

Abstract
Pd-catalyzed β-C–H functionalizations of carboxylic acid derivatives using an auxiliary as a directing group have been extensively explored in the past decade. In comparison to the most widely used auxiliaries in asymmetric synthesis, the simplicity and practicality of the auxiliaries developed for C–H activation remains to be improved. We previously developed a simple N-methoxyamide auxiliary to direct β-C–H activation, albeit this system was not compatible with carboxylic acids containing α-hydrogen atoms. Herein we report the development of a pyridine-type ligand that overcomes this limitation of the N-methoxyamide auxiliary, leading to a significant improvement of β-arylation of carboxylic acid derivatives, especially α-amino acids. The arylation using this practical auxiliary is applied to the gram-scale syntheses of unnatural amino acids, bioactive molecules and chiral bis(oxazoline) ligands.
1. Background
Aliphatic acids belong to an important class of useful building blocks due to their availability and the diverse reactivity of the carboxyl group. Among many methods for the preparation of α- or β-substituted carboxylic acids, α-enolate chemistry using chiral auxiliaries1 and asymmetric conjugate additions2 are the most powerful tools (Eqs 1–2). To seek an alternative synthetic disconnection, we initiated a research program centered on the β-C–H activation and subsequent carbon–carbon bond and carbon–heteroatom bond forming reactions in 2002 (Eq 3). Our early studies employed an oxazoline auxiliary as a directing group3 to investigate the reactivity and mechanism of β-C–H insertion by Pd(II) (Figure 1). We utilized the stereochemistry obtained in the C–H insertion step to deduce the pre-transition state structure of directed C–H insertions using chiral oxazoline auxiliaries. With hindsight and recent in-depth computational and kinetic studies,4 the primitive, but important insights we obtained from these studies regarding the conformation and structure of the C–H insertion precursors paved the way for our subsequent design of more efficient auxiliaries (Figure 1). In the past decade, while Daugulis’ bidentate 8-aminoquinoline auxiliary has emerged as a powerful directing group,5 we have focused on the development of mono-dendate simple amide auxiliaries, hoping to achieve ligand-accelerated and -controlled β-C–H functionalization reactions.
 |
(1) |
 |
(2) |
 |
(3) |
Figure 1.

Advantages and disadvantages of different directing groups developed in our laboratory
Due to the moderate reactivity of sodium or potassium carboxylates in β-C–H arylation,6 we developed an N-methoxyamide auxiliary to mimic the carboxylate while allowing improved coordination with Pd(II).7 The simple rationale behind this design was to best mimic the conformation of the coordination structure of Pd(II) with carboxylates while at the same time slightly increase the binding strength. This new auxiliary (CONHOMe) displayed excellent efficiency in directing β-C–H activation (Figure 1). For example, β-arylation of the amide derived from pivalic acid with Ph–I using this auxiliary proceeds at room temperature. We have also successfully exploited this reactivity to accomplish an unprecedented coupling of β-C–H bonds with alkyl boronic acids.7a Numerous applications of this powerful auxiliary in directed C(sp2)–H activation have also been reported with Pd(II), Rh(III), and Ru(II) catalysts.8 Unfortunately, C(sp3)–H activation of aliphatic acids using this auxiliary has been limited to substrates containing α-quaternary centers under current conditions. Apart from the known Thorpe-Ingold effect in cyclopalladation, we suspected that the acidic α-hydrogen of aliphatic acid substrates could be responsible for the lack of reactivity. This reasoning has led us to develop another acidic amide auxiliary (CONHArF, ArF = p-CF3C6F4) that is compatible with aliphatic acid substrates containing α-hydrogen atoms (Figure 1).9 Despite the broad utility of this new directing group, the simplicity of CONHOMe10 in terms of installation and removal prompted us to develop new conditions that may overcome the limitation of this potentially broadly useful auxiliary. Our recent collaboration with Bristol-Myers Squibb to establish a robust and scalable method for the preparation of a wide range of unnatural amino acids through C–H functionalization of readily available amino acids such as alanine provided a further incentive for this endeavor.
Herein we report the development of pyridine-type ligands that promote selective mono- and di-β-arylation of a broad range of carboxylic acids using a simple N-methoxyamide auxiliary as the directing group. 2-Picoline ligand (L7) promotes the selective mono-arylation of primary C(sp3)–H bonds and 2,6-lutidine ligand (L13) enables the subsequent arylation of secondary C(sp3)–H bonds in one pot. Sequential arylation of alanine derivatives with two different aryl iodides using these ligands enables the introduction of two distinct aryl groups to produce a variety of β-Ar-β-Ar′-α-amino acids with excellent levels of diastereoselectivity. Arylation of the N-methoxyamide derived from alanine with a variety of heterocyclic aryl iodides on gram scales to make various unnatural amino acids was also demonstrated. These unnatural amino acid intermediates were further transformed to drug molecules such as a human kynurenine aminotransferase (KAT) II inhibitor11 and Doxanthrine,12 as well as chiral hydroxamic acid ligands13 and a variety of new chiral pyridine-2,6-bis(oxazolines) (PyBOX) ligands14 (Figure 2).
Figure 2.
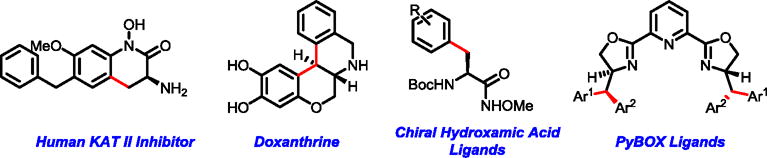
Synthetic applications
2. Results and Discussion
2.1. β-Mono-arylation directed by N-methoxyamide
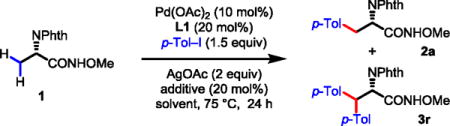
β-C–H functionalizations of amino acid derivatives using various auxiliaries have been extensively studied since the first report from the Corey group.15,16 It was established in this early study that the use of the phthalimide protecting group was crucial for achieving β-C–H activation.15a Recently, we have discovered that pyridine- and quinoline-based ligands promote activation of the C(sp3)–H bonds in alanine using CONHArF (ArF = p-CF3C6F4) auxiliary as the directing group.17 While the precise mechanistic origin of the ligand effects remains to be elucidated, a recent computational study suggests that the ligand is involved in every step of the catalytic cycle including the C–H activation step.18 This new development encouraged us to revisit whether a simpler N-methoxyamide auxiliary, with the assistance of a ligand, can accommodate substrates derived from carboxylic acids containing α-hydrogen atoms. Thus, phthaloyl alanine amide 1 was reacted with 1.5 equiv. of p-Tol–I using Pd(OAc)2 and ligand L1 under various conditions. We found that the mono-arylation proceeded under the conditions shown in Scheme 1 to give the arylated products as a mixture of amide 2a and the corresponding ester in 45% yield. A substantial amount of the starting material was converted to corresponding unreactive ester. The conversion of the N-methoxyamide to the corresponding ester via a radical process is known to be promoted by silver salts.10 A control experiment showed that ligand L1 was required for the formation of the arylated product.
Scheme 1.
