

Abstract
A new tumor-seeking tridentate topology consisting of a phosphino dithioether ((HOCH2)2PCH2CH2S(CH2)nCH2SR; PS2) ligand framework for the production of kinetically inert and in vivo stable facial [99mTc(CO)3(PS2)]+ or [Re(CO)3(PS2)]+ is described. The X-ray crystal structure of fac-Re(CO)3(PS2)PF6 is reported. The bioconjugation strategies for incorporating bombesin (BBN) peptides on to the PS2 tripodal framework and, thereby, de novo designing of GRP receptor-seeking Tc(PS2–BBN)(CO)3 are developed.
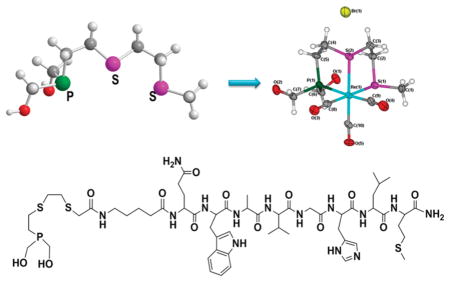
INTRODUCTION
Advances in metal-based molecular imaging and therapy agents will hinge upon discoveries in ligand architecture and coordination chemistry of specific transitionmetals.1 For example, phosphine-based ligating frameworks have been shown to impart in vivo stability to radioactive metals through chelate interactions, and such agents have been utilized for biomedical applications.2 It is very well established that the steric and electronic properties of phosphines exert a major influence on bonding interactions with transition metals of biological interest, thus, creating a compelling scenario for the de novo design of new functionalized phosphines.3 Specifically, water-soluble phosphines functionalized with peptides or other biomolecules serve as biological markers for the in vivo targeting of receptors overexpressed in tumors for diagnostic/therapeutic use.3 Surprisingly, despite the importance of peptide–phosphine conjugates in biomedical applications, little effort has been directed toward developing the synthetic strategies for the immobilization of biomolecules on water-soluble phosphine ligands.
Technetium-99m occupies a special position in inorganic radiopharmaceuticals1,4,5 because of the following reasons: (i) optimal nuclear properties (t1/2 = 6.02 h, E(γ) =140 keV) for molecular imaging; (ii) easy availability through the transportable 99Mo/99mTc generator system; and (iii) conversion to clinically useful, ligand exchangeable inorganic complexes for biomolecule conjugation. More importantly, 99mTc has a versatile inorganic chemistry, spanning different oxidation states, to produce a variety of ligand exchangeable coordination complexes.1 Among several precursor complexes of technetium, the kinetically inert [99mTc(CO)3(H2O)3]+ complex has attracted much attention in recent years due to its commercial availability as the Isolink kit and ease of generation under clinical settings.5 In fact, three water molecules in the complex can be replaced by a chelating ligand conjugated to a biomolecule such as a tridentate ligand or a “2+1” mixed ligand system, to yield a stable complex for targeted in vivo imaging applications. Alberto and co-workers have examined a library of N and S donor ligands for stabilizing the fac-[M- (CO)3]+ moiety.6 Their investigations showed that by judicious selection of the ligand system, effective labeling of biomolecules could be achieved.7 Likewise, Smith and co-workers have shown that the combination of bidentate N-donor ligands with a water-soluble monophosphine was effective in stabilizing the tricarbonyl core.8 On the inorganic chemistry front, novel tridentate N-donor ligand systems were developed by Marzilli and coworkers for the effective stabilization of the M(CO)3 moiety. They have shown that organic soluble metal precursors can serve as synthons for effective radiolabeling.9 In addition, electron-rich bidentate O-donor ligands (β-diketones acetylacetone and curcumin) along with monodentate nitrogen ligands have been explored to generate stable fac-[M(CO)3(OO)(N)]+ complexes. 10 However, precise knowledge of ligand design is not completely understood—more specifically, which combinations of hard bases vs soft donors would lead to enhanced in vivo stability of the [99mTc(CO)3]+ moiety in order to engineer the design and development of organ-specific (or tumor-specific) radiopharmaceuticals.
In this respect, as part of our ongoing drug discovery program,11 we have investigated pharmacophore motifs of a library of ligands encompassing hard nitrogen donors, phosphine π acids, and sulfur-containing soft donors. We, herein, report the development of ligand frameworks spanning the tripodal coordination sites of [99mTc(CO)3(H2O)3]+. The results reported herein encompass our successful chemistry for the synthesis of the phosphino dithioether ((HOCH2)2PCH2CH2S(CH2)nCH2-SCH3; PS2 ) tripodal ligand backbone, its conjugation with bombesin peptide, and the “proof of principle” that such a π-acid phosphine (P) and soft σ-donor thioether (S) topology will produce in vivo stable imaging agents. As a first step toward establishing the utility of the PS2 ligand framework for in vivo biomedical applications, we focused our efforts on the synthesis of bifunctional ligand systems, bioconjugation with receptor-avid bombesin peptide, complexation with 99mTc and Re carbonyl complexes, and in vivo stability studies in mice.
Bombesin (BBN), a 14-amino-acid peptide, was first isolated from the skin of the frog amphibian Bombina.12 BBN functions as a potent autocrine or paracrine growth factor for cells.13 In the past decade, a plethora of information has been generated on BBN/receptor expression and physiological processes.14 BBN shows high affinity for the gastrin-releasing peptide (GRP) receptor subtype BB2.14 GRP receptors are overexpressed in many cancers, including prostate, breast, and small cell lung cancer.15 Analogues of bombesin with modified structures have exhibited a similar or even higher affinity for these receptors. Synthetic peptides can be readily generated through automated solid phase techniques. For our studies, we have synthesized and utilized the seven-amino-acid truncated bombesin analogue (BBN) as a vehicle to target GRP receptors. The results reported in this paper include (i) synthesis and characterization of PS2 ligand systems, (ii) synthesis and X-ray crystallographic investigation of the [Re(CO)3(PS2)]+ complex, (iii) synthesis of the bifunctional PS2 ligand system and bioconjugation with the truncated bombesin peptide, (iv) synthesis and in vivo stability analysis of [99mTc(CO)3(PS2)]+ in mice, and (v) synthesis and in vivo stability analysis of [99mTc(CO)3(PS2–BBN)]+ in mice.
RESULTS AND DISCUSSION
Synthesis of (HOCH2)2PCH2CH2S(CH2)nCH2SCH3 (n = 1 (4a), 2 (4b))
The synthesis of the PS2 ligand system (4a and 4b) was accomplished with a multistep synthetic route as shown in Scheme 1. In the first step, mono S-alkylation of 1,2-ethanedithiol or 1,3-ethane dithiol was achieved by treating diethyl-2-bromoethylphosphonate with a large excess of the corresponding dithiol. Subsequently, the reduction of phosphonate groups in monothioether monothiol phosphonates (1a, 1b) using lithium aluminum hydride yielded monothioether monothiol phosphine (2a, 2b) in quantitative yields. The thiol groups in these phosphines were S-alkylated by treating them with methyl iodide in the presence of sodium hydride to yield 3a or 3b. It is noteworthy to recognize the fact that under the reaction conditions employed, no P alkylation was observed. In the final step, the P–H bonds in 3a and 3b were formylated with aqueous formaldehyde in the presence of 5 N HCl to yield the corresponding phosphonium chloride salt [(HOCH2)3PCH2CH2S(CH2)nCH2SCH3]Cl. These salts are highly stable and can be stored in a refrigerator for extended periods of time. Before utilization of the phosphonium salt for the metalation reaction, the salts were treated in situ with triethylamine to generate the corresponding bis-hydroxymethyl phosphine derivative (HOCH2)2PCH2CH2SCH2CH2-SCH3 (PS2 4a, 4b). All of the intermediate compounds and the final products were characterized using 1H, 13C, and 31P NMR spectroscopy. Only the final products were analyzed using mass spectrometry, as the intermediates were highly malodorous.
Scheme 1.

Synthesis of [M(CO)3(PS2)] +(M = Re (5), 99mTc (6))
Our next step was to evaluate the role of an increase in chelate size of the PS2 ligand systems with the fac-[M(CO)3]+ moiety. As a first step, we investigated the interactions of PS2 (4a, 4b) ligands with the [Re(CO)3Br3]2− core (Scheme 2). Typically, aqueous solutions of PS2 (4a, 4b) ligands were treated with [NEt4]2[Re(CO)3Br3] species in the presence of triethylamine, and the complexation products were analyzed using traditional analytical measurements. In the case of PS2 (4a), complex [Re(CO)3(PS2)]Br (5) was formed as a singular chemical product (Scheme 2). The chemical constitution of [Re(CO)3(PS2)]Br (5) was confirmed using NMR, IR, and single crystal X-ray diffraction techniques. In contrast, the reaction of 4b with [NEt4]2[Re(CO)3Br3] yielded multiple products as indicated by NMR and HPLC techniques; products were intractable even after several chromatographic purifications. It is important to note here that ligand 4a contains an ethyl linker whereas 4b contains a propyl linker between thioether groups. The formation of multiple products by ligand 4b may be attributed to the flexible three carbon linker between the two sulfur atoms. This topology of three carbon atoms, presumably, initiates the formation of polymeric products, connecting to more than one rhenium atom.
