapitulated by stimulation with various bacterial ligands, including those that trigger type I interferons. Overall, based on these data, we propose that during L. monocytogenes invasion P-gp is induced concomitantly with the type I interferon response and that P-gp enhances the type I interferon response. In summary, we hypothesize that P-gp is part of the host response to cytosolic infection that amplifies cytokine production.
The activation of type I interferons during L. monocytogenes infection has been the subject of extensive study over the last decade. The studies were premised on the model that a cytosolic surveillance system exists to detect replicating bacteria within cells (40). In line with this, several listerial ligands that could elicit a specific response were identified, all representing nucleic acids or their derivatives (such as RNA, dsDNA, and cyclic dinucleotides, primarily c-di-AMP) (44, 50, 62). Subsequently, multiple cognate innate immune receptors and adaptors (such as RIG-I, MDA5, STING, DDX41, LRRFIP1, and cGAS) that were shown to sense listerial ligands and activate signaling pathways leading to IFN-β activation were identified (29). Notably, most of these findings are based on experiments conducted in murine cells using murine knockout strains, and accordingly, our understanding of the activation of type I interferons in human cells, the natural host of L. monocytogenes, is somewhat limited. In this respect, several reports have indicated fundamental differences between murine and human cells regarding the activation of type I interferons by L. monocytogenes. For example, a study by Reimer et al. has demonstrated that IRF-3 and IRF-7, which are essential for IFN-β activation in murine cells, are dispensable in human cells, while p38 MAPK, ATF2, and NF-κB are critical (64). Furthermore, while in murine cells c-di-AMP is the prominent listerial ligand that triggers the type I interferon response, in PMA-treated THP-1 cells, listerial dsDNA, not c-di-AMP, was shown to be the prominent trigger (63). Of note, it is for this reason that we focused our study on non-PMA-treated THP-1 cells, as this protocol recapitulates the induction of the type I interferon response to L. monocytogenes infection and c-di-AMP that was observed in murine cells (50). More generally, such reports contradict fundamental knowledge gained using the murine model over the years and highlight potential disparities that can be resolved only by conducting more experiments in human-derived cells (activated and nonactivated). Indeed, in this study, we found a role for P-gp in enhancing the type I interferon response during L. monocytogenes infection that is specific to human cells that express the protein. Therefore, the phenomenon described here represents another example of a host response to bacterial infection that is mediated differently in human and murine cells.
This is the first report linking P-gp to activation of innate immunity in response to bacterial infection. There is no evidence that P-gp is directly involved in sensing bacterial ligands; however, there is burgeoning evidence that the membrane transporter influences the development of immune responses, including cytokine production (15). We noted that P-gp is activated and mediates cytokine enhancement only when bacteria invade the cytosol of host cells. Stimulation of cells with purified bacterial ligands did not activate P-gp, and infection with an L. monocytogenes isogenic strain that becomes trapped within phagosomes also proved ineffective at inducing P-gp expression. These results suggest that activation of P-gp is part of a general host sensing machinery that detects invading bacteria. In this regard, an association between P-gp and bacterial-pathogen invasion has been documented (65, 66). P-gp was found to protect against L. monocytogenes invasion and dissemination within intestinal epithelial cells, as its overexpression led to reduced L. monocytogenes invasion, whereas inhibition enhanced bacterial invasion (67). Helicobacter pylori infection was also shown to be associated with P-gp overexpression in the intestine (68), whereas Salmonella enterica serovar Typhimurium was shown to dampen P-gp function in order to promote epithelial cell invasion (69). In the case of Pseudomonas aeruginosa, it was shown that the bacteria secrete a toxin, called Cif, which selectively inhibits P-gp expression in a variety of epithelial cells (70). Taken together, these examples indicate an intimate interaction between bacterial pathogens and P-gp, though the molecular mechanisms that govern this interaction and their association with the innate immune system are not clear.