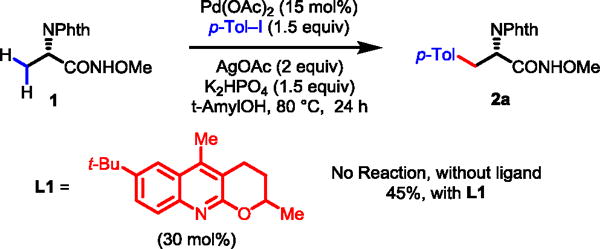
Preliminary Discovery for Ligand-Promoted C–H Arylation
Prior to undertaking further ligand screening, we needed to identify conditions under which the known decomposition of the N-methoxyamide to the unreactive ester via a radical process10a–c was minimized (Table 1). The use of (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) to inhibit the radical process led to a slight drop in product yield (Table 1, entry 2). The reaction did not proceed at a lower temperature of 60 °C (Table 1, entry 3). Switching to a range of commonly used solvents did not reduce the decomposition or improve the yields (Table 1, entries 4–8). We were pleased to find that the decomposition of the N-methoxyamide was effectively prevented by using acidic solvents, such as 2,2,2-trifluoroethanol (CF3CH2OH) or hexafluoro-2-propanol (HFIP) (Table 1, entries 9–10). Arylation proceeded in HFIP to give the mono-arylated product in 76% yield and the di-arylated product in 10% yield (Table 1, entry 10). Running the reaction in 1,2-dichloroethene (DCE) in the presence of 20 mol% trifluoroacetic acid (TFA) also significantly improved the yield to 75% (Table 1, entry 11).
Table 1.
Evaluation of Reaction Conditionsa
| |||
|---|---|---|---|
|
| |||
| Entry | Solvent | Additive | % Yield of 2ab |
| 1 | t-AmylOH | no | 45 |
| 2 | t-AmylOH | TEMPO | 40 |
| 3c | t-AmylOH | no | N.R. |
| 4 | Toluene | no | 30 |
| 5 | CH3CN | no | 35 |
| 6 | 1,4-Dioxane | no | 50 |
| 7 | DCE | no | 43 |
| 8 | DMF | no | 40 |
| 9 | CF3CH2OH | no | 60 |
| 10 | HFIP | no | 76(86)d |
| 11 | DCE | TFA | 75 |
Conditions: Substrate 1 (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), p-Tol–I (0.15 mmol), Ligand L1 (20 mol%), solvent (1.0 mL), 75 °C, 24 h.
Determined by 1HNMR analysis of the crude product using CH2Br2 as an internal standard, and the yield is based on the amide and ester.
Reaction run at 60 °C.
Combined 76% mono- and 10% di-arylated products determined by crude 1HNMR.
While further ligand screening using DCE/TFA solvent system did not provide noticeable improvement, dramatic ligand effects were observed for this reaction in HFIP (Table 2). A variety of quinoline-based ligands (L2–L4) afforded moderate to good yields (up to 83%). Further optimizations of quinoline-based ligands were not fruitful. We found a wide range of pyridines as suitable ligands for this reaction. 2-Picoline ligand (L7) was found to be highly effective affording both excellent yield (90%) and mono-selectivity (99%). Replacing the 2-methyl group by other substituents in 2-substituted pyridines (L8–L12) resulted in lower yields. Among the di-substituted and tri-substituted pyridines (L13–L18), L16 and L17 containing a 2-methyl group performed well with good yields (84% and 83%) and mono-selectivity. Interestingly, the use of 2,6-lutidine (L13) resulted in some loss of mono-selectivity affording the mono-arylated product 2a in 70% yield and the di-arylated product 3r in 10% yield. Arylation in the absence of ligand under these new conditions gave the desired product in 36% yield, thus confirming the significant ligand acceleration effect. Mechanistically, the comparison of the most effective ligand L7 with the less effective ligands L10 and L11 is informative. The decrease in binding strength of the ligands via either electronic or steric effects reduces the efficiency of the catalysts.
Table 2.
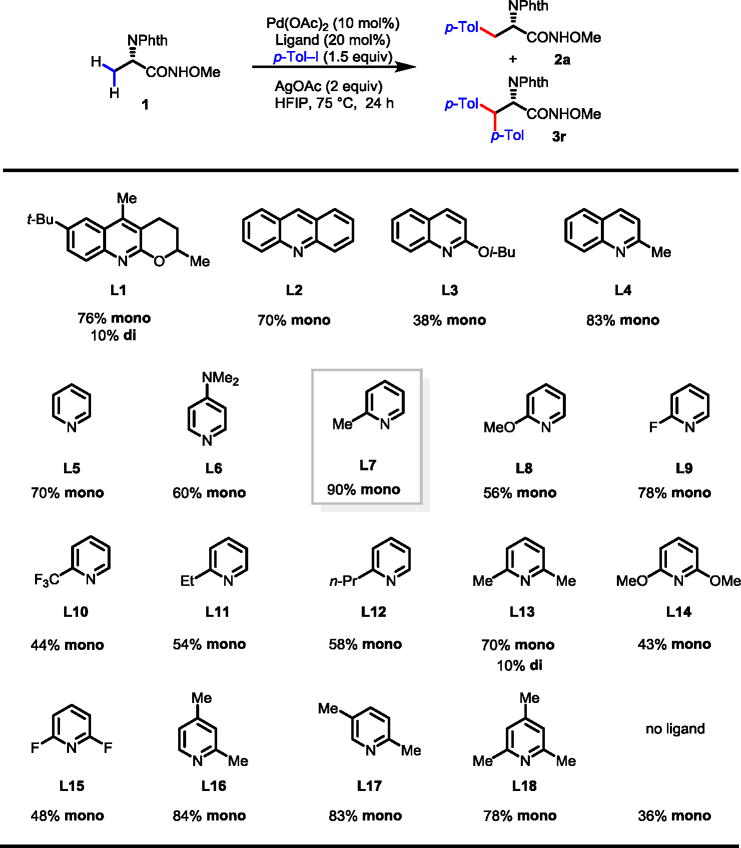
|
Conditions: Substrate 1 (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), p-Tol–I (0.15 mmol), Ligand (20 mol%), HFIP (1.0 mL), 75 °C, 24 h.
The yields were determined by 1H NMR analysis of the crude product using CH2Br2 as an internal standard. The mono:di ratio was determined by 1H NMR.
A broad range of variously substituted aryl iodides are compatible with this ligand-promoted β-C–H arylation reaction (Table 3). Aryl iodides containing methyl, phenyl and methoxy groups react with substrate 1 under the standard conditions to give the desired products in good to excellent yields (2a–f). Fluoro, chloro, bromo and iodo substituents are all tolerated and moderate to good yields are obtained (2g–k). Aryl iodides containing highly electron-withdrawing groups including acetyl and methoxycarbonyl are excellent coupling partners affording the arylation products in 71–82% yields (2l–p). Di- and tri-substituted aryl iodides display similar reactivity to the mono-substituted ones (2q–t). Most importantly, aryl iodides containing well-known directing groups such as acetamide, phosphonate and hydroxyls are also reactive coupling partners (2u–x), thus overcoming some limitations of previous protocols.16,17 These unnatural amino acids have been widely used as building blocks for the preparation of bioactive peptides.19,20 For example, the corresponding amino acid of 2v was used to replace tyrosine in a peptide to afford an improved β2 adrenergic receptor19 while a tetrapeptide containing the corresponding amino acid of 2w has been evaluated as a tyrosine kinase inhibitor.20
Table 3.
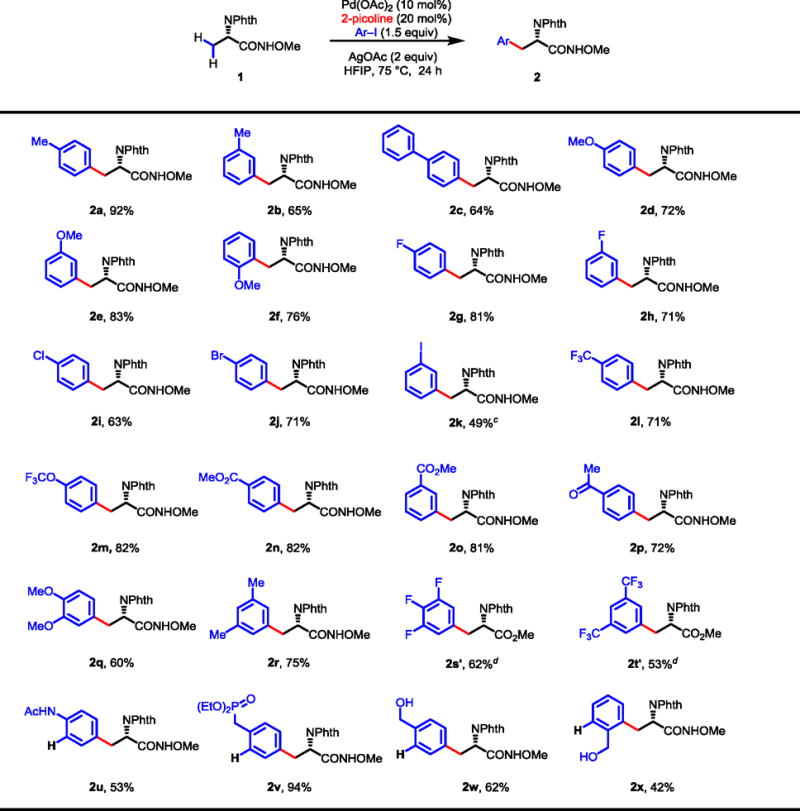
|
Conditions: Substrate 1 (0.1 mmol), Pd(OAc)2 (10 mol %), AgOAc (0.2 mmol), Ar–I (0.15 mmol), 2-picoline (20 mol%), HFIP (1.0 mL), 75 °C, 24 h.