Scheme 2.

Next, we investigated the interaction of PS2 (4a) with the organometallic technetium tricarbonyl precursor [99mTc(CO)3- (OH2)3]+, and this reaction yielded the corresponding complex [99mTc(CO)3PS2]+ (6) in near quantitative yield, as shown in Scheme 2. The formation of 6 was analyzed by reversed phase HPLC utilizing a radiometric (γ) detector. Gradient conditions are described in the Experimental Section, and complex 6 elutes with a retention time of 9.45 min, similar to rhenium analog 5, which has a retention time of 9.5 min (determined using a UV detector). Figure 1 shows the comparative HPLC chromatogram of complexes 5 and 6. The close lying retention times between complexes 5 and 6 further ascertain the structural similarity of these complexes. Also, we investigated the reaction of 4b with a technetium carbonyl precursor. Similar to the reaction of 4b with rhenium carbonyl, the reaction of 4b with [99mTc(CO)3- (OH2)3]+ yielded multiple species, as indicated by HPLC analysis.
Figure 1.
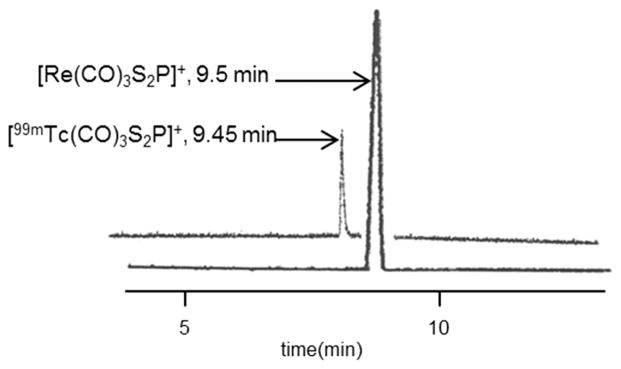
HPLC chromatograms of [M(CO)3(PS2)]+(M = Re (5) or 99mTc (6)) complexes.
In summary, the shorter reaction time and the formation of a single species, even under dilute conditions, indicate the high kinetic propensity of PS2 (4a) toward forming complexes with M(CO)3 + species. This property of 4a is attributed to the symbiosis of both a perfect chelate size and a perfect match between soft thioether and hydroxymethyl phosphine donors to the soft [M(CO)3]+ core.
In order to understand the molecular features of 5, single crystals were grown by slow evaporation of a methanolic solution of 5, and a detailed X-ray crystallographic investigation was performed. An ORTEP presentation of complex 5 is shown in Figure 2. X-ray crystallographic data for complex 5 are presented in Table 1. The geometry around the central rhenium atom is slightly distorted octahedral, with the carbonyls arranged facially around the metal center. The three remaining positions in the octahedron are occupied by two sulfur atoms and one phosphorus atom in the PS2 ligand.
Figure 2.

X-ray crystal structure of 5. Selected bond lengths (Å) and angles (deg): Re(1)–C(10) = 1.907(10), Re(1)–C(8) = 1.933(10), Re(1)–C(9) = 1.958(11), Re(1)–P(1) = 2.437 (2), Re(1)–S(1) = 2.487(2), Re(1)–S(2) = 2.475(2); P(1)–Re(1)–S(2) = 83.11(8), C(10)–Re(1)–C(8) = 86.9(4), C(10)–Re(1)–C(9) = 91.5(4), C(8)–Re(1)–C(9) = 89.3(4).
Table 1.
Crystallographic Data for Complex 5
| empirical formula | C10H17BrO5PReS2 |
| fw | 578.44 |
| temp | 173(2) K |
| wavelength | 0.71073 Å |
| cryst syst, space group | triclinic, P1̄ |
| unit cell dimensions |
a = 6.5967(4) Å, α = 89.0590(10)°, b = 8.9236(5) Å, β = 79.3080(10)°, c = 14.1771(8) Å, γ = 87.8210(10)° |
| volume | 819.43(8) Å3 |
| Z, calcd density | 2, 2.344 Mg/m3 |
| abs coeff | 10.217 mm−1 |
| F(000) | 548 |
| cryst size | 0.45 × 0.15 × 0.15 mm |
| θ range for data collection | 2.28–27.11° |
| limiting indices | −8 ≤ h ≤ 7, −11 ≤ k ≤ 11, −18 ≤ l ≤ 12 |
| reflns collected/unique | 5882/3538 [R(int) = 0.0346] |
| completeness to θ = 27.11 | 97.50% |
| abs correction | semiempirical from equivalents |
| max and min transm | 0.26 and 0.08 |
| refinement method | full-matrix least-squares on F2 |
| data/restraints/params | 3538/0/183 |
| GOF on F2 | 1.082 |
| final R indices [I > 2σ(I)] | R1 = 0.0549, wR2 = 0.1520 |
| R indices (all data) | R1 = 0.0590, wR2 = 0.1554 |
| extinction coefficient | 0.0007(8) |
| largest diff. peak and hole | 3.364 and −4.033 e.A-3 |
In Vitro Stability Studies
Complex 5 was challenged with a large excess of cysteine or human serum albumin (HSA) to establish the in vitro stability characteristics. Cysteine is a typical model chosen for these particular studies, as a number of potential metal-chelating thiol functionalities are present in vivo (e.g., cysteine, serum proteins, and glutathione), and HSA is the major constituent in the blood, which is rich in various hetero-atom donors. The 31P NMR spectroscopic data of aliquots of each sample were taken at different time intervals, and these studies indicated that complex 5 remained intact even after 6 months of incubation, demonstrating that the PS2 core provides in vitro stability to the Re(I) complex.
The stability of complex [99mTc(CO)3PS2]+ (6) under different pH conditions was investigated, and the stability was analyzed using HPLC (Table 2). We next investigated the in vitro stability of 6 by incubating the purified complex with a solution of 0.2 M cysteine, 0.2 M histidine, or 3 × 10−5 M HSA (thiol content 0.04 equiv/mol) at 37 °C at physiological pH for up to 24 h. No detectable decomposition of 6 was observed under these conditions, indicating the high in vitro stability.
Table 2.
Stability of [99mTc(CO)3 (PS2)]+ Complex (6) at Different pH’s with Time
| pH | percentage (%) of complex remaining
|
||
|---|---|---|---|
| 1 h | 4 h | 24 h | |
| 2 | 98±1 | 96±3 | 94±3 |
| 7 | 98±2 | 98±1 | 97± 2 |
| 11 | 97±2 | 96±2 | 95±2 |
The stability studies on 5 and 6 have further confirmed that complexes derived from the PS2 ligand framework 4a with two carbon linkers between sulfur atoms impart stability to the resulting technetium or rhenium complexes. Therefore, complex 6 bearing a ligand with tripodal architecture with two carbon linkers between the sulfur atoms was selected for performing detailed in vivo biodistribution studies in CF-1 mice.
In Vivo Biodistribution Studies of 6
Biodistribution studies on normal CF-1 mice were performed by intravenous administration of 6 followed by an analysis of radioactivity in various organs at various different time points. Results of this study as a function of percent injected dose (%ID) and the percent injected dose per unit mass (%ID/g) are given in Table 3. The data indicate rapid clearance of 6 from the blood, with only 0.59 ± 0.31%ID remaining in the whole blood after the 1 h post injection period. In addition, the data indicate that complex 6 clears through both the hepatobiliary and renal urinary pathways. However, the hydrophobic nature of 6 makes it preferentially excrete through the hepatobiliary pathway, with radioactivity of 52.41 ± 17.06 and 3.06 ± 1.37% of the injected dose in the large and small intestines, respectively, after the 4 h post injection period. Accordingly, the excretions through the renal pathway reach 37.43 ± 18.83% of the injected dose 4 h after injection. More importantly, there was no significant accumulation of radioactivity in the kidneys (0.32 ±0.05%ID 4 h post-injection). Thus, this study indicates that PS2 ligand systems are effective in interacting with the [99mTc(CO)3]+ core to produce complexes that demonstrate kinetic inertness under both in vitro and in vivo conditions.
Table 3.
