

Abstract
Cancer-associated muscle weakness is an important paraneoplastic syndrome for which there is currently no treatment. Tumor cells commonly metastasize to bone in advanced cancer to disrupt normal bone remodeling and result in morbidity that includes muscle weakness. Tumor in bone stimulates excessive osteoclast activity, which causes the release of growth factors stored in the mineralized bone matrix. These factors fuel a feed-forward vicious cycle of tumor growth in bone and bone destruction. Recent evidence indicates that these bone-derived growth factors can act systemically to cause muscle weakness. Muscle weakness can be caused by reduced muscle mass or reduced muscle function; in advanced disease, it is likely due to a combination of both reduced quantity and quality of muscle. In this review, we discuss possible mechanisms that lead to skeletal muscle weakness due to bone metastases.
Introduction
Cancer-associated muscle dysfunction is a major paraneoplastic syndrome, the spectrum of which ranges from muscle weakness in the absence of weight loss to profound muscle wasting and cachexia.1 Cancer-associated muscle dysfunction is a large research challenge and a deadly clinical problem; mortality is high (80%) and there is increased toxicity from cancer treatment.1,2,3 Skeletal muscle weakness is a major clinical problem for advanced cancer patients as they also often have bone metastases and associated bone pain, fractures, hypercalcemia and nerve compression syndromes.4 Muscle weakness in the setting of bone fragility likely increases the fracture risk even more than bone metastases alone.
Normal muscle contraction is dependent on precise calcium signaling in the muscle cell.5 During excitation–contraction (E–C) coupling in skeletal muscle, sequestered calcium in the sarcoplasmic reticulum is released through activated ryanodine receptor/calcium release channel (RyR1) into the cytoplasm, permitting calcium-dependent actin–myosin cross-bridging and muscle contraction.6 Cytosolic calcium is then transferred back to the lumen of the sarcoplasmic reticulum via a calcium-ATPase pump (SERCA) (Figure 1). Maladaptive modifications of RyR1 (nitrosylation and oxidation) resulting from chronic oxidative stress have been linked to pathologic sarcoplasmic reticulum calcium leak in diseases characterized by contractile dysfunction and muscle weakness, including heart failure,7,8,9 muscular dystrophy10 and age-related sarcopenia.11 RyR1 oxidation disrupts a critical interaction between RyR1 and its stabilizing subunit calstabin1, resulting in leaky channels with impaired calcium handling and weakened muscle force production.10,11 It is likely that similar mechanisms are involved in cancer-associated muscle weakness, as persistent increased oxidative stress is associated with cancer.12 Further, transforming growth factor β (TGFβ), which is a critical factor for bone remodeling,13 can mediate oxidative stress;14 hence, it should be no surprise that bone metastases could be associated with muscle dysfunction.
Figure 1.
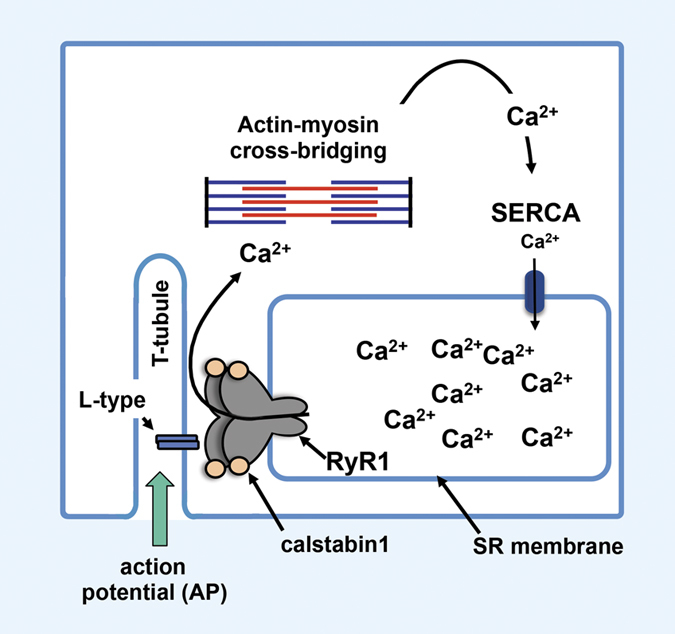
Skeletal muscle contraction. Skeletal muscle contraction begins with an action potential (AP) from the nervous system that activates L-type voltage-dependent calcium channels in the T-tubule system. Ryanodine receptor receptor/calcium release channel (RyR1) then releases calcium from the sarcoplasmic reticulum (SR). High cytosolic calcium causes actin–myosin cross-bridging and muscle contraction.6 Cytosolic calcium is then transferred back to the lumen of the SR via calcium-ATPase pump (SERCA) during myocyte relaxation. Oxidation of RyR1 disrupts a critical interaction between RyR1 and its stabilizing subunit calstabin1 leading to calcium ‘leak.' Pathologic calcium leak reduces tetanic calcium release, the key determinant of muscle contraction.