Isolated yields are shown.
Ar–I (0.3 mmol).
After C–H activation, the reaction mixture was subjected to PhI(OAc)2 (0.1 mmol), MeOH (1 mL), 80 °C, 3 h. Yields for two steps.
2.2. Arylation with heteroaryl iodides
Heteroatoms in heterocycles coordinate strongly to Pd(II) catalysts and result in catalyst poisoning. This detrimental effect often prevents the use of heteroaryl iodides as coupling partners in C–H activation reactions. We reasoned that the acidic solvent HFIP used in our new protocol could weaken the coordinating ability of the heterocycles. Furthermore, the pyridine-type ligand picoline could potentially also outcompete the coordination of the heteroaryl iodides. We thus proceeded to investigate the reactivity of a wide range of heterocyclic iodides under the standard conditions. We found that aryl iodides containing dioxane and chromonyl moieties were coupled with alanine substrate 1 successfully to give the desired products 4a and 4b in 76% and 70% yields respectively (Table 4). Tosyl-protected indolyl and indazolyl iodides also afforded 4c–f in synthetically useful yields (58–65%). These unnatural amino acids containing heteroaryls are not readily accessible via other methods21 and are often desirable in medicinal chemistry.
Table 4.
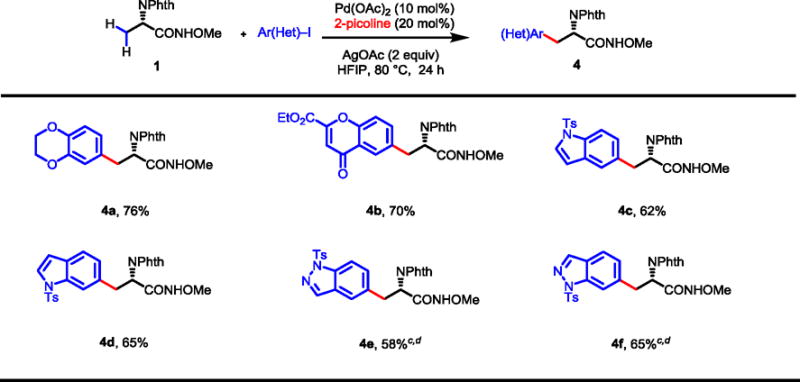
|
Conditions: Substrate 1 (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), Ar–I (0.15 mmol), 2-picoline (20 mol%), HFIP (1.0 mL), 80 °C, 24 h.
Isolated yields are shown.
Pd(OAc)2 (15 mol %), 2-picoline (30 mol%).
Yields based on the amide and ester.
However, arylation with the more coordinative pyridyl iodide gave poor yields (less than 10%). Considering that a halogen substituent at the 2-position of a pyridine can be readily removed or transformed to other functional groups, we tested the reactivity of 2-fluoro, 2-chloro and 2-bromopyridyl iodides. These coupling partners reacted with substrate 1 to give the desired products in approximately 20% yields under the standard conditions. Through further ligand screening, 2,6-lutidine was identified as a more efficient ligand. Thus, arylation with a range of pyridyl iodides were carried out under the optimized conditions (Table 5). Although 4-pyridyl iodide gave poor yield (4g′), all 2-halo-pyridyl iodides afford synthetically useful yields (4h′–4m′). The arylated products were obtained as a mixture of amides and esters which are treated with PhI(OAc)2 in one pot to give the pure esters as the isolated products. Arylation with pyridyl iodides containing 2-CF3, 2-Me and 2-OMe substituents proceeded to give the desired products in 42–66% yields. The presence of 3-CH2OH group reduced the yield to 32% (4r′). Various quinolinyl and quinoxalinyl iodides are also compatible with protocol affording heterocycle-containing amino acids (4s′–4v′) in moderate yields.
Table 5.

|
Conditions: Substrate 1 (0.1 mmol), Pd(OAc)2 (15 mol%), AgOAc (0.2 mmol), Ar–I (0.15 mmol), Ligand (30 mol%), HFIP (1.0 mL), 85 °C, 36 h, and then PhI(OAc)2 (0.1 mmol), MeOH (1 mL), 80 °C, 3h.
Isolated yields are shown.
Substrate 1 (0.2 mmol), Ar–I (0.1 mmol), yield based on the Ar–I.
Lactone was first formed, then was converted to 4r′ with MeSO3H. For details, see supporting information.
2.3. Removal of the auxiliary
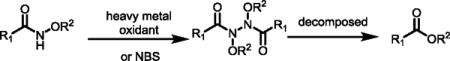 |
(4) |
 |
(5) |
Since N-alkoxyamides are used as masked esters, a number of deprotection procedures based on radical pathways have been developed (Eq 4).10, 22 N, N-Bis-heteroatom-substituted amides formed from N-alkoxyamides can thermally decompose to give the corresponding esters. A wide range of metal oxidants including silver oxide (Ag2O), nickel(IV) peroxide hydrate (NiO2•H2O), ceric ammonium nitrate (CAN), and lead(IV) acetate [Pb(OAc)4] have been used to convert N-alkoxyamides into esters.10 One-step conversion of N-alkoxyamides to esters by reacting with N-bromosuccinimide (NBS) in toluene has also been reported.22 N-Chlorohydroxamates generated from N-alkoxyamides can be converted to the esters by treating with sodium azide via Heron rearrangement (Eq 5).23 However, all of these protocols convert the amino acid-derived amides to esters in only moderate yields. We found that reacting N-methoxyamides with iodosobenzene diacetate [PhI(OAc)2] in methanol at 80 °C afforded esters in excellent yields excluding indole-containing amides (4c, 4d) which suffered from partial intramolecular radical lactamization. Lewis acid boron trifluoride diethyl etherate (Et2O•BF3) was also identified as an efficient reagent to convert N-methoxyamides into esters in methanol at 90 °C. This latter protocol is also compatible with the indole-containing amides. Importantly, no racemization of the α-chiral center was observed during the C–H arylation and the subsequent removal of the auxiliary (Scheme 2).
Scheme 2.
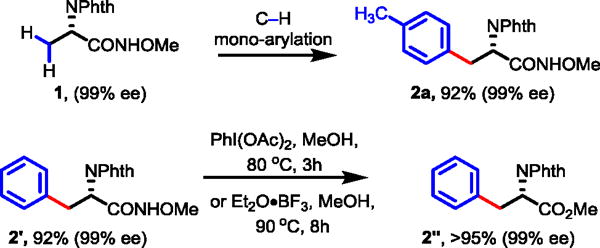
Removal of the Directing Group
2.4. Arylation of methylene C–H bonds
Following the development of mono-arylation of the primary C(sp3)–H bonds directed by N-methoxyamide, we began to search for ligands that would promote further arylation of the methylene C–H bonds. The formation of 10% of the di-arylated product with the quinoline ligand L1 (Table 1) led us to investigate whether these conditions can be optimized for arylation of methylene C–H bonds (Table 6). Considering the previously observed significant effects of inorganic bases on the β-C–H functionalizations of N-methoxyamides,7a we investigated the arylation with a wide range of base additives. Among the various potassium, sodium and lithium salts, monohydrogen phosphate (Table 6, entries 5 and 8) and dihydrogen phosphate (Table 6, entries 6, 9–11) significantly improved the yields of the arylation. Sodium dihydrogen phosphate was especially effective, affording the arylated product in 50% yield (Table 6, entries 9, 10). Increasing the amount of sodium dihydrogen phosphate monohydrate (NaH2PO4•H2O) to three equivalents improved the yield to 72% (Table 6, entry 15).