Biodistribution (% ID/organa and % ID/ga) of [99mTc(CO)3S2P]+ (6) in Normal CF-1 Mice 1 h, 4 h, and 24 h, post-IV injection
| organ | time
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 h
|
4 h
|
4 h
|
||||||||||
| %ID | %ID/g | %ID | %ID/g | %ID | %ID/g | |||||||
| brain | 0.01 | (0.01) | 0.03 | (0.03) | 0.02 | (0.02) | 0.05 | (0.05) | 0.01 | (0.00) | 0.02 | (0.01) |
| bloodb | 0.6 | (0.3) | 0.22 | (0.13) | 0.4 | (0.1) | 0.17 | (0.06) | 0.00 | (0.00) | 0.00 | (0.00) |
| heart | 0.03 | (0.02) | 0.20 | (0.14) | 0.03 | (0.01) | 0.2 | (0.1) | 0.03 | (0.02) | 0.2 | (0.1) |
| lung | 0.09 | (0.02) | 0.40 | (0.13) | 0.08 | (0.03) | 0.3 | (0.1) | 0.04 | (0.02) | 0.2 | (0.1) |
| liver | 6.9 | (1.3) | 3.8 | (0.8) | 2.9 | (0.9) | 1.8 | (0.5) | 0.6 | (0.1) | 0.4 | (0.1) |
| spleen | 0.08 | (0.12) | 0.5 | (0.7) | 0.02 | (0.01) | 0.17 | (0.02) | 0.02 | (0.02) | 0.2 | (0.1) |
| stomach | 0.60 | (0.21) | 1.4 | (0.6) | 0.4 | (0.2) | 1.0 | (0.6) | 0.3 | (0.2) | 0.4 | (0.3) |
| l. int | 16.7 | (4.3) | 19.1 | (11.0) | 52.4 | (17.1) | 58.2 | (13.2) | 2.7 | (2.2) | 2.7 | (2.1) |
| sm. int | 23.3 | (8.7) | 13.5 | (5.5) | 3.1 | (1.4) | 2.2 | (1.4) | 0.4 | (0.3) | 0.3 | (0.2) |
| kidney | 1.0 | (0.2) | 2.2 | (0.6) | 0.3 | (0.1) | 0.8 | (0.2) | 0.10 | (0.02) | 0.21 | (0.04) |
| urine | 46.3 | (13.3) | 37.4 | (18) | 68.1 | (8) | ||||||
| muscle | 0.02 | (0.01) | 0.10 | (0.06) | 0.02 | (0.01) | 0.12 | (0.04) | 0.03 | (0.02) | 0.2 | (0.2) |
| bone | 0.01 | (0.01) | 0.13 | (0.06) | 0.02 | (0.00) | 0.2 | (0.06) | 0.02 | (0.02) | 0.2 | (0.2) |
| pancreas | 0.4 | (0.2) | 1.1 | (0.5) | 0.3 | (0.1) | 1.0 | (0.4) | 0.02 | (0.02) | 0.06 | (0.07) |
| carcass | 4.5 | (1.3) | 2.9 | (0.6) | 1.0 | (0.4) | ||||||
| feces | 26 | (10) | ||||||||||
Values represent the mean (±SD), N= 5 of the% ID/organ or the %ID/g of tissue. Body weights of mice ranged from 21 to 28 g.
Total blood volume is estimated to be 6.5% of the total body weight.
Synthesis of PS2–BBN (10)
We have subsequently explored further organic chemistry in the PS2 core toward bioconjugation with receptor-specific peptides. First, our efforts were focused on incorporating a “–COOH” functionality on the PS2 ligand framework, as summarized in Scheme 3. The monothiolphosphine intermediate 2 was utilized as the starting material for the synthesis of the carboxylate functionalized PS2 system. PS2–COOH (8) was synthesized in a two-step procedure. In the first step, compound 2 was S-alkylated with bromo-t-butyl acetate in the presence of NaHin THF, followed by deprotection of the t-butyl group by bubbling dry HCl to yield 8 in quanitative yields.
Scheme 3.
To demonstrate the utility of 8 in synthesizing receptor-avid peptide–phosphine conjugates, a bombesin 5-Ava-BBN[7–14] analogue, was chosen. The eight-amino-acid-containing bombesin analogue can be used as a vehicle to target GRP receptors, which are overexpressed on a variety of tumor tissues.15 The conjugation of phosphine 8 to the BBN analog was successfully achieved by combinatorial solid phase peptide synthesis (SPPS) as well as employing 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) as the activating agent in the presence of triethylamine to yield the bombesin peptide conjugate 9, as described in Scheme 4. The BBN analog was synthesized using automated-SPPS, cleaved from the resin, and coupling with 8 was achieved in solution. The crude product obtained was purified by reversed phase HPLC using a UV detector. The HPLC purified compound 9 was characterized by 31P{1H} NMR spectroscopy and mass spectrometry. The 31P- {1H} NMR spectrum of compound 9 shows a singlet at δ = −143.9 ppm, which confirmed the presence of a primary phosphine group. The Electrospray Ionization Mass Spectrum (ESI-MS) of conjugate 9 shows a peak at 1233.5 (calculated M+, 1232.5) corresponding to the M+H+ peak and further establishing the identity of 9. The phosphine–peptide conjugate 9 was converted into the corresponding hydroxymethyl phosphonium salt 10, as shown in Scheme 4. The phosphonium salt 10 was purified by reversed phase HPLC to yield the pure product as a fluffy white powder. The 31P NMR spectrum of 10 in water shows a sharp singlet at δ = 24.2 ppm, which is typical for the tetrakis(hydroxymethyl) phosphonium group. Electrospray mass spectrometry of the product showed a peak at 1323.0 (m/z), further confirming the presence of the phosphonium salt 10.
Scheme 4.

Synthesis and in Vitro Stability of [99mTc(CO3)PS2–BBN] +(11)
The [99mTc(CO)3]+ complex of PS2-5-Ava-BBN- [7–14]NH2 (PS2 –BBN) (11) was synthesized by incubation of PS2–BBN (10) with [99mTc(CO)3(OH2)3]+ at pH 7.5–8.0 for 30 min at room temperature (25 °C) to yield the corresponding complex [99mTc(CO)3(PS2 –BBN)]+ (11) in greater than 95% yields, as determined via radiometric detection on HPLC analysis. In vitro stability of complex 11 was studied by incubating the HPLC purified complex with a solution of 0.2 M cysteine, 0.2 Mhistidine, or 2% HSA solution in saline at pH 7.4 at 37 °C, and the radiochemical purity of the complex at different time points was analyzed by HPLC. The study indicated no detectable decomposition even at 24 h post-incubation.
In Vivo Biodistribution Studies of 11
In vivo biodistribution studies were performed in normal CF-1 mice by intravenous administration of the [99mTc(CO)3PS2–BBN]+ (11) complex. Results of this study as a function of the percent injected dose (%ID) and the percent injected dose per unit mass (%ID/g) are given in Table 4. The [99mTc(CO)3PS2–BBN]+ complex showed rapid clearance from the blood pool with activities reaching 1.9 ± 2.5% injected dose within 1 h post-injection and 0.43 ± 0.12% injected dose within 4 h post-injection. In addition, data on the accumulation of radioactivity in blood (0.67% ID at 1 h and 0.0% ID at 24 h) and the stomach (0.93% ID/g at 1 h and 1.11% ID/g at 24 h) provide pharmacokinetic evidence for the in vivo stability of 11. [99mTc(CO)3PS2–BBN]+ (11) exhibited renal clearance with radioactivity reaching 0.49 ± 0.03% injected dose within 1 h post-injection. The amount of radioactivity found in the stomach, an indication of in vivo decomposition of a 99mTc conjugate, was negligible at 1 h (0.52 ± 0.22%ID) and 24 h (0.84 ± 0.67%ID) post-injection. There was retention of the conjugate in the liver (3.30 ± 0.74% ID, 4 h PI). The retention in the liver is due to the high hydrophobic nature of conjugate 11.
Table 4.
Biodistribution (%ID/organa and %ID/ga) of [99mTc(CO)3S2P-BBN]+ (11) in Normal CF-1 Mice at 1 h, 4 h, and 24 h, Post-IV-Injection
| organ | time
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 h
|
4 h
|
24 h
|
||||||||||
| % ID | %ID/g | % ID | %ID/g | % ID | %ID/g | |||||||
| tail | ||||||||||||
| brain | 0 | (0.00) | 0 | (0.00) | 0 | (0.00) | 0 | (0.00) | 0 | (0.00) | 0 | (0.00) |
| bloodb | 1 | (2) | 0.67 | (0.84) | 0.43 | (0.12) | 0.15 | (0.04) | 0 | (0.00) | 0 | (0.00) |
| heart | 0.02 | (0.01) | 0.14 | (0.03) | 0.01 | (0.01) | 0.04 | (0.02) | 0.06 | (0.11) | 0.4 | (0.6) |
| lung | 0.08 | (0.06) | 0.33 | (0.25) | 0.03 | (0.01) | 0.15 | (0.04) | 0.02 | (0.01) | 0.07 | (0.05) |
| liver | 6.5 | (0.8) | 3.4 | (0.36) | 3.3 | (0.74) | 1.6 | (0.4) | 0.9 | (0.1) | 0.4 | (0.06) |
| spleen | 0.09 | (0.02) | 0.66 | (0.17) | 0.03 | (0.01) | 0.15 | (0.07) | 0.02 | (0.02) | 0.12 | (0.17) |
| stomach | 0.52 | (0.22) | 0.93 | (0.42) | 0.8 | (0.7) | 1.1 | (0.8) | ||||
| l. int | 16.4 | (14) | 19.0 | (21) | 30 | (19) | 33 | (22) | 4 | (2) | 3 | (2) |
| sm. int | 42.0 | (14) | 26.3 | (7) | 9.3 | (6) | 5.6 | (4.1) | 1.1 | (0.7) | 0.6 | (0.4) |
| kidney | 0.49 | (0.03) | 1.0 | (0.1) | 0.22 | (0.04) | 0.44 | (0.05) | 0.07 | (0.01) | 0.13 | (0.02) |
| urine | 20.3 | (6) | 45.7 | (14) | 38 | (3) | ||||||
| muscle | 0.03 | (0.01) | 0.15 | (0.04) | 0.04 | (0.07) | 0.2 | (0.4) | 0.01 | (0.02) | 0.02 | (0.05) |
| bone | 0.01 | (0.01) | 0.1 | (0.06) | 0.04 | (0.07) | 0.5 | (1) | 0 | (0.01) | 0.04 | (0.08) |
| pancreas | 3.0 | (1) | 8.7 | (2) | 0.47 | (0.10) | 1.1 | (0.15) | 0.11 | (0.02) | 0.27 | (0.06) |
| carcass | 10.2 | (2) | 3.7 | (1) | 1.5 | (0.9) | ||||||
| feces | 52.8 | (6.6) | ||||||||||
Values represent the mean (±SD), N = 5 of the %ID/organ or the %ID/g of tissue. Body weights of mice ranged from 21 to 28 g.