There are few reports that suggest potential mediators of the association between P-gp and the type I interferon response to L. monocytogenes infection. One such mediator is p38 MAPK, a critical player in the induction of IFN-β against L. monocytogenes bacteria in both human and murine cells (40, 64), which was shown to activate the transcription of P-gp in cancer cells and thus mediate multidrug resistance (71, 72). Another is phosphoinositide 3-kinase (PI3K), which is also activated during L. monocytogenes infection (73) and was shown to regulate P-gp under different conditions (74). Though both look promising, inhibiting the functions of the two kinases, using the specific inhibitors SB202190 and wortmannin, respectively, did not alter P-gp transcription during L. monocytogenes infection, though, as expected, it did reduce IFN-β transcription (see Fig. S3 in the supplemental material). So far, our knowledge of the regulation of P-gp transcription in vivo, and particularly during bacterial infection, is very limited, and further investigation is warranted in order to understand the phenomenon described here.
Lastly, the ability of P-gp to enhance the type I interferon response may be related to its documented role in secretion of cytokines and host immunomodulatory molecules (15). Alternatively, P-gp could potentially export molecules of bacterial origin outside infected cells and thus lead to immune activation of neighboring cells, essentially enhancing inflammation. In this respect, it was suggested that mammalian and bacterial MDRs share substrates and thus could potentially propagate innate immune responses by cotransporting bacterial ligands (60). Altogether, this study raises important questions regarding the function of P-gp in the development of innate immune responses and in interaction with bacterial pathogens. Our findings are particularly relevant to the development of anticancer and antimicrobial treatments, since many include MDR inhibitors. For example, in cancer patients, inhibition of P-gp could lead to increased sensitivity to bacterial infections and, potentially, to reduced positive antitumor effects of the immune system. A deeper understanding of the role of human MDR transporters in innate immune responses is critical for improvement of current therapeutic strategies.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Herskovits laboratory members for critical reading of the manuscript.
This work was funded by a European commission FP7 framework-INFECT-ERA (Israel Ministry of Health [MOA]) grant to A.A.H.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00380-15.
REFERENCES
- 1.Deeley RG, Westlake C, Cole SP. 2006. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev 86:849–899. doi: 10.1152/physrev.00035.2005. [DOI] [PubMed] [Google Scholar]
- 2.Borst P, Elferink RO. 2002. Mammalian ABC transporters in health and disease. Annu Rev Biochem 71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman MM, Ambudkar SV. 2001. Overview: ABC transporters and human disease. J Bioenerg Biomembr 33:453–458. doi: 10.1023/A:1012866803188. [DOI] [PubMed] [Google Scholar]
- 4.Simon SM, Schindler M. 1994. Cell biological mechanisms of multidrug resistance in tumors. Proc Natl Acad Sci U S A 91:3497–3504. doi: 10.1073/pnas.91.9.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dean M, Rzhetsky A, Allikmets R. 2001. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res 11:1156–1166. doi: 10.1101/gr.GR-1649R. [DOI] [PubMed] [Google Scholar]
- 6.Gottesman MM, Ling V. 2006. The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett 580:998–1009. doi: 10.1016/j.febslet.2005.12.060. [DOI] [PubMed] [Google Scholar]