Table 6.

| ||
|---|---|---|
|
| ||
| Entry | Additives (equiv) | % Yield |
| 1 | no | 10 |
| 2 | K2CO3 | 0 |
| 3 | KHCO3 | 8 |
| 4 | K3PO4(0.5) | 14 |
| 5 | K2HPO4 | 40 |
| 6 | KH2PO4 | 30 |
| 7 | KF | 16 |
| 8 | Na2HPO4 | 32 |
| 9 | NaH2PO4•H2O | 50 |
| 10 | NaH2PO4 | 49 |
| 11 | LiH2PO4 | 36 |
| 12 | LiOAc | 0 |
| 13 | NaH2PO4•H2O(1.5) | 56 |
| 14 | NaH2PO4•H2O (2) | 66 |
| 15 | NaH2PO4•H2O (3) | 72 |
| 16 | NaH2PO4•H2O (4) | 70 |
Conditions: Substrate 2′ (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), p-Tol–I (0.3 mmol), L1 (20 mol%), HFIP (1.0 mL), 100 °C, 36 h.
Determined by 1H NMR analysis of the crude product using CH2Br2 as an internal standard.
With this promising result in hand, we began to further improve the arylation of phenylalanine 2′ by screening ligands (Table 7). The dependence of this reaction on ligand was confirmed by the complete loss of reactivity in the absence of a ligand. While other quinoline-based ligands (L2, L3, L19) were all inferior to L1, 2,6-lutidine (L13) and 2,4,6-trimethylpyridine (L18) proved to be superior ligands, affording the desired product in 92% and 86% yields respectively.
Table 7.

|
Conditions: Substrate 2′ (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), p-Tol–I (0.3 mmol), Ligand (20 mol%), HFIP (1.0 mL), 100 °C, 36 h.
Determined by 1H NMR analysis of the crude product using CH2Br2 as an internal standard.
The scope of this newly developed methylene C–H arylation was examined with a board range of electron-rich and electron-poor aryl iodides (Table 8). Aryl iodides containing electron-donating substituents at the ortho-, meta- and para-positions served as efficient coupling partners, affording the β-arylated phenylalanine products in good to excellent yields (3a–f). Arylation of 2′ with fluoro- and chloro-substituted aryl iodides proceeded to afford the desired products in 70–75% yields (3g–i). This reaction is also compatible with aryl iodides containing electron-withdrawing groups (3j–m). Arylation with naphthalene iodide and di-substitued aryl iodides also afforded synthetically useful yields (3n–p). In all cases, this arylation reaction affords excellent diastereoselectivity which can be explained by a previously isolated C–H cleavage intermediate from a related amide substrate.17
Table 8.
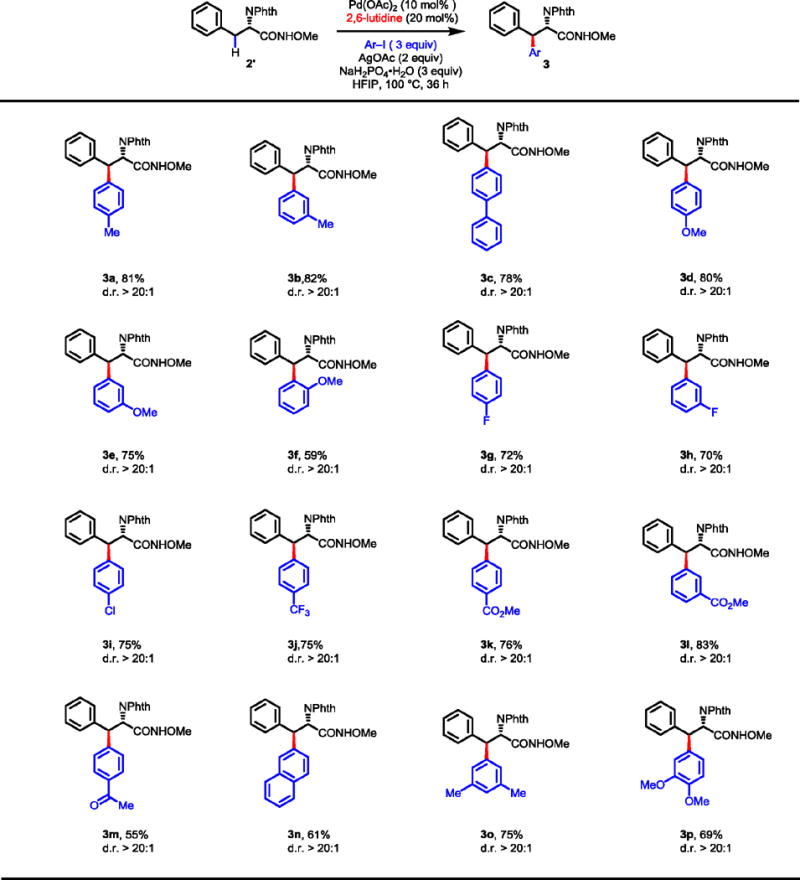
|
Conditions: Substrate 2′ (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), Ar–I (0.3 mmol), 2,6-lutidine (20 mol%), NaH2PO4•H2O (0.3 mmol), HFIP (1.0 mL), 100 °C, 36 h.
Isolated yields are shown.
The removal of the auxiliary from the β-diaryl alanine products using PhI(OAc)2 resulted in the formation of a substantial amount of a lactamization side product. The use of Lewis acid Et2O•BF3 proved to be effective in removing the auxiliary in high yields. Importantly, the enantiopurity of the α-chiral center was retained during both the C–H arylation step and the subsequent conversion of the amide to the corresponding ester (Scheme 3).
Scheme 3.
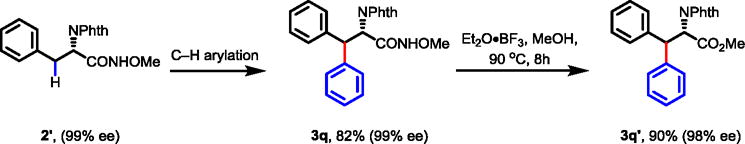
Removal of the Directing Group
2.5. Homo-diarylation of alanine
The recently identified 2,6-lutidine ligand was also applied to the arylation of alanine substrate 1 to obtain homo-diarylation products in one pot (Table 9). Various β-diaryl-α-amino acids (3q-t) were synthesized in 63–75% yields. Considering the steric hindrance on the β-carbon of these diarylated amino acids, we anticipate that the corresponding chiral amino alcohols are highly valuable for the synthesis of bulky chiral bis(oxazoline) ligands.
Table 9.
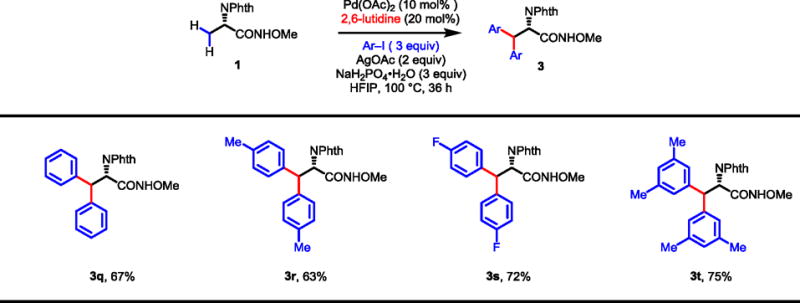
|
Conditions: Substrate 1 (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), Ar–I (0.3 mmol), 2,6-lutidine (20 mol%), NaH2PO4•H2O (0.3 mmol), HFIP (1.0 mL), 100 °C, 36 h.