Total blood volume is estimated to be 6.5% of the total body weight.
The results provide important evidence that the PS2–BBN ligand is able to produce in vivo stable complexes with [99mTc(CO)3(OH2)3]+. An uptake of radioactivity was observed in the pancreas with activity reaching 3.06 ± 1.08% ID (8.69 ± 2.07%ID/g) 1 h post-injection, as compared to [99mTc(CO)3- PS2]+ complex 6, which had very limited uptake in the pancreas (0.37 ± 0.18% injected dose (1.13 ± 0.48% ID/g) 1 h post-injection (Figure 3)). This result demonstrates the ability of the [99mTc(CO)3PS2–BBN]+ complex to target GRP-receptor expressing cells in vivo. Acini cells in the pancreas express GRP receptors that are accessible to the bloodstream, and hence complex 11 is able to target the pancreas. The pancreas to muscle and pancreas to blood ratios (based on %ID per gram) reached a maximum of 58 and 13, respectively, at 1 h post-injection. The uptake of complex 11 in the pancreas is lower than values previously reported. For example, 99mTc(H2O)(CO)3-Dpr- (SSS)-BBN[7–14]NH2 showed an uptake of 23.3%ID/g and 10.2%ID/g in the pancreas and intestines, respectively, 1 h post-injection. 8a In a similar fashion, our previous investigations utilizing BBN conjugated N-donor ligands labeled with 99mTc- (CO)3 + have shown increased GRP receptor recognition under in vivo conditions.8 In the present study, a five carbon spacer (5-aminovaleric acid) was used between the radioisotope and bombesin peptide, and it is possible that the closer proximity of the radionuclide and binding region may be one of the reasons for decreased uptake in the GRP receptor-avid pancreas. However, it is very well accepted that the spacer length between the radionuclide and binding region plays a key role in determining the in vivo affinity.8 Our future studies will focus on two important aspects: (i) modulating the spacer length between the ligand and biomolecule and (ii) establishing that the accumulation of conjugate 11 is receptor-mediated by blocking with free peptides. The results presented in the paper establish that the “PS2” ligand system is novel and effective in stabilizing the [M(CO)3] core under in vitro and in vivo conditions.
Figure 3.
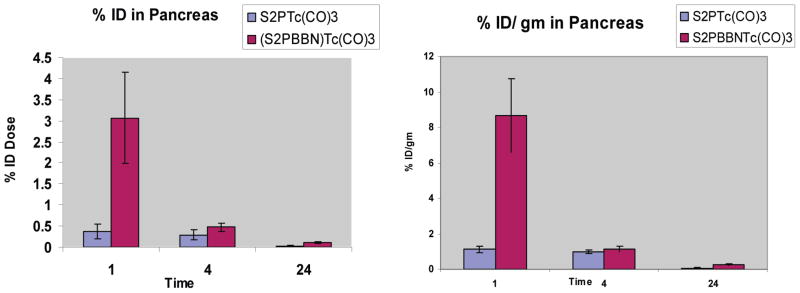
Comparison of pancreatic uptake between [99mTc(CO)3(PS2)]+ and [99mTc(CO)3(PS2–BBN)]+ in normal CF-1 mice.
CONCLUSION
Among a library of hard nitrogen base donors, π-acid phosphine, and soft sulfur donor ligands screened so far, the kinetic inertness and thermodynamic stability displayed by “facially” spanning tridentate phosphinodithio ether (PS2)-stabilized [Tc(CO)3(PS2)]+/[Re(CO)3(PS2)]+ complexes is an intriguing discovery. Indeed, several previous reports involving facially spanning 1,4,7-trithia cyclononane (9S3) complexes [M(9S3)2]+ (M = Tc, Re, Ru, Os) provide evidence for the susceptibility of these systems for ethene loss.16 While the incorporation of a phosphine center within the macrocyclic framework (e.g., 9PS2 ligands) improved the kinetic inertness of [Mo(9PS2)(CO)3]+ in comparison with [Mo(9S3)(CO)3]+, the reverse has been found to be true in a comparative study of [Fe(9S3)2]2+ and [Fe(9PS2)2]2+.16 In this context, results from our current studies have demonstrated that the compact PS2 ligand system possesses topology for facial tridentate coordination with Tc(CO)3 +/Re(CO)3 + cores, thereby producing five-membered [M(CO)3(PS2)]+ (M = Tc/Re) compounds that are both kinetically robust and in vivo stable.
The aforementioned results presented herein demonstrate an effective and practically useful chemical basis for the stabilization of [Tc(CO)3]/[Re(CO)3]+ cores and also for further conjugations of biologically useful metalated cores with tumor seeking peptides. Indeed, the results reported herein have wider implications in the areas of diagnostic and therapeutic biomedicine. The new approach of conjugating transition metals to biomolecules provides a straightforward route to the design and development of cancer diagnostic agents. Additionally, the incorporation of phosphines onto peptides (and proteins) will also help to engineer metal binding sites, which may eventually provide conformational integrity, biospecificity, and enhanced enzymatic activities. The combination of the dithioether with a phosphine moiety is unprecedented in technetium carbonyl chemistry. The in vivo stability of technetium complexes 6 and 11 imparted by the tripodal framework 4a has provided a new pharmacophore to deliver the diagnostic or the therapeutic dose at the tumor site for potential applications in the care and treatment of cancer patients.
EXPERIMENTAL SECTION
All reactions were carried out under nitrogen with standard Schlenk techniques. All chemicals were obtained from Aldrich Chemical Co. and were used without further purification. NMR spectra were recorded on a Bruker ARX-300 spectrometer using the specified solvent; 1H and 13C chemical shifts are reported in ppm, downfield from internal standard SiMe4, 31PNMR(121.5 MHz) with 85%H3PO4 as an external standard, and positive chemical shifts downfield of the standard. Combustion analysis was done by Oneida Research Services, Whitesboro, New York.
Synthesis of (EtO)2P(O)CH2CH2S(CH2)nCH2SH (1a, 1b)
To an ice cold (0 °C) solution of 1,2-ethanedithiol (2.80 mL, 33.38 mmol) in THF (5 mL) was added dropwise a 60% suspension of sodium hydride (0.212 g, 5.305 mmol) in THF, and the resulting solution was stirred for 10 min. Bromoethyldiethylphosphonate (1.00 g, 4.08 mmol) was added dropwise, and the stirring was continued for 2 h. Excess sodium hydride was quenched by the addition of 4 mL of water. The solution was extracted into ethylacetate (3 × 20 mL), and the organic layer was dried over anhydrous sodium sulfate. Solvent was removed in vacuo to afford (EtO)2P(O)CH2CH2SCH2CH2SH (1a) in 96% yield (1.02 g). 1HNMR (CDCl3, 300 MHz): δ 4.11 (m, 4H, P(O)CH2CH3), 2.78 (m, 6H, SCH2), 2.03 (m, 2H, PCH2), 1.72 (t, 1H, SH), 1.33 (t, 6H, P(O)CH2CH3). 13C {1H} NMR (CDCl3, 75 MHz): δ 61.70 (d, P(O)CH2CH3), 35.94 (s, SCH2CH2S), 26.71 (d, JP–C = 136.80 Hz, P-CH2), 24.71 (d, 2JP–C = 3.69 Hz, PCH2CH2S), 24.34 (S, CH2), 16.32 (d, P(O)CH2CH3). 31P{1H}NMR (CDCl3, 121 MHz): δ 25.30 (s).