- 7.Doyle LA, Ross DD. 2003. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22:7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z-S, Tiwari AK. 2011. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J 278:3226–3245. doi: 10.1111/j.1742-4658.2011.08235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lage H. 2008. An overview of cancer multidrug resistance: a still unsolved problem. Cell Mol Life Sci 65:3145–3167. doi: 10.1007/s00018-008-8111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juliano RL. 1976. The role of drug delivery systems in cancer chemotherapy. Prog Clin Biol Res 9:21–32. [PubMed] [Google Scholar]
- 11.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. 1987. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A 84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. 1999. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol 39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 13.Frank MH, Denton MD, Alexander SI, Khoury SJ, Sayegh MH, Briscoe DM. 2001. Specific MDR1 P-glycoprotein blockade inhibits human alloimmune T cell activation in vitro. J Immunol 166:2451–2459. doi: 10.4049/jimmunol.166.4.2451. [DOI] [PubMed] [Google Scholar]
- 14.Langmann T, Mauerer R, Schmitz G. 2006. Human ATP-binding cassette transporter TaqMan low-density array: analysis of macrophage differentiation and foam cell formation. Clin Chem 52:310–313. doi: 10.1373/clinchem.2005.059774. [DOI] [PubMed] [Google Scholar]
- 15.van de Ven R, Oerlemans R, van der Heijden JW, Scheffer GL, de Gruijl TD, Jansen G, Scheper RJ. 2009. ABC drug transporters and immunity: novel therapeutic targets in autoimmunity and cancer. J Leukoc Biol 86:1075–1087. doi: 10.1189/jlb.0309147. [DOI] [PubMed] [Google Scholar]
- 16.Johnstone RW, Ruefli AA, Smyth MJ. 2000. Multiple physiological functions for multidrug transporter P-glycoprotein? Trends Biochem Sci 25:1–6. doi: 10.1016/S0968-0004(99)01493-0. [DOI] [PubMed] [Google Scholar]
- 17.Fletcher JI, Haber M, Henderson MJ, Norris MD. 2010. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer 10:147–156. doi: 10.1038/nrc2789. [DOI] [PubMed] [Google Scholar]
- 18.Barnes KM, Dickstein B, Cutler GB Jr, Fojo T, Bates SE. 1996. Steroid treatment, accumulation, and antagonism of P-glycoprotein in multidrug-resistant cells. Biochemistry 35:4820–4827. doi: 10.1021/bi952380k. [DOI] [PubMed] [Google Scholar]
- 19.Drach J, Gsur A, Hamilton G, Zhao S, Angerler J, Fiegl M, Zojer N, Raderer M, Haberl I, Andreeff M, Huber H. 1996. Involvement of P-glycoprotein in the transmembrane transport of interleukin-2 (IL-2), IL-4, and interferon-gamma in normal human T lymphocytes. Blood 88:1747–1754. [PubMed] [Google Scholar]
- 20.Ernest S, Bello-Reuss E. 1999. Secretion of platelet-activating factor is mediated by MDR1 P-glycoprotein in cultured human mesangial cells. J Am Soc Nephrol 10:2306–2313. [DOI] [PubMed] [Google Scholar]
- 21.Raggers RJ, Vogels I, van Meer G. 2001. Multidrug-resistance P-glycoprotein (MDR1) secretes platelet-activating factor. Biochem J 357:859–865. doi: 10.1042/0264-6021:3570859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raghu G, Park SW, Roninson IB, Mechetner EB. 1996. Monoclonal antibodies against P-glycoprotein, an MDR1 gene product, inhibit interleukin-2 release from PHA-activated lymphocytes. Exp Hematol 24:1258–1264. [PubMed] [Google Scholar]
- 23.Kooij G, Backer R, Koning JJ, Reijerkerk A, van Horssen J, van der Pol SMA, Drexhage J, Schinkel A, Dijkstra CD, den Haan JMM, Geijtenbeek TBH, de Vries HE. 2009. P-Glycoprotein acts as an immunomodulator during neuroinflammation. PLoS One 4:e8212. doi: 10.1371/journal.pone.0008212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ludescher C, Pall G, Irschick EU, Gastl G. 1998. Differential activity of P-glycoprotein in normal blood lymphocyte subsets. Br J Haematol 101:722–727. doi: 10.1046/j.1365-2141.1998.00751.x. [DOI] [PubMed] [Google Scholar]
- 25.Ho EA, Piquette-Miller M. 2006. Regulation of multidrug resistance by pro-inflammatory cytokines. Curr Cancer Drug Targets 6:295–311. doi: 10.2174/156800906777441753. [DOI] [PubMed] [Google Scholar]