Bone Metastases in Advanced Cancer
Bone remodeling, coordinately balanced by bone-destroying osteoclasts and bone-forming osteoblasts, maintains bone strength in healthy adults. This process is driven by the coupled activity of osteoclasts that resorb mineralized matrix and osteoblasts that lay down new bone.15,16 Bone metastases are common in patients with advanced malignancy, especially those with breast, prostate and lung cancer. Tumor cells in the bone microenvironment disrupt normal bone remodeling to result in excess bone destruction or bone formation. Tumor cells produce factors that directly or indirectly stimulate osteoclastic bone resorption, which releases growth factors from the bone matrix, such as TGFβ that stimulate tumor invasion, growth and further osteolysis.17 This reciprocal interaction between cancer cells and the bone microenvironment results in a feed-forward ‘vicious cycle' that increases both bone destruction and the tumor burden (Figure 2).17
Figure 2.

Vicious cycle of osteolytic bone metastasis. Osteolytic bone destruction due to dysregulation of normal bone remodeling is predominant in breast cancer metastasis. Breast cancer cells colonizing the bone secrete osteolytic factors: parathyroid hormone-related protein (PTHrP), cyclooxygenase-2 (COX-2), IL-8 and IL-11. Tumor cells also express transcription factors GLI2, runt-related transcription factor 2 (RUNX2) and hypoxia-induced growth factor 1α (HIF1α) that promote osteolysis. Jagged1 (Jag1) expressed on tumor cells activates osteoclast differentiation by inducing Notch signaling in pre-osteoclasts. Bone resorption releases TGFβ from the bone matrix, which enhances tumor cell proliferation and survival, thus feeding a vicious cycle leading to further bone destruction. Reviewed in.17,65
Bone metastases are classified on the basis of radiographic appearance as either osteolytic or osteoblastic (osteosclerotic). Breast cancer is typically associated with osteolytic or mixed lesions. Despite the radiographic appearance, most tumors in bone have uncoupled components of both bone destruction and new bone formation. Perhaps most devastating is the fact that once the primary tumor has spread to the bone it is almost always incurable.4 The current standard of care for patients with bone metastases of any type includes bone-targeted anti-resorptive therapy, such as zoledronic acid or denosumab, in addition to chemotherapy, hormonal therapy, radiation and surgery. These effectively reduce skeletal-related events but do not cure the disease.4,17
Cancer Cachexia
A significant co-morbidity of bone metastases is muscle weakness that is often associated with cancer cachexia. Cachexia is a common paraneoplastic syndrome that is characterized by severe wasting due to loss of skeletal muscle mass (with or without loss of fat mass) due to a negative protein balance caused by abnormal metabolism.18,19 Although the age and chemotherapeutic treatment regimens of patients with advanced disease and bone metastases make it difficult to assess the true incidence of malignancy-induced muscle weakness,20 a clinical perspective suggests that many patients do experience severe muscle weakness and fatigue. Cancer cachexia is a multifactorial syndrome that is common in advanced malignancy occurring in ∼80% of patients, which cannot be reversed by nutritional support and leads to significant function deficits. Cancer cachexia is estimated to be responsible for 20% of cancer-related deaths.18,19 However, there is a large heterogeneity in clinical presentation of cachexia that can vary according to tumor type, site and individual patient factors. In fact, the true incidence of cancer cachexia is likely to be greatly underestimated.20 Reducing cachexia has been shown to extend life span even without affecting tumor growth in mice.21 Improving muscle function and mobility of cancer patients should thus have a positive impact on adherence to treatment regimens and overall health.20 Therefore, a better understanding of the mechanisms of muscle weakness associated with bone metastases and cancer cachexia will lead to improved therapy. Moreover, refocusing attention to determine muscle quality in addition to improving muscle mass will likely provide the most beneficial treatment options for this devastating complication of malignancy.