Synthesis of H2PCH2CH2SCH2CH2SH (2)
To a stirring solution of (EtO)2P(O)CH2CH2SCH2CH2SH (1; 0.95 g, 3.6 mmol) in anhydrous diethyl ether (15 mL) was added dropwise a 1 M solution of lithium aluminum hydride (4.7 mL, 4.7 mmol) in diethyl ether, and the stirring continued for 60 min. Excess lithium aluminum hydride was destroyed by the careful addition of wet ether, and finally 4 mL of 5 N HCl was added to dissolve the precipitate. The organic layer was diluted with diethyl ether (50 mL), washed with brine (2 × 30 mL), and dried using anhydrous sodium sulfate. Solvent was removed under reduced pressure to afford pure product H2PCH2CH2SCH2CH2SH (2) in 90% yield (492 mg). 1H NMR (CDCl3, 300 MHz): δ 2.77 (dt, 2H, PH2, 1JP–H = 196.50 Hz, 3JH–H = 9.00 Hz), 2.73 (m, 6H, SCH2), 1.73–1.80 (m, 2H, P-CH2). 13C{1H}NMR (CDCl3, 75 MHz): δ 35.84 (s, SCH2), 34.97 (s, CH2), 24.61 (s, HSCH2), 14.83 (d, JP–C = 11. 45 Hz, P-CH2). 31P {1H} NMR (CDCl3, 121 MHz): δ −140.65 (s).
Synthesis of H2PCH2CH2SCH2CH2SCH3 (3)
To a suspension of sodium hydride (115 mg, 4.8 mmol) in anhydrous THF (3 mL) was added dropwise a solution of H2PCH2CH2SCH2CH2SH (2; 570.5 mg, 3.7 mmol) in THF (5 mL), and it was stirred for 20 min. To this suspension, methyl iodide (0.5 mL, 7.3 mmol) was added and stirred for 2 h. Excess sodium hydride was destroyed by slow addition of water (4 mL). The compound was extracted into ethyl acetate (3 × 20 mL), and the organic layer was dried using anhydrous sodium sulfate. Solvent was removed under reduced pressure to afford the pure product PS2H2 (3) in 60% yield (369 mg). 1H NMR (CDCl3, 300 MHz): δ 2.85 (dt, 2H, PH2, 1JP–H = 196.83 Hz, 3JH–H = 6.07 Hz), 2.74 (m, 6H, SCH2), 2.14 (s, 3H, SCH3) 1.79 (m, 2H, P-CH2). 13C {1H} NMR (CDCl3, 75 MHz): δ 35.14 (s, SCH2), 34.03 (s, CH2), 31.34(s, SCH2), 15.40 (s, SCH3), 14.77 (d, JP–C = 11.20 Hz, P-CH2). 31P{1H} NMR (CDCl3, 121 MHz): δ −141.13 (s).
Synthesis of Cl(HOCH2)3PCH2CH2SCH2CH2SCH3 (4a)
To a vigorously stirring suspension of H2PCH2CH2SCH2CH2SCH3 (3) (0.35 g, 2.10 mmol) in degassed ethanol (3 mL) was added a 37% w/w aqueous solution of formaldehyde (0.14 g, 4.90 mmol) and 2 N HCl (0.2 mL). The stirring was continued for 1 h and solvent removed under vacuum conditions to yield Cl(HOCH2)3PCH2CH2SCH2CH2SCH3 (4a) in 80% yield (0.50 g). 1H NMR (D2O, 300 MHz): δ 4.76 (s, 6H, PCH2O), 2.87–2.76 (m, 8H, CH2), 2.09 (s, 3H, S-CH3). 13C {1H} NMR (D2O, 75 MHz): δ 50.21 (d, JP–C = 53.74 Hz, P-CH2), 32.58 (s, SCH2), 30.25 (s,CH2), 22.74 (d, JP–C = 5.24 Hz, SCH2CH2P), 14.87 (d, JP–C = 38.19 Hz, SCH2CH2P), 13.85 (s, SCH3). 31P{1H} NMR (D2O, 121 MHz): δ 24.69 (s).
Synthesis of (HOCH2)2PCH2CH2SCH2CH2SCH3 (4b)
Triethyl amine (20.2 mg, 0.20 mmol) was added to a vigorously stirring solution of 4a (20 mg, 0.07 mmol) in degassed water (0.5 mL), and the resulting solution was stirred for 30 min. Solvent and excess triethylamine were removed in vacuo to obtain the crude product as a viscous oil. The crude product was purified on a C-18 reversed phase SepPak column, by elution with methanol–water (1:99) to obtain 13.5 mg of pure product 4b (85% yield) as a clear viscous oil. (M+H+) calcd., 229.0486; obsd., 229.0482. 1H NMR (D2O, 300 MHz): δ 3.92 (t, 4H, JP–H = 6 Hz, PCH2O), 2.74–2.66 (m, 6H, CH2), 2.00 (s, 3H, S-CH3), 1.77 (bm, 2H, PCH2CH2). 31P {1H} NMR (D2O, 121 MHz): δ −29.32 (s).
Synthesis of [Re(CO)3{(CH2OH)2PCH2CH2SCH2CH2SCH3}]Br (5)
[NEt4][ReBr3(CO)3] (84 mg, 0.109 mmol) was dissolved in distilled water (1.5 mL), and the solution was stirred for 5 min to quantitatively generate [Re(OH2)3(CO)3]Br. To this solution was added a solution of PS2 (4; 24.8 mg, 0.109 mmol)8 in 4 mL of water and 20 μL of triethylamine, and the resulting solution was stirred for 3 h. The progress of the reaction was monitored by 31P NMR spectroscopy, and after the consumption of phosphine was complete, stirring was stopped and the solvent was removed under reduced pressure. The remaining solid was washed thrice with diethyl ether (3 × 5 mL) and dried. The solid obtained was washed several times with dichloromethane (4 × 3 mL) to remove traces of tetraethylammonium bromide and triethylammonium hydrochloride. The solid obtained was dried under vacuum conditions and dissolved in methanol to obtain crystals suitable for the X-ray diffraction study. ESI-MS (m/z): [M+ – Br] calcd., 498.9; obsd., 499. 1H (D2O, 300 MHz, δ): 4.23 (s, 4H), 3.47–3.04 (m, 6H), 3.94–4.09 (m, 1H), 1.67–1.52 (m, 2H). 31P{1H}(D2O, 121 MHz, δ): 39.65. Analysis calculated for C10H17S2O5BrPRe: C, 22.22%; O, 3.56%. Found: 22.01%; 3.07%.
In Vitro Stability Studies of 5
In order to determine the in vitro stability of the rhenium S2P–rhenium complex 5, 1 mg of 5 was dissolved in saturated solutions of cysteine and human serum albumin (HSA) in water at 25 °C. Cysteine is a typical model chosen for these particular studies, as a number of potential metal-chelating thiol functionalities are present in vivo (e.g., serum proteins and glutathione), and HSA is a major constituent in the blood. 31P NMR spectroscopic data for aliquots of each sample were taken at different time intervals. The studies indicate the complex was stable even after 6 months of incubation.
Synthesis of [99mTc(CO)3(PS2)] + (6)
Complex 6 was prepared by adding a 200 μL solution of [99mTc(CO)3(OH2)3]+ (0.2–0.5 GBq) in saline to a solution of 50 μL of 4b (0.05 mg/mL = 1.6 × 10−4 M) and 100 μL of 1 M NaHCO3. The resulting solution was vortexed for 15 s and incubated for 30 min at RT to give the complex in >99% yield. The effective concentration of the PS2 ligand is 2.4 × 10−5 M. The mobile phase for the HPLC consisted of a gradient system with solvent A corresponding to water with 0.1% trifluoroacetic acid (TFA) and solvent B corresponding to acetonitrile with 0.1% TFA. The HPLC gradient started with 95% A/5% B followed by a linear gradient from 95% A/5% B to 20% A/80% B for 2–18 min. The gradient remained at 20% A/80% B for 6 min between 18 and 24 min. The flow rate was 1.5 mL/min and the chart speed 0.5 cm/min. The formation of the complex was analyzed by reversed phase HPLC utilizing a radiometric (γ) detector. The retention time for the complex was 9.45 min and corresponds to the rhenium complex, which has a retention time of 9.5 min.
pH Stability Studies of [99mTc(CO)3(PS2)] + (6)
The crude [99mTc(CO)3(PS2)]+ was peak purified on HPLC by collecting the fraction corresponding only to the complex. The acetonitrile in the solvent was removed by bubbling nitrogen gas for 1 h. The pH of the above solution was adjusted to different pH’s with either 0.1 M NaOH or 0.1M HCl. The percent radiochemical stability (% RCP) of the resulting complex was analyzed by HPLC at 1, 4 ,and 24 h post-complexation.
In Vitro Stability Studies of [99mTc(CO)3(PS2)] + (6)
The in vitro stability of complex [99mTc(CO)3(PS2)]+ (6) was measured as a function of time. The complex obtained was peak purified on HPLC by collecting the fraction corresponding only to the complex. The acetonitrile in the solvent was removed by bubbling nitrogen gas for 1 h. The purified complex was incubated with 0.2 M cysteine, 0.2 M histidine, and 2% HSA solution in saline at pH 7.4, and the radiochemical purity of the complex at different time points was analyzed by HPLC. No detectable decomposition of the chelate was observed under these conditions, indicating the high in vitro stability of the technetium complex 6.