Isolated yields are shown.
2.6. One-Pot Sequential Hetero-diarylation of Alanine
The exclusive mono-selectivity of 2-picoline-promoted β-arylation of primary C–H bonds provides a possibility for achieving sequential arylation of alanine substrate 1 with two different aryl iodides in one pot. Thus, 1 was subjected to the mono-arylation conditions with 1.2 equiv. of 4-iodotoluene. After 1 was completely arylated as shown by thin layer chromatography (TLC), 3 equiv. of phenyl iodide (Ph–I) as well as other reagents (2,6-lutidine, AgOAc, NaH2PO4•H2O) required for the methylene C–H activation were added to the reaction to initiate the second arylation (Table 10). This one-pot procedure afforded the hetero-diarylated product 3u in 71% yield. The formation of homo-arylated product with 4-iodotoluene was not observed suggesting that the remaining aryl iodide from the first step was outcompeted by the excess phenyl iodide introduced in the second step. This protocol is also compatible with other combinations of a variety of aryl iodides, affording diverse range of hetero-diarylated products in 43–71% yields with excellent diastereoselectivity (3u–ab). The switching of the arylation order to access different diastereomers was also demonstrated with the preparation of 3w and 3x. Given the availability of both enantiomers of the starting amino acids, all four diasteromeric products can be obtained by switching the order of addition of the two different aryl iodides. Overall, this protocol offers a highly versatile approach for the preparation of chiral β-Ar-β-Ar′-α-amino acids. A sequential hetero-diarylation using a strongly coordinating bidentate directing group has also been demonstrated,16a although the mono-arylated product needs to be isolated and subjected to different conditions to perform the second arylation. A recent report24 on an improved synthesis of differentially substituted dehydro-β,β-diarylalanine derivatives and subsequent asymmetric hydrogenation also speaks to the need for efficient methods for preparing chiral β-Ar-β-Ar′-α-amino acids.
Table 10.

|
Conditions: Substrate 1 (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), Ar1–I (0.12 mmol), 2-picoline (20 mol%), HFIP (1.0 mL), 75 °C, 24 h. Then Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), Ar2–I (0.3 mmol), 2,6-lutidine (20 mol%), NaH2PO4•H2O (0.3 mmol), 100 °C, 36 h.
Isolated yields are shown.
2.7. Arylation of Other Carboxylic Acids
To demonstrate the generality of N-methoxyamide as a directing group for the arylation of C(sp3)–H bond, we also examined the arylation of other aliphatic acid substrates. Under the conditions for the β-arylation of primary C–H bonds using 2-picoline as the ligand, amides derived from 2-methyl butyric acid, β-hydroxy acid, β-amino acid, and 2-aminoisobutyric acid afforded the arylated products in 53–72% yields (Table 11, 6a–d). Interestingly, arylation of the cyclobutyl C–H bonds in the amide substrate derived from 1-aminocyclobutane-1-carboxylic acid afforded 86% yield under these conditions (6g). As expected, β-arylation of amide substrates derived from tyrosine and L-2-aminobutyric acid only proceeded under the conditions developed for methylene C–H bonds (6e-f). To our surprise, cyclopropyl C–H bond in 5h is less reactive under these conditions and requires the use of ligand L1 to allow the arylation to proceed in moderate yield (45%).
Table 11.
Arylation of Other Amino Acids and Carboxylic Acidsa
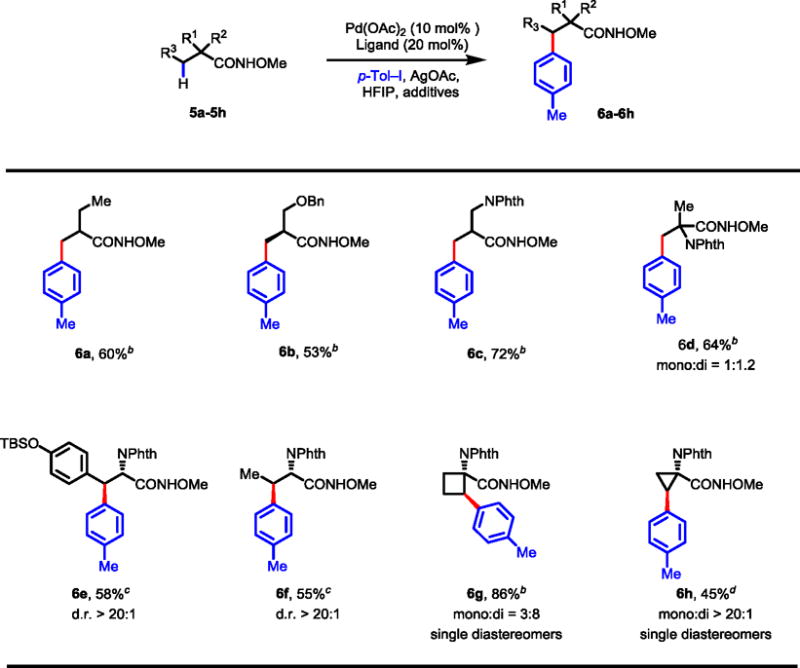
|
Isolated yields are shown.
Conditions: Substrate (0.2 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.4 mmol), p-Tol–I (0.3 mmol), 2-picoline (20 mol%), HFIP (2.0 mL), 80 °C, 24 h.
Conditions: Substrate (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.2 mmol), p-Tol–I (0.3 mmol), 2,6-lutidine (20 mol%), NaH2PO4•H2O (0.3 mmol), HFIP (1.0 mL), 90 °C, 36 h.
Substrate (0.1 mmol), Pd(OAc)2 (15 mol%), AgOAc (0.2 mmol), p-Tol–I (0.3 mmol), L1 (30 mol %), TFA (20 mol %), DCE (1.0 mL), 85 °C, 36 h.
2.8. Gram-scale syntheses of unnatural amino acids
The use of N-methoxyamide auxiliary has important advantages for gram-scale preparation of unnatural amino acids. Firstly, methoxyamine hydrochloride is inexpensive and has a low molecular weight. Secondly, the installation involves treatment of carboxylic acid with oxalyl chloride and methoxyamine hydrochloride at room temperature to afford the N-methoxyamides in nearly quantitative yields. Notably, the installation of other amide directing groups such as CONHArF (ArF = p-CF3C6F4) often requires refluxing conditions or proceeds in low yields. Furthermore, the removal of this auxiliary using Et2O•BF3 or PhI(OAc)2 is highly reliable and high yielding with retention of stereochemistry at the acidic α-carbon center.
In response to needs for bioactive peptides from Bristol-Myers Squibb, alanine substrate 1 was coupled with fluorinated and trifluoromethylated aryl iodides (20 mmol scale) using 2-picoline as the ligand to give the mono-arylated products in excellent yields (Scheme 4). The resulting crude products were treated with PhI(OAc)2 in methanol to afford the esters in 85–98% yields over two steps. The phthalimide group was removed in presence of ethylenediamine to generate the free amines that were subsequently converted to Fmoc-protected amino esters. Finally, the esters were hydrolyzed in presence of lithium hydroxide to give 7.5–9.0 grams of the desired amino acids (7–9) in 50–55% overall yields. The less reactive heteroaryl iodides and trifluoro-aryl iodide were also successfully coupled with alanine 1 using 2,6-lutidine. Following a similar procedure, 1.1–6.0 grams of these desired Fmoc-protected amino acids were prepared (10–13) (Scheme 5, for details, see supporting information).
Scheme 4.

Gram-Scale Synthesis of Unnatural Amino Acids 7–9
Scheme 5.