Muscle–bone Cross-talk
Muscle and bone anabolism are tightly coupled during growth and development.22,23 Conversely, muscle and bone catabolism occur during aging.24 Yet, the cellular and molecular mechanisms linking these two tissues are not well understood. Nor is it known which tissue influences the metabolism of the other.
Muscle secretes many factors that can act on other tissues, including bone. These factors, collectively termed myokines, include the bone active molecules insulin-like growth factor 1 (IGF-1), fibroblast growth factor 2 (FGF-2), myostatin (also called growth and differentiation factor 8 [GDF8]) and IL-6.25 IGF-1 and FGF-2 at the muscle–bone interface stimulate bone formation.26,27 Myostatin is a mediator of cachexia,28 and myostatin deficiency increases bone density.29 Conversely, bone-derived factors are known to modulate muscle. For example, Indian hedgehog (Ihh) promotes myoblast survival and myogenesis in both mouse and chick embryos,22 thus indicating bidirectional bone–muscle cross-talk. It seems likely that, in cases of abnormal physiology, such as with the bone destruction that occurs from osteolytic bone metastases, the signals may also originate in bone and act on muscle.
Preclinical data from our laboratory show that mice with MDA-MB-231 breast cancer bone metastases have a significant reduction in forelimb grip strength and ex vivo maximum specific force generation of the extensor digitorum longus muscle due to improper calcium handling and that is independent of reduced muscle mass. Ex vivo-specific force calculations compensate for the differences in size and weight of individual muscles. Further muscle dysfunction is systemic and dependent on tumor-induced osteolytic bone resorption without tumor cell involvement in the muscle. Mice with primary MDA-MB-231 breast tumors do not get bone metastases and do not have muscle dysfunction.30 In a mouse model of osteolytic multiple myeloma, we observed systemic muscle dysfunction in the absence of cachexia.31 In both of these mouse models of tumor-induced osteolytic bone destruction, the severity of muscle dysfunction correlated with increased osteolysis. In these mice with muscle weakness, there is evidence of oxidation of the calcium-handling protein, RyR1, and calcium leak in the skeletal muscle, which is responsible for the muscle weakness. Collectively, these observations suggest that the bone microenvironment could mediate these effects.
Bone-derived Factor(s) that may lead to Skeletal Muscle Weakness
Which bone-derived factors can induce systemic muscle dysfunction? Bone matrix is a rich storehouse of growth factors that have known effects on muscle, such as activin, TGFβ, IGF-1 and bone morphogenic protein 2 (BMP-2).32,33 Some of these factors are released and activated as a consequence of osteoclastic bone resorption and have the potential to act systemically to promote muscle dysfunction. Skeletal muscle weakness is observed in other clinical conditions associated with bone loss, such as in patients with hyperparathyroidism. Patients with hyperparathyroidism fatigue quickly and have skeletal muscle atrophy, suggesting a link between bone loss and muscle weakness.34
The bone is a large storehouse for growth factors, such as TGFβ, which are deposited in the mineralized bone matrix by osteoblasts. In fact, bone is the largest storehouse of TGFβ in the body13,35 and has a central role to promote tumor osteolysis due to bone metastases.36,37,38,39 TGFβ is released in high concentrations from the mineralized bone matrix during osteoclastic bone resorption,38 a process activated in all types of bone metastases. TGFβ acts on tumor cells to enhance secretion of osteolytic factors40 that increase bone destruction and prometastatic factors, driving the feed-forward cycle of tumor growth and further bone destruction.17 Human breast cancer bone metastases show active TGFβ signaling by nuclear accumulation of phosphoSMAD2/336 and TGFβ signaling blockade via stable expression of dominant-negative TGFβ receptor 2 (DNTβRII), or a using a TGFβ receptor 1 kinase inhibitor, suppresses bone metastasis in mice.39,41
TGFβ is a potent regulator of wound healing in muscle, and persistent exposure leads to altered extracellular matrix architecture and formation of fibrotic tissue in muscle.42 Increased TGFβ signaling in muscle also inhibits satellite cell activation, impairs myocyte differentiation43,44 and is associated with skeletal muscle dysfunction in many of the muscular dystrophies.45,46 In a direct assessment of the effect of TGFβ on muscle function, the contractile properties of the extensor digitorum longus muscle were examined from limbs exposed to recombinant TGFβ via subcutaneous injection directly into the lower hindlimb. Muscle function in TGFβ-treated limbs showed a significant reduction in specific force without changes in muscle mass47 in contrast to other TGFβ family members that lead to reduced muscle mass (see below). These experiments suggest that TGFβ can reduce muscle function via a variety of mechanisms, which include fibrosis, myocyte differentiation and alteration in calcium-handling proteins in the sarcoplasmic reticulum, such as RyR1 and SERCA, which is independent of changes in muscle mass.