- 26.Liptrott NJ, Penny M, Bray PG, Sathish J, Khoo SH, Back DJ, Owen A. 2009. The impact of cytokines on the expression of drug transporters, cytochrome P450 enzymes and chemokine receptors in human PBMC. Br J Pharmacol 156:497–508. doi: 10.1111/j.1476-5381.2008.00050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pendse SS, Behjati S, Schatton T, Izawa A, Sayegh MH, Frank MH. 2006. P-glycoprotein functions as a differentiation switch in antigen presenting cell maturation. Am J Transplant 6:2884–2893. doi: 10.1111/j.1600-6143.2006.01561.x. [DOI] [PubMed] [Google Scholar]
- 28.Pamer EG. 2004. Immune responses to Listeria monocytogenes. Nat Rev Immunol 4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 29.Dussurget O, Bierne H, Cossart P. 2014. The bacterial pathogen Listeria monocytogenes and the interferon family: type I, type II and type III interferons. Front Cell Infect Microbiol 4:50. doi: 10.3389/fcimb.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stavru F, Archambaud C, Cossart P. 2011. Cell biology and immunology of Listeria monocytogenes infections: novel insights. Immunol Rev 240:160–184. doi: 10.1111/j.1600-065X.2010.00993.x. [DOI] [PubMed] [Google Scholar]
- 31.Stockinger S, Decker T. 2008. Novel functions of type I interferons revealed by infection studies with Listeria monocytogenes. Immunobiology 213:889–897. doi: 10.1016/j.imbio.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 32.Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. 2001. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cossart P, Vicente MF, Mengaud J, Baquero F, Perez-Diaz JC, Berche P. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect Immun 57:3629–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith GA, Marquis H, Jones S, Johnston NC, Portnoy DA, Goldfine H. 1995. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect Immun 63:4231–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabinovich L, Sigal N, Borovok I, Nir-Paz R, Herskovits AA. 2012. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 150:792–802. doi: 10.1016/j.cell.2012.06.036. [DOI] [PubMed] [Google Scholar]
- 36.Dussurget O, Pizarro-Cerda J, Cossart P. 2004. Molecular determinants of Listeria monocytogenes virulence. Annu Rev Microbiol 58:587–610. doi: 10.1146/annurev.micro.57.030502.090934. [DOI] [PubMed] [Google Scholar]
- 37.Portnoy DA, Auerbuch V, Glomski IJ. 2002. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J Cell Biol 158:409–414. doi: 10.1083/jcb.200205009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perry AK, Chen G, Zheng D, Tang H, Cheng G. 2005. The host type I interferon response to viral and bacterial infections. Cell Res 15:407–422. doi: 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi K, Hernandez LD, Galán JE, Janeway CA Jr, Medzhitov R, Flavell RA. 2002. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110:191–202. doi: 10.1016/S0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 40.O'Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. 2002. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci U S A 99:13861–13866. doi: 10.1073/pnas.202476699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stockinger S, Reutterer B, Schaljo B, Schellack C, Brunner S, Materna T, Yamamoto M, Akira S, Taniguchi T, Murray PJ, Muller M, Decker T. 2004. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J Immunol 173:7416–7425. doi: 10.4049/jimmunol.173.12.7416. [DOI] [PubMed] [Google Scholar]
- 43.O'Connell RM, Vaidya SA, Perry AK, Saha SK, Dempsey PW, Cheng G. 2005. Immune activation of type I IFNs by Listeria monocytogenes occurs independently of TLR4, TLR2, and receptor interacting protein 2 but involves TNFR-associated NF kappa B kinase-binding kinase 1. J Immunol 174:1602–1607. doi: 10.4049/jimmunol.174.3.1602. [DOI] [PubMed] [Google Scholar]