Gram-Scale Synthesis of Unnatural amino Amino Acids 10–13
2.9. Diverse Synthetic Applications
The practical advantage of the N-methoxyamide auxiliary is further demonstrated by its versatile transformations to various biologically active compounds (Scheme 6). Radical cyclization of 2e with [bis(trifluoroacetoxy)iodo]benzene (PIFA) led to a lactam25 and subsequent deprotection of the phthalamide using ethylenediamine afforded 14 as a key intermediate for the synthesis of glycogen phosphorylase inhibitors.26 Conversion of 2e to carboxylic acid and subsequent treatment with oxalyl chloride and aluminum trichloride gave the 2-amino-1-indanone 15,27 a key intermediate of α1-adrenoceptor antagonists.28 The amino ester 16 derived from 2e was readily converted to Fmoc-protected unnatural amino acid 17 as a useful building block for peptide synthesis. Through the Pictet-Spengler reaction, 16 could be also converted to a chiral tetrahydroisoquinoline 18, a known N-methyl-D-aspartate (NMDA) agonist.29 A chiral bioactive indoline 1930 could also be synthesized via our previously developed intramolecular C–H amination reaction of sulfonyl protected 16.31
Scheme 6.

Application to Synthesis of a Variety of Biologically Active Compounds
The N-methoxyamide auxiliary also gives access to N-hydroxy-3-amino-3,4-dihydroquinolinone class of compounds. First reported by Davis and co-workers over thirty years ago, they were shown to exhibit antibacterial activity.32 More recently, similar scaffolds have been identified as potent inhibitors of KAT II, an enzyme currently being investigated as a therapeutic target for cognitive impairment associated with schizophrenia, among other disorders.11 Using the above protocols, arylation followed by a known radical cyclization, medicinally important analogues of N-methoxy-3-amino-3,4-dihydroquinolines 14′ and 20–22 were prepared in a straightforward manner (Table 12).
Table 12.
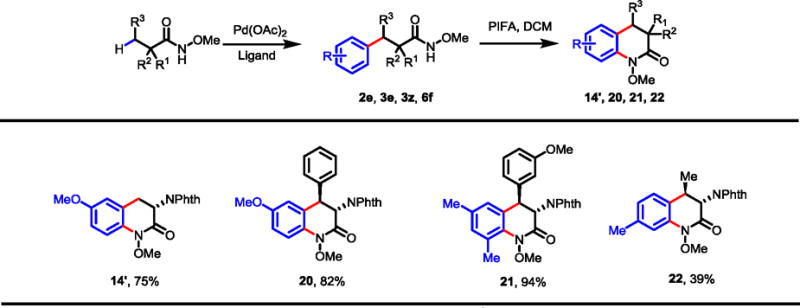
|
Conditions for cyclization: Substrate, PIFA (2 equiv.), DCM, 0 °C to r.t.
Isolated yields are shown.
Chiral amino alcohols derived from chiral amino acids are essential building blocks for the preparation of chiral ligands. For example, the sterically hindered chiral amino alcohol derived from tert-leucine is a key precursor for the synthesis of one of the most effective chiral oxazoline ligands in asymmetric catalysis. β-Ar-β-Ar′-α-amino acids prepared via the recently developed procedure can be readily reduced to the corresponding chiral amino alcohols containing two chiral centers that are previously difficult to make (Scheme 7). Thus, employing our hetero-diarylation protocol above, 3z was obtained in moderate yields via a two steps, one-pot procedure on a 10 mmol scale. Hydrolysis of the amide group to ester followed by removal of the phthalimide protecting group led to the amino ester 24 which was reduced to the amino alcohol 25 in 75% yield. From this common intermediate 25, PyBox ligand 26 and Box ligand 27 were successfully prepared.33 We anticipate that these novel chiral bis(oxazoline) ligands will display interesting and useful properties in asymmetric catalysis. The potential impact of the additional chiral centers of ligands 26 and 27 on asymmetric catalysis is also intriguing.
Scheme 7.
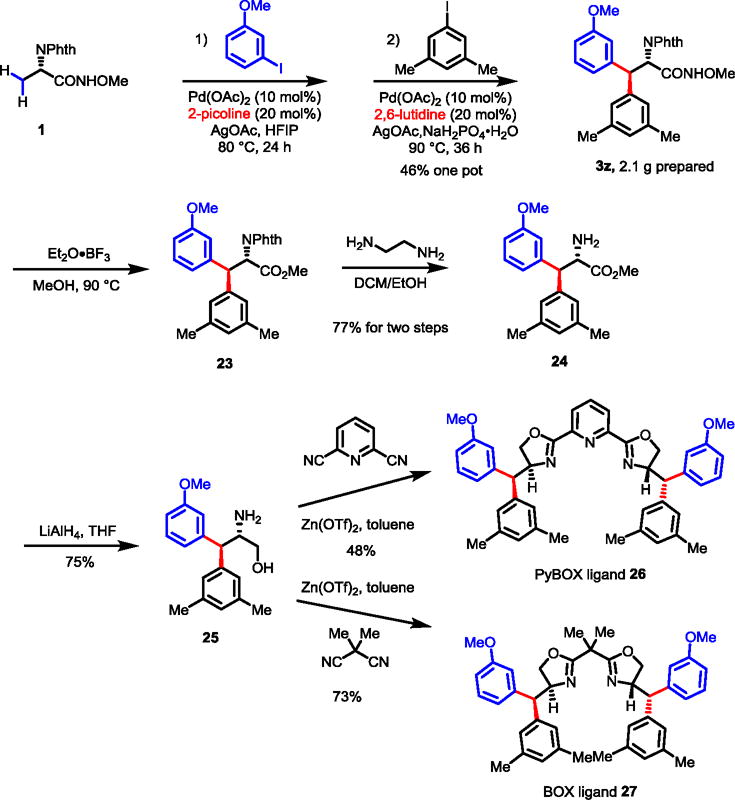
Synthesis of PyBOX and BOX Ligands
3. Conclusion
During the past decade-long efforts to develop a simple and practical auxiliary or directing group for β-C–H fucntionalizations of carboxylic acids, we have focused on the use of relatively weak coordination from the substrates to direct metalation and match this weak coordination with ligand development to enhance the reactivity. The simple N-methoxyamide group, initially used as a masked ester, has been reinvented as a broadly useful directing group for β-C–H arylation with the assistance of two pyridine-type ligands. 2-Picoline promotes the mono-selective arylation of primary C(sp3)–H bonds while 2,6-lutidine enables the subsequent arylation of secondary C(sp3)–H bonds in one pot. This new method is extensively applied to gram-scale synthesis of novel, unnatural amino acids, as well as bioactive compounds and chiral bis(oxazoline) ligands.
4. Experimental Section
4.1 General procedure for monoarylation with aryl iodides (Table 3)
The starting material 1 (0.1 mmol, 24.8 mg), Pd(OAc)2 (10 mol%, 2.2 mg), and AgOAc (0.2 mmol, 33.4 mg) were weighed in air and placed in a sealed tube (10 mL) with a magnetic stir bar. To the reaction mixture, aryl iodide (0.15 mmol), 2-picoline (20 mol%, 2 μL), HFIP (1.0 mL) were added. The reaction mixture was first stirred at room temperature for 10 min and then heated to 75 °C for 24 hours under vigorous stirring. Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was purified by a silica gel-packed flash chromatography column using DCM/EtOAc (1/0 to 4/1 to 2/1) as the eluent.
4.2 General procedure for monoarylation with heterocyclic iodides (Table 4 and 5)
Method A (Table 4)
The starting material 1 (0.1 mmol, 24.8 mg), Pd(OAc)2 (10 mol%, 2.2 mg), and AgOAc (0.2 mmol, 33.4 mg) were weighed in air and placed in a sealed tube (10 mL) with a magnetic stir bar. To the reaction mixture, aryl iodide (0.15 mmol), 2-picoline (20 mol%, 2 μL), HFIP (1.0 mL) were added. The reaction mixture was first stirred at room temperature for 10 min and then heated to 80 °C for 24 hours under vigorous stirring. Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was purified by a silica gel-packed flash chromatography column using DCM/EtOAc (1/0 to 4/1 to 2/1) as the eluent.