Other TGFβ family members may have a role in cancer-associated muscle dysfunction. The high-affinity activin receptor type 2B, ActRIIB, mediates signaling of a small group of TGFβ family members, including activin, myostatin and GDF-11, and is important in regulating muscle mass.48 Pharmacological blockade of ActRIIB prevents muscle wasting, induces muscle satellite cell mobilization and differentiation and significantly prolongs survival in murine models of cachexia.21 In addition, blockade of ActRIIB markedly improves muscle function in a Duchenne muscular dystrophy model (mdx mice).49 However, in these studies, it is not possible to determine whether the effect is due to blocking activin, myostatin or GDF-11 signaling because of receptor promiscuity. Myostatin antagonist has been investigated as a way to improve muscle wasting due to cachexia, as myostatin is a potent inhibitor of skeletal muscle differentiation and growth.50 GDF-11 shares 90% sequence homology with myostatin and in skeletal muscle inhibits myoblast differentiation,51 suggesting that GDF-11 may act in a very similar manner as myostatin. It is likely that these proteins promote the muscle wasting characteristic of cachexia, whereas other mediators contribute to muscle dysfunction by altering calcium handling (RyR1 and SERCA) or myofibrillar proteins in muscle cells (Figure 3).
Figure 3.

Bone–muscle cross-talk. Bone and muscle are physically and functionally tightly coupled. During osteolytic bone resorption due to tumor metastases in bone, bone-derived factors may be responsible for reduced skeletal muscle mass, whereas TGFβ is responsible for systemic muscle dysfunction via oxidation of RyR1.30 Muscle weakness occurring via atrophy involves induction of skeletal muscle-specific ubiquitin ligase expression (atrogin-1/MAFbx and MuRF1/TRIM63) and reduced protein synthesis.66 Muscle weakness can also occur through disruption of calcium signaling (RyR1 or SERCA oxidation) that reduces SR calcium and increases cytosolic calcium30,31 or by interference with actin-myosin cross-bridging.67 Myofibrillar protein oxidation has also been shown to lead to contractile dysfunction in heart failure and represents another possible mechanism of muscle weakness in cancer.68 In certain settings, muscle atrophy and muscle dysfunction can occur together furthering overall weakness.
In contrast to the negative effects possible from activin, myostatin and TGFβ signaling in muscle, IGF-1 and BMP-2 signaling results in muscle hypertrophy.42,52,53 IGF-1 is a major regulator of muscle mass because of its effect on myogenic cell proliferation and differentiation.54 Likewise, BMP signaling leads to muscle hypertrophy, but interestingly specific force (corrected for muscle mass) is significantly lower when BMP signaling is constitutively activated.52 This result demonstrates the importance of interpreting muscle-specific function (quality) not merely muscle mass (quantity) in murine models of skeletal muscle weakness. The contribution of BMP and TGFβ signaling in skeletal muscle that ultimately leads to a positive or a negative protein balance is unclear and remains to be determined.
In the setting of cancer cachexia without bone metastases, tumor-derived factors may also lead to muscle wasting. Tumor-derived parathyroid hormone-related protein has been shown to have a role in cancer cachexia and muscle weakness in a model of Lewis lung carcinoma (LLC).55 In bone metastases from breast cancer, blocking TGFβ prevents parathyroid hormone-related protein secretion and inhibits bone destruction39 that could theoretically lead to improved muscle function. However, in mice with bone metastases and cachexia the skeletal muscle-specific force (which takes into account the reduction in muscle size) is reduced. As noted above, TGFβ, when injected directly into the hindlimb of mice, has been shown to reduce skeletal muscle-specific force and indicate a direct role for TGFβ to reduce muscle function independent of muscle mass.47 These data show that careful consideration must be given to studies of muscle force and muscle mass.