- 44.Abdullah Z, Schlee M, Roth S, Mraheil MA, Barchet W, Bottcher J, Hain T, Geiger S, Hayakawa Y, Fritz JH, Civril F, Hopfner KP, Kurts C, Ruland J, Hartmann G, Chakraborty T, Knolle PA. 2012. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J 31:4153–4164. doi: 10.1038/emboj.2012.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Archer KA, Durack J, Portnoy DA. 2014. STING-dependent type I IFN production inhibits cell-mediated immunity to Listeria monocytogenes. PLoS Pathog 10:e1003861. doi: 10.1371/journal.ppat.1003861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, Zaver SA, Schenk M, Zeng S, Zhong W, Liu ZJ, Modlin RL, Liu YJ, Cheng G. 2012. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol 13:1155–1161. doi: 10.1038/ni.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCaffrey RL, Fawcett P, O'Riordan M, Lee KD, Havell EA, Brown PO, Portnoy DA. 2004. A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc Natl Acad Sci U S A 101:11386–11391. doi: 10.1073/pnas.0403215101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crimmins GT, Herskovits AA, Rehder K, Sivick KE, Lauer P, Dubensky TW Jr, Portnoy DA. 2008. Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc Natl Acad Sci U S A 105:10191–10196. doi: 10.1073/pnas.0804170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaplan Zeevi M, Shafir NS, Shaham S, Friedman S, Sigal N, Nir Paz R, Boneca IG, Herskovits AA. 2013. Listeria monocytogenes multidrug resistance transporters and cyclic di-AMP, which contribute to type I interferon induction, play a role in cell wall stress. J Bacteriol 195:5250–5261. doi: 10.1128/JB.00794-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woodward JJ, Iavarone AT, Portnoy DA. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witte CE, Whiteley AT, Burke TP, Sauer JD, Portnoy DA, Woodward JJ. 2013. Cyclic di-AMP is critical for Listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. mBio 4:e00282–00213. doi: 10.1128/mBio.00282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herskovits AA, Auerbuch V, Portnoy DA. 2007. Bacterial ligands generated in a phagosome are targets of the cytosolic innate immune system. PLoS Pathog 3:e51. doi: 10.1371/journal.ppat.0030051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ. 2003. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 54.Cohen K, Emmanuel R, Kisin-Finfer E, Shabat D, Peer D. 2014. Modulation of drug resistance in ovarian adenocarcinoma using chemotherapy entrapped in hyaluronan-grafted nanoparticle clusters. ACS Nano 8:2183–2195. doi: 10.1021/nn500205b. [DOI] [PubMed] [Google Scholar]
- 55.Drinberg V, Bitcover R, Rajchenbach W, Peer D. 2014. Modulating cancer multidrug resistance by sertraline in combination with a nanomedicine. Cancer Lett 354:290–298. doi: 10.1016/j.canlet.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 56.Regev R, Assaraf YG, Eytan GD. 1999. Membrane fluidization by ether, other anesthetics, and certain agents abolishes P-glycoprotein ATPase activity and modulates efflux from multidrug-resistant cells. Eur J Biochem 259:18–24. doi: 10.1046/j.1432-1327.1999.00037.x. [DOI] [PubMed] [Google Scholar]
- 57.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. 1981. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 41:1967–1972. [PubMed] [Google Scholar]
- 58.Seral C, Carryn S, Tulkens PM, Van Bambeke F. 2003. Influence of P-glycoprotein and MRP efflux pump inhibitors on the intracellular activity of azithromycin and ciprofloxacin in macrophages infected by Listeria monocytogenes or Staphylococcus aureus. J Antimicrob Chemother 51:1167–1173. doi: 10.1093/jac/dkg223. [DOI] [PubMed] [Google Scholar]
- 59.Leitner I, Nemeth J, Feurstein T, Abrahim A, Matzneller P, Lagler H, Erker T, Langer O, Zeitlinger M. 2011. The third-generation P-glycoprotein inhibitor tariquidar may overcome bacterial multidrug resistance by increasing intracellular drug concentration. J Antimicrob Chemother 66:834–839. doi: 10.1093/jac/dkq526. [DOI] [PubMed] [Google Scholar]