Method B (Table 5)
The starting material 1 (0.1 mmol, 24.8 mg), Pd(OAc)2 (15 mol%, 3.3 mg), and AgOAc (0.2 mmol, 33.4 mg) were weighed in air and placed in a sealed tube (10 mL) with a magnetic stir bar. To the reaction mixture, aryl iodide (0.15 mmol), 2,6-lutidine (30 mol%, 3 μL), HFIP (1.0 mL) were added. The reaction mixture was first stirred at room temperature for 10 min and then heated to 80 °C for 36 hours under vigorous stirring. Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was added PhI(OAc)2 (0.1 mmol, 32.2 mg) and MeOH (1 mL) in a sealed tube (10 mL) with a magnetic stir bar, The reaction mixture was heated to 80 °C for 3 hours, Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was purified by a silica gel-packed flash chromatography column using hexanes/EtOAc (5/1 to 4/1 to 2/1) as the eluent.
Method C (Table 5)
The starting material 1 (0.2 mmol, 49.6 mg), Pd(OAc)2 (15 mol%, 3.3 mg), and AgOAc (0.2 mmol, 33.4 mg) were weighed in air and placed in a sealed tube (10 mL) with a magnetic stir bar. To the reaction mixture, aryl iodide (0.1 mmol), 2,6-lutidine (30 mol%, 3 μL), HFIP (1.0 mL) were added. The reaction mixture was first stirred at room temperature for 10 min and then heated to 80 °C for 36 hours under vigorous stirring. Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was added PhI(OAc)2 (0.1 mmol, 32.2 mg) and MeOH (1 mL) in a sealed tube (10 mL) with a magnetic stir bar. The reaction mixture was heated to 80 °C for 3 hours. Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was purified by a silica gel-packed flash chromatography column using hexanes/EtOAc (5/1 to 4/1 to 2/1) as the eluent.
4.3 General procedure for arylation of phenylalanine (Table 8)
The substrate 2′ (0.1mmol, 32.4 mg), Pd(OAc)2 (10 mol%, 2.2 mg), NaH2PO4•H2O (0.3 mmol, 42 mg) and AgOAc (0.2 mmol, 33.4 mg) were weighed in air and placed in a sealed tube (10 mL) with a magnetic stir bar. To the reaction mixture, aryl iodide (0.3 mmol), 2,6-lutidine (20 mol%, 2 μL), HFIP (1 mL) were added. The reaction mixture was first stirred at room temperature for 10 min and then heated to 100 °C for 36 hours under vigorous stirring. Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was purified by a silica gel-packed flash chromatography column using hexanes/EtOAc (5/1 to 3/1 to 2/1) as the eluent.
4.4 General procedure for large scale of mono-arylation step
The starting material 1 (10.0 mmol, 2.48 g), Pd(OAc)2 (1.50 mmol, 337 mg), and AgOAc (20.0 mmol, 3.34 g) were weighed in air and placed in a sealed tube (350 mL) with a magnetic stir bar. To the reaction mixture, aryl iodide (15 mmol), ligand (3.00 mmol), and HFIP (100 mL) were added. The reaction mixture was first stirred at room temperature for 10 min and then heated to 90 °C for 36 hours under vigorous stirring. Upon completion, the reaction mixture was cooled to room temperature. The solvents were removed under reduced pressure and recovered for next time use. The resulting mixture was added PhI(OAc)2 (10 mmol, 3.22 g) and MeOH (100 mL) in a sealed tube (350 mL) with a magnetic stir bar. The reaction mixture was heated to 80 °C for 3 hours. Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was purified by a silica gel-packed flash chromatography column using hexanes/EtOAc (5/1 to 4/1 to 2/1) as the eluent.
4.5 General procedure for one pot synthesis of β-Ar-β-Ar′-α-amino acids (Table 10)
The starting material (0.1 mmol, 24.8 mg), Pd(OAc)2 (10 mol%, 2.2 mg), and AgOAc (0.2 mmol, 33.4 mg) were weighed in air and placed in a sealed tube (10 mL) with a magnetic stir bar. To the reaction mixture, the first aryl iodide (0.12 mmol), 2-picoline (20 mol%, 2 μL), HFIP (1.0 mL) were added. The reaction mixture was first stirred at room temperature for 10 min and then heated to 75 °C for 24 hours under vigorous stirring. Upon completion, the reaction mixture was cooled to room temperature, to the reaction mixture, Pd(OAc)2 (0.01 mmol, 2.2 mg), NaH2PO4•H2O (0.3 mmol, 42 mg), AgOAc (0.2 mmol, 33.4 mg), the second aryl iodide (0.3 mmol) and 2,6-lutidine (0.2 mmol, 2 μL), were added. The reaction mixture was first stirred at room temperature for 10 min and then heated to 100 °C for 36 hours under vigorous stirring. Upon completion, the reaction mixture was cooled to room temperature, filtered with celite and washed with DCM. The solvents were removed under reduced pressure and the resulting mixture was purified by a silica gel-packed flash chromatography column using hexanes/EtOAc (5/1 to 3/1 to 2/1) as the eluent.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute (TSRI) and the NIH (NIH (NIGMS, 2R01GM084019) for financial support. We thank SIOC, Zhejiang Medicine and Pharmaron (fellowships to G.C.). We thank Drs Joel Barrish, John Kadow and Percy Carter for helpful discussions on the amino acid synthesis.
Footnotes
Supporting Information Avialable. Detailed experimental procedures, characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737. [Google Scholar]; (b) Evans DA. Aldrichim Acta. 1982;15:23. [Google Scholar]; (c) Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J Am Chem Soc. 1997;119:6496. [Google Scholar]; (d) Corey EJ, Enders D. Tetrahedron Lett. 1976;17:3. [Google Scholar]; (e) Meyers AI. Acc Chem Res. 1978;11:375. [Google Scholar]; (f) Job A, Janeck CF, Bettray W, Peters R, Enders D. Tetrahedron. 2002;58:2253. [Google Scholar]
- 2.(a) Feringa BL. Acc Chem Res. 2000;33:346. doi: 10.1021/ar990084k. [DOI] [PubMed] [Google Scholar]; (b) Mizutani H, Degrado SJ, Hoveyda AH. J Am Chem Soc. 2002;124:779. doi: 10.1021/ja0122849. [DOI] [PubMed] [Google Scholar]; (c) Hayashi T, Yamasaki K. Chem Rev. 2003;103:2829. doi: 10.1021/cr020022z. [DOI] [PubMed] [Google Scholar]
- 3.(a) Giri R, Chen X, Yu J-Q. Angew Chem Int Ed. 2005;44:2112. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]; (b) Giri R, Liang J, Lei J-G, Li J-J, Wang D-H, Chen X, Naggar IC, Guo C, Foxman BM, Yu J-Q. Angew Chem Int Ed. 2005;44:7420. doi: 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]; For a palladium-catalyzed β-C–H oxygenation of ketones using imine directing groups, see:; (c) Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 4.Giri R, Lan Y, Liu P, Houk KN, Yu J-Q. J Am Chem Soc. 2012;134:14118. doi: 10.1021/ja304643e. [DOI] [PubMed] [Google Scholar]