In addition to factors released from bone matrix during osteoclast-driven resorption, other insults that affect muscle and bone in patients with malignancy may promote muscle weakness. Serum 25-hydroxyvitamin D concentrations are often low in breast cancer patients with osteoporosis or bone metastases who receive bisphosphonate therapy.56 Functional muscle tests in vitamin D receptor knock-out (VDRKO) mice showed an increase in sinking episodes in a forced swim test57 and reduced time before falling from a vertical screen test.58 These results indicate an overall muscle weakness in mice lacking vitamin D receptor that may involve reduced muscle mass, as well as reduced muscle function. In humans, the bone mineralization defects associated with rickets and osteomalacia are associated with muscle weakness measured by reduced timed up and go, 6-minute walk, stair climbing and object lifting.59,60 It should be noted that myopathies reported with vitamin D deficiency might also involve calcium and phosphate deficiencies, thus complicating the assessment of individual factors.61 Despite these studies, the exact mechanism by which vitamin D deficiency causes muscle weakness is unknown.
MicroRNA (miRNA) profiling has identified signatures associated with cancer, bone and muscle. Human miRNA Let-7 was recently shown to be increased in serum of mice with breast cancer bone metastases.62 miRNA Let-7 is also increased in serum of elderly patients with muscle weakness and has been suggested to reduce regenerative capacity in aging.63 Further studies are needed to show the mechanism by which Let-7 may affect muscle.
Other unexplored mechanisms of muscle weakness include the role of the sympathetic nervous system due to bone metastases. The sympathetic nervous system modulates skeletal muscle metabolism, ion transport and contractility. Recent evidence has shown that the sympathetic nervous system is capable of promoting breast cancer bone metastasis through stimulation of marrow stromal cells;64 yet, a connection to muscle weakness has not been investigated.
Summary
Bone and muscle functions are tightly coupled in normal physiology. Most studies have focused on muscle as an endocrine organ with a predominant role in bone cell function. However, recent evidence suggests that events in bone may alter muscle function as well. The example discussed here, bone metastases, represents a severe disruption of normal bone remodeling. Bone is a rich storehouse of growth factors that have activity in bone (as a part of normal remodeling) and in other organs, including muscle. It is therefore possible that during accelerated bone resorption, such as that which occurs in bone metastases, bone might have a predominant role to alter muscle function and become a source of ‘osteokines' that affect muscle function. Likewise, factors released from muscle may have an important role in bone metabolism that could further exacerbate the role of bone as a driver of muscle dysfunction.
Whatever factors are identified that transmit signals between bone and muscle, it is clear that bone-derived factors are capable of impacting muscle and that the effects can manifest as reduction in muscle mass (quantity) or reduction in myocyte function (quality) or both, as is likely in advanced disease (Figure 3). Identification and characterization of factors involved in bone–muscle cross-talk will provide new possibilities for therapeutic intervention in muscle weakness and cachexia associated with malignancy.
Acknowledgments
This work was supported by NIH grants (U01CA143057 from the NCI Tumor Microenvironment Network; R01CA69158), Susan G. Komen Foundation, the Indiana Economic Development Grant, the Jerry and Peggy Throgmartin Endowment of the IU Simon Cancer Center, IU Simon Cancer Center Breast Cancer Program and a generous donation from the Withycombe family.
Footnotes
T.A. Guise was a consultant/advisory board member of Novartis. D.L. declares no conflict of interest.