- 60.Lismond A, Tulkens PM, Mingeot-Leclercq MP, Courvalin P, Van Bambeke F. 2008. Cooperation between prokaryotic (Lde) and eukaryotic (MRP) efflux transporters in J774 macrophages infected with Listeria monocytogenes: studies with ciprofloxacin and moxifloxacin. Antimicrob Agents Chemother 52:3040–3046. doi: 10.1128/AAC.00105-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leber JH, Crimmins GT, Raghavan S, Meyer-Morse NP, Cox JS, Portnoy DA. 2008. Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog 4:e6. doi: 10.1371/journal.ppat.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stetson DB, Medzhitov R. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 63.Hansen K, Prabakaran T, Laustsen A, Jorgensen SE, Rahbaek SH, Jensen SB, Nielsen R, Leber JH, Decker T, Horan KA, Jakobsen MR, Paludan SR. 2014. Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J 33:1654–1666. doi: 10.15252/embj.201488029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reimer T, Schweizer M, Jungi TW. 2007. Type I IFN induction in response to Listeria monocytogenes in human macrophages: evidence for a differential activation of IFN regulatory factor 3 (IRF3). J Immunol 179:1166–1177. doi: 10.4049/jimmunol.179.2.1166. [DOI] [PubMed] [Google Scholar]
- 65.Mercado-Lubo R, McCormick BA. 2010. The interaction of gut microbes with host ABC transporters. Gut Microbes 1:301–306. doi: 10.4161/gmic.1.5.12925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ho G.-T, Moodie FM, Satsangi J. 2003. Multidrug resistance 1 gene (P-glycoprotein 170): an important determinant in gastrointestinal disease? Gut 52:759–766. doi: 10.1136/gut.52.5.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neudeck BL, Loeb JM, Faith NG, Czuprynski CJ. 2004. Intestinal P glycoprotein acts as a natural defense mechanism against Listeria monocytogenes. Infect Immun 72:3849–3854. doi: 10.1128/IAI.72.7.3849-3854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Omar M, Crowe A, Parsons R, Ee H, Tay CY, Hughes J. 2012. P-glycoprotein expression in Helicobacter pylori-positive patients: the influence of MDR1 C3435T polymorphism. J Dig Dis 13:414–420. doi: 10.1111/j.1751-2980.2012.00606.x. [DOI] [PubMed] [Google Scholar]
- 69.Siccardi D, Mumy KL, Wall DM, Bien JD, McCormick BA. 2008. Salmonella enterica serovar Typhimurium modulates P-glycoprotein in the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol 294:G1392–G1400. doi: 10.1152/ajpgi.00599.2007. [DOI] [PubMed] [Google Scholar]
- 70.Ye S, MacEachran DP, Hamilton JW, O'Toole GA, Stanton BA. 2008. Chemotoxicity of doxorubicin and surface expression of P-glycoprotein (MDR1) is regulated by the Pseudomonas aeruginosa toxin Cif. Am J Physiol Cell Physiol 295:C807–C818. doi: 10.1152/ajpcell.00234.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barancik M, Bohacova V, Kvackajova J, Hudecova S, Krizanova O, Breier A. 2001. SB203580, a specific inhibitor of p38-MAPK pathway, is a new reversal agent of P-glycoprotein-mediated multidrug resistance. Eur J Pharm Sci 14:29–36. doi: 10.1016/S0928-0987(01)00139-7. [DOI] [PubMed] [Google Scholar]
- 72.Guo X, Ma N, Wang J, Song J, Bu X, Cheng Y, Sun K, Xiong H, Jiang G, Zhang B, Wu M, Wei L. 2008. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 8:375. doi: 10.1186/1471-2407-8-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ireton K, Payrastre B, Cossart P. 1999. The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositide 3-kinase. J Biol Chem 274:17025–17032. doi: 10.1074/jbc.274.24.17025. [DOI] [PubMed] [Google Scholar]
- 74.Kuo MT, Liu Z, Wei Y, Lin-Lee YC, Tatebe S, Mills GB, Unate H. 2002. Induction of human MDR1 gene expression by 2-acetylaminofluorene is mediated by effectors of the phosphoinositide 3-kinase pathway that activate NF-kappaB signaling. Oncogene 21:1945–1954. doi: 10.1038/sj.onc.1205117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.