In Vivo Stability Studies of [99mTc(CO)3(PS2)] + (6)
The complex obtained was peak purified on HPLC by collecting the fraction corresponding only to the complex. The acetonitrile in the solvent was removed by bubbling nitrogen gas for 1 h. The biodistribution studies of the complex [99mTc(CO)3(PS2)]+ were determined by injecting the peak purified complex in 4–6-week-old normal CF-1 mice (Charles River Laboratories, Wilmington, MA). The mice were injected via tail vein with 80–100 μL of the complex in a pH7 phosphate buffered saline containing 55–80 kBq of complex [99mTc(CO)3(PS2)]+. The animals were sacrificed 0.5, 1, 4, and 24 h post-injection. The tissues and organs were excised from animals (N = 5 per group), rinsed in saline, weighed, and counted in a NaI(Tl) well counter. Percent injected dose (%ID) and %ID/g were calculated. Between 0.7 and 1.0 mL of blood was withdrawn from the heart via a cardiac puncture immediately after sacrifice and counted. It was assumed that the whole blood constituted 6.5% of the total body weight.
Synthesis of H2PCH2CH2SCH2CH2SCH2COOtBu (7)
To a suspension of sodium hydride (19 mg, 0.8 mmol) in anhydrous THF (3 mL) was added 2 (100 mg, 0.65 mmol) very slowly, and it was stirred for 30 min. To this suspension, t-butylbromoacetate (128 mg, 0.65 mmol) in anhydrous THF (2 mL) was added dropwise and stirred for 4 h. Excess sodium hydride was destroyed by the slow addition of water (3–4 mL). The compound was extracted into diethyl ether (4 × 20 mL), and the organic layer was dried with sodium sulfate. Solvent was removed under vacuum conditions to yield the crude product. The crude reaction mixture was purified on a silica gel column by eluting the product with a 1% ethylacetate/hexane solvent combination to yield the pure product in 80% yield (172 mg). 1H NMR (CDCl3, 300 MHz): δ 3.08 (s, 2H, SCH2CO), 2.67 (dt, 2H, PH2, 1JP–H = 195.00 Hz, 3JH–H = 6.01 Hz), 2.78–2.63 (m, 6H, SCH2), 1.72–1.70 (m, 2H, P-CH2), 1.41 (s, 9H, CCH3). 13C{1H}NMR (CDCl3, 75 MHz): δ 169.30 (s, COOtBu), 81.32 (S, COOC(CH3)3), 34.92 (s, SCH2CO), 34.56 (s, SCH2), 32.18 (s, SCH2), 27.80 (s, SCH2), 14.71 (d, JP–C = 11.25 Hz, P-CH2). 31P{1H} NMR (CDCl3, 121 MHz): δ −140.92 (s).
Synthesis of H2PCH2CH2SCH2CH2SCH2COOH (8)
Dry HCl gas, generated in situ by the dropwise addition of sulfuric acid to sodium chloride, was bubbled through a solution of H2PCH2CH2SCH2CH2- SCH2COOtBu (7; 120 mg, 0.44 mmol) in anhydrous methylene chloride for 30 min. The solution was stirred for 3 h and the solvent removed under reduced pressure to quantitatively yield H2PS2COOH (H2PCH2CH2SCH2CH2SCH2COOH) (8; 93 mg, 0.43 mmol). 1H NMR (CDCl3, 300 MHz): δ 3.31 (s, 2H, SCH2CO), 2.77 (dt, 2H, PH2, 1JP–H = 195.00 Hz, 3JH–H = 9.01 Hz), 2.92–2.43 (m, 6H, SCH2), 1.89–1.41 (m, 2H, P-CH2). 13C {1H} NMR (CDCl3, 75 MHz): δ 175.62 (s, COOH), 35.06 (s, SCH2CO), 32.45 (s, SCH2), 31.14 (s, SCH2), 27.84 (s, SCH2), 14.71 (d, JP–C = 11.25 Hz, P-CH2). 31P {1H} NMR (CDCl3, 121 MHz): δ −141.26 (s).
Synthesis of H2PS2-5-Ava-BBN[7–14]NH2(H2PS2BBN) (9)
A solution of 5-Ava-BBN[7–14]NH2 (mol.wt.: 1024.21; 12 mg; 12 μmol) in DMF (0.5 mL) was added to a reaction mixture containing H2PS2- COOH(8; 3.8 mg; 18 μmol), HBTU(mol. wt.: 379.25; 6.8 mg; 18 μmol), and triethylamine (30 μL) in DMF (0.5 mL). The reaction mixture was stirred for 30 min at room temperature. The resultant phosphine–BBN conjugate was purified by reversed-phase HPLC and analyzed by 31P NMR spectroscopy and electrospray mass spectrometry. The pure product (5.6 mg; 4.6 μmol; % yield ~ 38%) was obtained as a white powder. 31P NMR (D2O, 121 MHz, δ): δ −143.9 (s). ESI-MS (m/z) calcd., 1232.5 (M+); found, 1233.5 (M+ + H+).
Synthesis of S2P-5-Ava-BBN[7–14]NH2(PS2–BBN) (10)
To a solution of phosphine PH2S2-5-Ava-BBN[7–14]NH2 (9; 5 mg, 4 μmol) in ethanol (500 μL) and DMF (500 μL) were added 5 N hydrochloric acid (30 μL) and 37% aqueous formaldehyde (30 μL). The reaction mixture was stirred at room temperature (25 °C) for 30 min. The resultant phosphine–BBN phosphonium salt conjugate S2P-5-Ava- BBN[7–14]NH2(PS2 –BBN) (10) was purified by reversed-phase HPLC and analyzed by 31P NMR spectroscopy and electrospray mass spectrometry. The pure product (~55%) was obtained as a white powder. 31P NMR (D2O): δ 24.2 (s). ESI-MS (m/z) calcd., 1323.6 (M+ − Cl); found, 1323.0.
Synthesis of [99mTc(CO)3(PS2–BBN)] + (11)
The Tc-99m complex was prepared by adding a 250 μL solution of [99mTc(CO)3(OH2)3]+ (0.3–0.9 GBq) in saline to a solution of 50μL of PS2–BBN(0.26 mg/mL = 1.9 × 10−4 M) and 50 μL of 1 M NaHCO3. The resulting solution was vortexed for 15 s and incubated for 30 min at RT to give the complex in >99% yield. The mobile phase for the HPLC consisted of a gradient system with solvent A corresponding to water with 0.1% trifluoroacetic acid (TFA) and solvent B corresponding to acetonitrile with 0.1% TFA. The HPLC gradient started with 95% A/5% B followed by a linear gradient from 95% A/5% B to 30% A/70% B for 2–25 min. The gradient changed from 30% A/70% B to 5% A/95% B over a 5 min period between 25 and 30 min. The flow rate was 1.5 mL/min and the chart speed 0.5 cm/min. Retention time for the complex was 17.33 min.
pH Studies of [99mTc(CO)3(PS2–BBN)] + (11)
The complex obtained was peak purified on HPLC by collecting the fraction corresponding only to the complex. The acetonitrile in the solvent was removed under reduced pressure by applying a vacuum for 1 h. The pH of the above solution was adjusted to different pH’s with either 0.1M NaOH or 0.1 M HCl. The percent radiochemical stability of the resulting complex was analyzed by HPLC at 1, 4, and 24 h post-complexation.
In Vitro Studies of [99mTc(CO)3(PS2–BBN)] + (11)
The in vitro stability of the complex [99mTc(CO)3(PS2–BBN)]+ was measured as a function of time. The complex obtained was peak purified on HPLC by collecting the fraction corresponding only to the complex. The acetonitrile in the solvent was removed under reduced pressure by applying a vacuum for 1 h. The purified complex was incubated with 0.2 Mcysteine, 0.2 M histidine, or 2% HSA solution in saline at pH 7.4, and the radiochemical purity of the complex at 25 °C was analyzed at different time points using radiometric HPLC.
In Vivo Studies of [99mTc(CO)3(PS2–BBN)] + (11)
Animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee. The biodistribution studies of the complex [99mTc(CO)3(PS2 –BBN)]+ were determined by injecting complex 11 in normal 4–6-week-old CF-1 female mice (Charles River Laboratories, Wilmington, MA). The mice were injected via tail vein with 80–100 μL of the complex in pH 7 phosphate buffered saline containing 55–80 kBq of complex [99mTc(CO)3(PS2 –BBN)]+. The animals were sacrificed at 1, 4, and 24 h post-injection. The tissues and organs were excised from the animals (N = 5 per group), rinsed with saline, weighed, and counted in a NaI(Tl) well counter. The percent injected dose (%ID) and %ID/g were calculated. Between 0.7 and 1.0 mL of blood was withdrawn from the heart via cardiac puncture immediately after sacrifice and counted. It was assumed that the whole blood constituted 6.5% of the total body weight.