References
- Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 2013; 10: 90–99. [DOI] [PubMed] [Google Scholar]
- Giordano A, Calvani M, Petillo O, Carteni M, Melone MR, Peluso G. Skeletal muscle metabolism in physiology and in cancer disease. J Cell Biochem 2003; 90: 170–186. [DOI] [PubMed] [Google Scholar]
- von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers-update 2014. J Cachexia, Sarcopenia Muscle 2014; 5: 261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodman GD. Mechanisms of bone metastasis. N Eng J Med 2004; 350: 1655–1664. [DOI] [PubMed] [Google Scholar]
- Huxley AF. Muscular contraction. J Physiol 1974; 243: 1–43. [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW et al. Remodeling of ryanodine receptor complex causes "leaky" channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci USA 2008; 105: 2198–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005; 123: 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 2000; 101: 365–376. [DOI] [PubMed] [Google Scholar]
- Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol 2003; 160: 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 2009; 15: 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W et al. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 2011; 14: 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 2010; 49: 1603–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonewald LF, Mundy GR. Role of transforming growth factor-beta in bone remodeling. Clin Orthop Relat Res 1990; 250: 261–276. [PubMed] [Google Scholar]
- Carmona-Cuenca I, Herrera B, Ventura JJ, Roncero C, Fernandez M, Fabregat I. EGF. blocks NADPH oxidase activation by TGF-beta in fetal rat hepatocytes, impairing oxidative stress, and cell death. J Cell Physiol 2006; 207: 322–330. [DOI] [PubMed] [Google Scholar]
- Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem 2010; 285: 25103–25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MR, Burr DB. Bisphosphonate effects on bone turnover, microdamage, and mechanical properties: what we think we know and what we know that we don't know. Bone 2011; 49: 56–65. [DOI] [PubMed] [Google Scholar]
- Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011; 11: 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011; 12: 489–495. [DOI] [PubMed] [Google Scholar]
- Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012; 16: 153–166. [DOI] [PubMed] [Google Scholar]
- Fox KM, Brooks JM, Gandra SR, Markus R, Chiou CF. Estimation of cachexia among cancer patients based on four definitions. J Oncol 2009; 2009: 693458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010; 142: 531–543. [DOI] [PubMed] [Google Scholar]
- Bren-Mattison Y, Hausburg M, Olwin BB. Growth of limb muscle is dependent on skeletal-derived Indian hedgehog. Dev Biol 2011; 356: 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch F, Bailey DA, Baxter-Jones A, Mirwald R, Faulkner R. The 'muscle-bone unit' during the pubertal growth spurt. Bone 2004; 34: 771–775. [DOI] [PubMed] [Google Scholar]
- Greenlund LJ, Nair KS. Sarcopenia–consequences, mechanisms, and potential therapies. Mech Ageing Dev 2003; 124: 287–299. [DOI] [PubMed] [Google Scholar]
- DiGirolamo DJ, Kiel DP, Esser KA. Bone and skeletal muscle: neighbors with close ties. J Bone Miner Res 2013; 28: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Pun S, Wronski TJ. Bone anabolic effects of basic fibroblast growth factor in ovariectomized rats. Endocrinology 1999; 140: 5780–5788. [DOI] [PubMed] [Google Scholar]
- Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest 2002; 110: 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J Cell Physiol 2006; 209: 501–514. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Samaddar T, Pennington C, McCormick J. Increased muscle mass with myostatin deficiency improves gains in bone strength with exercise. J Bone Miner Res 2006; 21: 477–483. [DOI] [PubMed] [Google Scholar]
- Waning DL, Mohammad KS, Andersson DC, John SK, Rieken S, Xie W et al. Role of the tumor-bone microenvironment in muscle weakness and cachexia. Cancer-Induced Bone Disease IBMS: Miami, FL, USA, 2013;. [Google Scholar]
- Waning DL, Mohammad KS, Reiken S, Marks AR, Roodman GD, Guise TA. Muscle weakness is associated with osteolysis in multiple myeloma. Cancer-Induced Bone Disease IBMS: Miami, FL, USA, 2013;. [Google Scholar]
- Sakai R, Eto Y. Involvement of activin in the regulation of bone metabolism. Mol Cell Endocrinol 2001; 180: 183–188. [DOI] [PubMed] [Google Scholar]
- Wildemann B, Kadow-Romacker A, Haas NP, Schmidmaier G. Quantification of various growth factors in different demineralized bone matrix preparations. J Biomed Mater Res A 2007; 81: 437–442. [DOI] [PubMed] [Google Scholar]
- Patten BM, Bilezikian JP, Mallette LE, Prince A, Engel WK, Aurbach GD. Neuromuscular disease in primary hyperparathyroidism. Ann Intern Med 1974; 80: 182–193. [DOI] [PubMed] [Google Scholar]
- Dallas SL, Rosser JL, Mundy GR, Bonewald LF. Proteolysis of latent transforming growth factor-beta (TGF-beta )-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J Biol Chem 2002; 277: 21352–21360. [DOI] [PubMed] [Google Scholar]
- Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA 2005; 102: 13909–13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003; 3: 537–549. [DOI] [PubMed] [Google Scholar]
- Korpal M, Yan J, Lu X, Xu S, Lerit DA, Kang Y. Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med 2009; 15: 960–966. [DOI] [PubMed] [Google Scholar]
- Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999; 103: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest 1996; 98: 1544–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn LK, Mohammad KS, Fournier PG, McKenna CR, Davis HW, Niewolna M et al. Hypoxia and TGF-beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS ONE 2009; 4: e6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano AL, Munoz-Canoves P. Regulation and dysregulation of fibrosis in skeletal muscle. Exp Cell Res 2010; 316: 3050–3058. [DOI] [PubMed] [Google Scholar]
- Allen RE, Boxhorn LK. Inhibition of skeletal muscle satellite cell differentiation by transforming growth factor-beta. J Cell Physiol 1987; 133: 567–572. [DOI] [PubMed] [Google Scholar]
- Allen RE, Boxhorn LK. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J Cell Physiol 1989; 138: 311–315. [DOI] [PubMed] [Google Scholar]
- Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R et al. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology 2005; 65: 826–834. [DOI] [PubMed] [Google Scholar]
- Kollias HD, McDermott JC. Transforming growth factor-beta and myostatin signaling in skeletal muscle. J Appl Physiol 2008; 104: 579–587. [DOI] [PubMed] [Google Scholar]
- Mendias CL, Gumucio JP, Davis ME, Bromley CW, Davis CS, Brooks SV. Transforming growth factor-beta induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve 2012; 45: 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA 2005; 102: 18117–18122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistilli EE, Bogdanovich S, Goncalves MD, Ahima RS, Lachey J, Seehra J et al. Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am J Pathol 2011; 178: 1287–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN et al. Induction of cachexia in mice by systemically administered myostatin. Science 2002; 296: 1486–1488. [DOI] [PubMed] [Google Scholar]
- Jeanplong F, Falconer SJ, Thomas M, Matthews KG, Oldham AM, Watson T et al. Growth and differentiation factor-11 is developmentally regulated in skeletal muscle and inhibits myoblast differentiation. Open J Mol Integr Physiol 2012; 2: 127–138. [Google Scholar]
- Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E et al. BMP signaling controls muscle mass. Nat Genet 2013; 45: 1309–1318. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle 2011; 1: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florini JR, Ewton DZ, Coolican SA. Growth hormone and the insulin-like growth factor system in myogenesis. Endocr Rev 1996; 17: 481–517. [DOI] [PubMed] [Google Scholar]
- Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014; 513: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang-Gillam A, Miles DA, Hutchins LF. Evaluation of vitamin D deficiency in breast cancer patients on bisphosphonates. Oncologist 2008; 13: 821–827. [DOI] [PubMed] [Google Scholar]
- Burne TH, Johnston AN, McGrath JJ, Mackay-Sim A. Swimming behaviour and post-swimming activity in Vitamin D receptor knockout mice. Brain Res Bull 2006; 69: 74–78. [DOI] [PubMed] [Google Scholar]
- Kalueff AV, Lou YR, Laaksi I, Tuohimaa P. Impaired motor performance in mice lacking neurosteroid vitamin D receptors. Brain Res Bull 2004; 64: 25–29. [DOI] [PubMed] [Google Scholar]
- Russell JA. Osteomalacic myopathy. Muscle nerve 1994; 17: 578–580. [DOI] [PubMed] [Google Scholar]
- Schott GD, Wills MR. Muscle weakness in osteomalacia. Lancet 1976; 1: 626–629. [DOI] [PubMed] [Google Scholar]
- Aono Y, Hasegawa H, Yamazaki Y, Shimada T, Fujita T, Yamashita T et al. Anti-FGF-23 neutralizing antibodies ameliorate muscle weakness and decreased spontaneous movement of Hyp mice. J Bone Miner Res 2011; 26: 803–810. [DOI] [PubMed] [Google Scholar]
- Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell 2013; 24: 542–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond MJ, McCarthy JJ, Sinha M, Spratt HM, Volpi E, Esser KA et al. Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiol Genomics 2011; 43: 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JP, Karolak MR, Ma Y, Perrien DS, Masood-Campbell SK, Penner NL et al. Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol 2012; 10: e1001363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waning DL, Guise TA. Molecular mechanisms of bone metastasis and associated muscle weakness. Clin Cancer Res 2014; 20: 3071–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev 2009; 89: 381–410. [DOI] [PubMed] [Google Scholar]
- Miller MS, Callahan DM, Toth MJ. Skeletal muscle myofilament adaptations to aging, disease, and disuse and their effects on whole muscle performance in older adult humans. Front Physiol 2014; 5: 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton M, Menazza S, Sheeran FL, Polverino de Laureto P, Di Lisa F, Pepe S. Oxidation of myofibrillar proteins in human heart failure. J Am Coll Cardiol 2011; 57: 300–309. [DOI] [PubMed] [Google Scholar]