Crystallographic Data Collection and Refinement of Structure for 5
A suitable crystal was chosen and mounted on a glass fiber with epoxy resin. The crystal data and refinement results are given in the Supporting Information. Data reduction and processing followed routine procedures. Structures were solved by direct methods and refined on Fo 2. Absorption corrections were done by semiempirical equivalents. Crystal structure data for 5: triclinic, space group P1̄, a = 6.5967(4), b = 8.9236(5), c = 14.1771(8) Å, α = 89.0590(10), β = 79.3080(10), γ = 87.8210(10)°, V = 819.43(8)Å3, Z = 2, ρcalcd = 2.344 Mg cm−3, 2θmax = 54.2°, MoKα radiation (λ = 0.71073 Å), T = 173 K. A colorless crystal with dimensions 0.45 × 0.15 × 0.15 mm3 was grown by slow evaporation of an aqueous solution of 5. No. reflns = 5882 (3538 > 2σ(I)), refinement of F2, R1 = 0.0548, wR2 = 0.1508, GOF = 1.068.
Supplementary Material
Acknowledgments
This work was supported by funds from the U.S. Department of Energy (DE-FG02-01ER63192), Department of Radiology and the University of Missouri Research Reactor.
Footnotes
Supporting Information. 13C NMR data and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Heeg MJ, Jurisson S. Acc Chem Res. 1999;32:1053–1060. [Google Scholar]; (b) Liu S. Chem Soc Rev. 2004;33:445–461. doi: 10.1039/b309961j. [DOI] [PubMed] [Google Scholar]; (c) Tweedle MF. Acc Chem Res. 2009;42:958–968. doi: 10.1021/ar800215p. [DOI] [PubMed] [Google Scholar]; (d) Katayev EA, Kolesnikov GV, Sessler JL. Chem Soc Rev. 2009;38:1572–1586. doi: 10.1039/b806468g. [DOI] [PubMed] [Google Scholar]
- 2.(a) Katti KV, Gali H, Smith CJ, Berning DE. Acc Chem Res. 1999;32:9–17. [Google Scholar]; (b) Kannan R, Nagavarakishore P, Volkert WA, Barnes C, Jurisson S, Katti KV. J Am Chem Soc. 2002;124:7276–7277. doi: 10.1021/ja025987e. [DOI] [PubMed] [Google Scholar]; (c) Kothari KK, Gali H, Prabhu KR, Pillarsetty N, Owen NK, Katti KV, Hoffman TJ, Volkert WA. Nucl Med Biol. 2002;29:83–89. doi: 10.1016/s0969-8051(01)00280-3. [DOI] [PubMed] [Google Scholar]; (d) Gali H, Hoffman TJ, Sieckman GL, Owen NK, Katti KV, Volkert WA. Bioconjugate Chem. 2001;12:354–363. doi: 10.1021/bc000077c. [DOI] [PubMed] [Google Scholar]; (e) Pillarsetty N, Katti KK, Hoffman TJ, Volkert WA, Katti KK, Kamei H, Koide T. J Med Chem. 2003;46:1130–1132. doi: 10.1021/jm025615g. [DOI] [PubMed] [Google Scholar]; (f) Deutsch E. Radiochim Acta. 1993;63:195–7. [Google Scholar]
- 3.(a) Fernandes C, Correia JDG, Gano L, Santos I, Seifert S, Syhre R, Bergmann R, Spies H. Bioconjugate Chem. 2005;16:660–668. doi: 10.1021/bc049718k. [DOI] [PubMed] [Google Scholar]; (b) Schiller E, Seifert S, Tisato F, Refosco F, Kraus W, Spies H, Pietzsch HJ. Bioconjugate Chem. 2005;16:634–643. doi: 10.1021/bc049745a. [DOI] [PubMed] [Google Scholar]; (c) Karra SR, Schibli R, Gali H, Katti KV, Hoffman TJ, Higginbotham C, Sieckman GL, Volkert WA. Bioconjugate Chem. 1999;10:254–260. doi: 10.1021/bc980096a. [DOI] [PubMed] [Google Scholar]; (d) Pillasetty N, Kannan R, Barnes CL, Katti KV. J Am Chem Soc. 2005;127:331–336. doi: 10.1021/ja047238y. [DOI] [PubMed] [Google Scholar]; (e) Kannan RK, Katti KK, Barbour LJ, Pillarsetty N, Barnes CL, Katti KV. J Am Chem Soc. 2003;125:6955–6961. doi: 10.1021/ja034682c. [DOI] [PubMed] [Google Scholar]; (f) Prabhu KR, Pillarsetty N, Gali H, Katti KV. J Am Chem Soc. 2000;122:1554–1555. [Google Scholar]; (g) Gilbertson SR, Collibee SE, Agarkov A. J Am Chem Soc. 2000;122:6522–6523. [Google Scholar]
- 4.(a) Kereiakes JG. Biophysical aspects:medical use of technetium-99m. American Institute of Physics; Woodbury, NY: 1992. [Google Scholar]; (b) Helm L. Coord Chem Rev. 2008;252:2346–2361. [Google Scholar]; (c) Zolle I, editor. Technetium-99 Radiopharmaceuticals: Preparation and Quality Control in Nuclear Medicine. 1. Springer; New York, NY: 2006. [Google Scholar]; (d) International Atomic Energy Agency. Trends in radiopharmaceuticals (ISTR-2005). proceedings of an international symposium organized by the International Atomic Energy Agency; Vienna. November 14–18, 2005; Vienna, Austria: International Atomic Energy Agency; 2007. [Google Scholar]; (e) Nicolini M, Mazzi U. Technetium, Rhenium, And Other Metals in Chemistry and Nuclear Medicine, 6. SGEd.iali; Padova, Italy: 2002. [Google Scholar]; (f) Schwochau K. Technetium: Chemistry and Radiopharmaceutical Applications. Wiley-VCH; Weinheim, Germany: 2000. [Google Scholar]
- 5.(a) Xavier C, Giannini C, Dall’Angelo S, Gano L, Maiorana S, Alberto R, Santos I. J Biol Inorg Chem. 2008;13:1335. doi: 10.1007/s00775-008-0419-y. [DOI] [PubMed] [Google Scholar]; (b) Maria L, Paulo A, Santos IC, Santos I, Kurz P, Spingler B, Alberto R. J Am Chem Soc. 2006;128:14590. doi: 10.1021/ja0644226. [DOI] [PubMed] [Google Scholar]; (c) Hafliger P, Agorastos N, Spingler B, Georgiev O, Viola G, Alberto R. Chembiochem. 2005;6:414. doi: 10.1002/cbic.200400210. [DOI] [PubMed] [Google Scholar]; (d) Rattat D, Schubiger PA, Berke HG, Schmalle H, Alberto R. Cancer Biother Radiopharm. 2001;16:339. doi: 10.1089/108497801753131426. [DOI] [PubMed] [Google Scholar]; (e) Amann A, Decristoforo C, Ott I, Wenger M, Bader D, Alberto R, Putz G. Nucl Med Biol. 2001;28:243. doi: 10.1016/s0969-8051(01)00192-5. [DOI] [PubMed] [Google Scholar]; (f) Schibli R, La Bella R, Alberto R, Garcia-Garayoa E, Ortner K, Abram U, Schubiger PA. Bioconjugate Chem. 2000;11:345. doi: 10.1021/bc990127h. [DOI] [PubMed] [Google Scholar]; (g) Pietzsch HJ, Gupta A, Reisgys M, Drews A, Seifert S, Syhre R, Spies H, Alberto R, Abram U, Schubiger PA, Johannsen B. Bioconjugate Chem. 2000;11:414. doi: 10.1021/bc990162o. [DOI] [PubMed] [Google Scholar]; (h) Waibel R, Alberto R, Willuda J, Finnern R, Schibli R, Stichelberger A, Egli A, Abram U, Mach JP, Pluckthun A, Schubiger PA. Nat Biotechnol. 1999;17:897. doi: 10.1038/12890. [DOI] [PubMed] [Google Scholar]
- 6.(a) Alberto R. Eur J Nucl Med Mol Imaging. 2003;30:1299. doi: 10.1007/s00259-003-1292-0. [DOI] [PubMed] [Google Scholar]; (b) Alberto R, Ortner K, Wheatley N, Schibli R, Schubiger AP. J Am Chem Soc. 2001;123:3135. doi: 10.1021/ja003932b. [DOI] [PubMed] [Google Scholar]; (c) Schibli R, La Bella R, Alberto R, Garcia-Garayoa E, Ortner K, Abram U, Schubiger PA. Bioconjugate Chem. 2000;11:345. doi: 10.1021/bc990127h. [DOI] [PubMed] [Google Scholar]; (d) Pietzsch HJ, Gupta A, Reisgys M, Drews A, Seifert S, Syhre R, Spies H, Alberto R, Abram U, Schubiger PA, Johannsen B. Bioconjugate Chem. 2000;11:414. doi: 10.1021/bc990162o. [DOI] [PubMed] [Google Scholar]; (e) Egli A, Alberto R, Tannahill L, Schibli R, Abram U, Schaffland A, Waibel R, Tourwe D, Jeannin L, Iterbeke K, Schubiger PA. J Nucl Med. 1999;40:1913. [PubMed] [Google Scholar]
- 7.(a) Liu Y, Oliveira BL, Correia JDG, Santos IC, Santos I, Spingler B, Alberto R. Org Biomol Chem. 2010;8:2829. doi: 10.1039/c002796k. [DOI] [PubMed] [Google Scholar]; (b) Xavier C, Pak JK, Santos I, Alberto R. J Organomet Chem. 2007;692:1332. [Google Scholar]; (c) Maria L, Paulo A, Santos IC, Santos I, Kurz P, Spingler B, Alberto R. J Am Chem Soc. 2006;128:14590. doi: 10.1021/ja0644226. [DOI] [PubMed] [Google Scholar]; (d) Mundwiler S, Kuendig M, Ortner K, Alberto R. Dalton Trans. 2004:1320. doi: 10.1039/b400220b. [DOI] [PubMed] [Google Scholar]; (e) Kurz P, Spingler B, Fox T, Alberto R. Inorg Chem. 2004;43:3789. doi: 10.1021/ic049774x. [DOI] [PubMed] [Google Scholar]; (f) Kunze S, Zobi F, Kurz P, Spingler B, Alberto R. Angew Chem, Int Ed. 2004;43:5025. doi: 10.1002/anie.200460923. [DOI] [PubMed] [Google Scholar]
- 8.(a) Smith CJ, Sieckman GL, Owen NK, Hayes DL, Mazuru DG, Kannan R, Volkert WA, Hoffman TJ. Cancer Res. 2003;63:4082. [PubMed] [Google Scholar]; (b) Retzloff LB, Heinzke L, Figureoa SD, Sublett SV, Ma L, Sieckman GL, Rold TL, Santos I, Hoffman TJ, Smith CJ. Anticancer Res. 2010;30:19. [PubMed] [Google Scholar]; (c) Lane SR, Veerendra B, Rold TL, Sieckman GL, Hoffman TJ, Jurisson SS, Smith CJ. Nucl Med Biol. 2008;35:263. doi: 10.1016/j.nucmedbio.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Alves S, Correia JDG, Gano L, Rold TL, Prasanphanich A, Haubner R, Rupprich M, Alberto R, Decristoforo C, Santos I, Smith CJ. Bioconjugate Chem. 2007;18:530. doi: 10.1021/bc060234t. [DOI] [PubMed] [Google Scholar]; (e) Alves S, Correia JDG, Santos I, Veerendra B, Sieckman GL, Hoffman TJ, Rold TL, Figueroa SD, Retzloff L, McCrate J, Prasanphanich A, Smith CJ. Nucl Med Biol. 2006;33:625. doi: 10.1016/j.nucmedbio.2006.03.007. [DOI] [PubMed] [Google Scholar]; (f) Alves S, Paulo A, Correia JDG, Gano L, Smith CJ, Hoffman TJ, Santos I. Bioconjugate Chem. 2005;16:438. doi: 10.1021/bc0497968. [DOI] [PubMed] [Google Scholar]
- 9.(a) Perera T, Marzilli PA, Fronczek FR, Marzilli LG. Inorg Chem. 2010;49:2123. doi: 10.1021/ic901705x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Perera T, Fronczek FR, Marzilli PA, Marzilli LG. Inorg Chem. 2010;49:7035. doi: 10.1021/ic100714m. [DOI] [PubMed] [Google Scholar]; (c) Lipowska M, He H, Xu X, Taylor AT, Marzilli PA, Marzilli LG. Inorg Chem. 2010;49:3141. doi: 10.1021/ic9017568. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Christoforou AM, Marzilli PA, Fronczek FR, Marzilli LG. Inorg Chem. 2007;46:11173. doi: 10.1021/ic701576u. [DOI] [PubMed] [Google Scholar]; (e) Christoforou AM, Fronczek FR, Marzilli PA, Marzilli LG. Inorg Chem. 2007;46:6942. doi: 10.1021/ic700594a. [DOI] [PubMed] [Google Scholar]
- 10.(a) Sagnou M, Benaki D, Triantis C, Tsotakos T, Psycharis V, Raptopoulou CP, Pirmettis I, Papadopoulos M, Pelecanou M. Inorg Chem. 2011;50:1295. doi: 10.1021/ic102228u. [DOI] [PubMed] [Google Scholar]; (b) Sagnou M, Tsoukalas C, Triantis C, Raptopoulou CP, Terzis A, Pirmettis I, Pelecanou M, Papadopoulos M. Inorg Chim Acta. 2010;363:1649. [Google Scholar]
- 11.(a) Chanda N, Shukla R, Zambre A, Mekapothula S, Kulkarni RR, Katti K, Bhattacharyya K, Fent GM, Casteel SW, Boote EJ, Viator JA, Upendran A, Kannan R, Katti KV. Pharm Res. 2011;28:279. doi: 10.1007/s11095-010-0276-6. [DOI] [PubMed] [Google Scholar]; (b) Cagnolini A, Ballard B, Engelbrecht HP, Rold TL, Barnes C, Cutler C, Hoffman TJ, Kannan R, Katti K, Jurisson SS. Nucl Med Biol. 2011;38:63. doi: 10.1016/j.nucmedbio.2010.06.013. [DOI] [PubMed] [Google Scholar]; (c) Chanda N, Kattumuri V, Shukla R, Zambre A, Katti K, Upendran A, Kulkarni RR, Kan P, Fent GM, Casteel SW, Smith CJ, Boote E, Robertson JD, Cutler C, Lever JR, Katti K, Kannan R. Proc Natl Acad Sci USA. 2010;107:8760. doi: 10.1073/pnas.1002143107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chanda N, Shukla R, Katti KV, Kannan R. Nano Lett. 2009;9:1798. doi: 10.1021/nl8037147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Anastasi A, Erspamer V, Bucci M. Experientia. 1971;27:166–167. doi: 10.1007/BF02145873. [DOI] [PubMed] [Google Scholar]; (b) Erspamer GF, Severini C, Erspamer V, Melchiorri P, Delle Fave G, Nakajima T. Regul Pept. 1988;21:1–11. doi: 10.1016/0167-0115(88)90085-7. [DOI] [PubMed] [Google Scholar]
- 13.(a) Qu X, Xiao D, Weber HC. Curr Opin Endocrinol Diabetes. 2003;10:60. [Google Scholar]; (b) Xiao D, Qu X, Weber HC. Regul Pept. 2002;109:141. doi: 10.1016/s0167-0115(02)00197-0. [DOI] [PubMed] [Google Scholar]; (c) Houben H, Denef C. Front Horm Res. 1991;19:176. [Google Scholar]
- 14.(a) Spindel ER. Handbook of Biologically Active Peptides. Elsevier; New York: 2006. p. 277. [Google Scholar]; (b) Flores DG, Lenz G, Roesler R, Schwartsmann G. Cancer Ther. 2009;7:332. [Google Scholar]; (c) Li X, Lv Y, Yuan A, Li Z. J Cancer Res Clin Oncol. 2010;136:483. doi: 10.1007/s00432-010-0766-2. [DOI] [PubMed] [Google Scholar]; (d) Hohla F, Schally AV. Cell Cycle. 2010;9:1738. doi: 10.4161/cc.9.9.11347. [DOI] [PubMed] [Google Scholar]
- 15.(a) Bartholdi MF, Wu JM, Pu H, Troncoso P, Eden PA, Feldman RI. Int J Cancer. 1998;79:82–90. doi: 10.1002/(sici)1097-0215(19980220)79:1<82::aid-ijc16>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]; (b) Markwalder R, Reubi JC. Cancer Res. 1999;59:1152–1159. [PubMed] [Google Scholar]; (c) Sun BD, Halmos G, Schally AV, Wang XE, Martinez M. Prostate. 2000;42:295–303. doi: 10.1002/(sici)1097-0045(20000301)42:4<295::aid-pros7>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; (d) Halmos Wittliff JL, Schally AV. Cancer Res. 1995;55:280–287. [PubMed] [Google Scholar]; (e) Gugger M, Reubi JC. Am J Pathol. 1999;155:2067–2076. doi: 10.1016/S0002-9440(10)65525-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Reubi JC, Korner M, Waser B, Mazzucchelli L, Guillou L. Eur J Nucl Med Mol Imaging. 2004;31:803. doi: 10.1007/s00259-004-1476-2. [DOI] [PubMed] [Google Scholar]; (g) Reubi JC, Waser B. Eur J Nucl Med Mol Imaging. 2003;30:781–793. doi: 10.1007/s00259-003-1184-3. [DOI] [PubMed] [Google Scholar]
- 16.(a) Sowrey FE, Blower PJ, Jeffery C, MacLean EJ, Went MJ. Inorg Chem Commun. 2002;5:832–836. [Google Scholar]; (b) Mullen GED, Blower PJ, Price DJ, Powell AK, Howard MJ, Went MJ. Inorg Chem. 2000;39:4093–4098. doi: 10.1021/ic991240m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.