- 5.Examples of Daugulis’ bidentate 8-aminoquinoline auxiliary in C(sp3)–H activation of simple aliphatic acids using different metals. Pd:; (a) Zaitsev VG, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]; (b) Daugulis O, Do H-Q, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shabashov D, Daugulis O. J Am Chem Soc. 2010;132:3965. doi: 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Feng Y, Wang Y, Landgraf B, Liu S, Chen G. Org Lett. 2010;12:3414. doi: 10.1021/ol101220x. [DOI] [PubMed] [Google Scholar]; Ni:; (e) Aihara Y, Chatani N. J Am Chem Soc. 2014;136:898. doi: 10.1021/ja411715v. [DOI] [PubMed] [Google Scholar]; (f) Wu X, Zhao Y, Ge H. J Am Chem Soc. 2014;136:1789. doi: 10.1021/ja413131m. [DOI] [PubMed] [Google Scholar]; (g) Li M, Dong J, Huang X, Li K, Wu Q, Song F, You J. Chem Commun. 2014;50:3944. doi: 10.1039/c4cc00716f. [DOI] [PubMed] [Google Scholar]; Cu:; (h) Wang Z, Ni J, Kuninobu Y, Kanai M. Angew Chem Int Ed. 2014;53:3496. doi: 10.1002/anie.201311105. [DOI] [PubMed] [Google Scholar]; (i) Wu X, Zhao Y, Zhang G, Ge H. Angew Chem Int Ed. 2014;53:3706. doi: 10.1002/anie.201311263. [DOI] [PubMed] [Google Scholar]; Fe:; (j) Shang R, Ilies L, Matsumoto A, Nakamura E. J Am Chem Soc. 2013;135:6030. doi: 10.1021/ja402806f. [DOI] [PubMed] [Google Scholar]
- 6.Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. J Am Chem Soc. 2007;129:3510. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]
- 7.Wang D-H, Wasa M, Giri R, Yu J-Q. J Am Chem Soc. 2008;130:7190. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]
- 8.Examples of application of CONHOMe in directed C(sp2)–H activation. Pd(II):; (a) Wasa M, Yu J-Q. J Am Chem Soc. 2008;130:14058. doi: 10.1021/ja807129e. [DOI] [PubMed] [Google Scholar]; (b) Wang G-W, Yuan T-T, Li D-D. Angew Chem Int Ed. 2011;50:1380. doi: 10.1002/anie.201005874. [DOI] [PubMed] [Google Scholar]; (c) Karthikeyan J, Cheng C-H. Angew Chem Int Ed. 2011;50:9880. doi: 10.1002/anie.201104311. [DOI] [PubMed] [Google Scholar]; (d) Wrigglesworth JW, Cox B, Lioyd-Jones GC, Booker-Milburn KI. Org Lett. 2011;13:5326. doi: 10.1021/ol202187h. [DOI] [PubMed] [Google Scholar]; (e) Zhong H, Yang D, Wang S, Huang J. Chem Commun. 2012;48:3236. doi: 10.1039/c2cc17859a. [DOI] [PubMed] [Google Scholar]; (f) Liu Y-J, Xu H, Kong W-J, Shang M, Dai H-X, Yu J-Q. Nature. 2014;515:389. doi: 10.1038/nature13885. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rh(III):; (g) Guimond N, Gouliaras C, Fagnou K. J Am Chem Soc. 2010;132:6908. doi: 10.1021/ja102571b. [DOI] [PubMed] [Google Scholar]; (h) Rakshit S, Grohmann C, Besset T, Glorius F. J Am Chem Soc. 2011;133:2350. doi: 10.1021/ja109676d. [DOI] [PubMed] [Google Scholar]; (i) Zeng R, Fu C, Ma S. J Am Chem Soc. 2012;134:9597. doi: 10.1021/ja303790s. [DOI] [PubMed] [Google Scholar]; (j) Hyster TK, Knörr L, Ward TR, Rovis T. Science. 2012;338:500. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Ye B, Cramer N. Science. 2012;338:504. doi: 10.1126/science.1226938. [DOI] [PubMed] [Google Scholar]; Ru(II):; (l) Ackermann L, Fenner S. Org Lett. 2011;13:6548. doi: 10.1021/ol202861k. [DOI] [PubMed] [Google Scholar]; (m) Li B, Feng H, Xu S, Wang B. Chem Eur J. 2011;17:12573. doi: 10.1002/chem.201102445. [DOI] [PubMed] [Google Scholar]; (n) Li B, Ma J, Wang N, Feng H, Xu S, Wang B. Org Lett. 2012;14:736. doi: 10.1021/ol2032575. [DOI] [PubMed] [Google Scholar]
- 9.Examples of using CONHArF as an auxiliary in C–H activation:; (a) Wasa M, Engle KM, Yu J-Q. J Am Chem Soc. 2009;131:9886. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wasa M, Engle KM, Yu J-Q. J Am Chem Soc. 2010;132:3680. doi: 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wasa M, Engle KM, Lin DW, Yoo EJ, Yu J-Q. J Am Chem Soc. 2011;133:19598. doi: 10.1021/ja207607s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wasa M, Chan KSL, Zhang X-G, He J, Miura M, Yu J-Q. J Am Chem Soc. 2012;134:18570. doi: 10.1021/ja309325e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhu R-Y, He J, Wang X-C, Yu J-Q. J Am Chem Soc. 2014;136:13194. doi: 10.1021/ja508165a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Crawford RJ, Raap R. J Org Chem. 1963;28:2419. [Google Scholar]; (b) Cooley JH, Mosher MW, Khan MA. J Am Chem Soc. 1968;90:1867. [Google Scholar]; (c) De Almeida MV, Barton DHR, Bytheway I, Ferreira JA, Hall MB, Liu W, Taylor DK, Thomson L. J Am Chem Soc. 1995;117:4870. [Google Scholar]; (d) Glover SA, Mo G, Rauk A. Tetrahedron. 1999;55:3413. [Google Scholar]; (e) Kawase M, Kitamura T, Kikugawa Y. J Org Chem. 1989;54:3394. [Google Scholar]; (f) Yus M, Radivoy G, Alonso F. Synthesis. 2001;914 [Google Scholar]; (g) Samanta R, Bauer JO, Strohmann C, Antonchick AP. Org Lett. 2006;14:5518. doi: 10.1021/ol302607y. [DOI] [PubMed] [Google Scholar]; For N-methoxyamide as ortho-lithiation directing group, see:; (h) Fisher LE, Caroon JM, Jahangir, Stabler SR, Lundberg S, Muchowski JM. J Org Chem. 1993;58:3643. [Google Scholar]
- 11.(a) McAllister LA, Bechle BM, Dounay AB, Evrard E, Gan X, Ghosh S, Kim J-Y, Parikh VD, Tuttle JB, Verhoest PR. J Org Chem. 2011;76:3484. doi: 10.1021/jo200530j. [DOI] [PubMed] [Google Scholar]; (b) Dounay AB, Anderson M, Bechle BM, Campbell BM, Claffey MM, Evdokimov A, Evrard E, Fonseca KR, Gan X, Ghosh S, Hayward MM, Horner W, Kim J-Y, McAllister LA, Pandit J, Paradis V, Parikh VD, Reese MR, Rong S, Salafia MA, Schuyten K, Strick CA, Tuttle JB, Valentine J, Wang H, Zawadzke LE, Verhoest PR. ACS Med Chem Lett. 2012;3:187. doi: 10.1021/ml200204m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tuttle JB, Anderson M, Bechle BM, Campbell BM, Chang C, Dounay AB, Evrard E, Fonseca KR, Gan X, Ghosh S, Horner W, James LC, Kim J-Y, McAllister LA, Pandit J, Parikh VD, Rago BJ, Salafia MA, Strick CA, Zawadzke LE, Verhoest PR. ACS Med Chem Lett. 2013;4:37. doi: 10.1021/ml300237v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Cueva JP, Giorgioni G, Grubbs RA, Chemel BR, Watts VJ, Nichols DE. J Med Chem. 2006;49:6848. doi: 10.1021/jm0604979. [DOI] [PubMed] [Google Scholar]; (b) Przybyla JA, Cueva JP, Chemel BR, Hsu KJ, Riese DJ, II, McCorvy JD, Chester JA, Nichols DE, Watts VJ. Eur Neuropsychopharmacol. 2009;19:138. doi: 10.1016/j.euroneuro.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cueva JP, Chemel BR, Juncosa JI, Jr, Lill MA, Watts VJ, Nichols DE. Eur J Med Chem. 2012;48:97. doi: 10.1016/j.ejmech.2011.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao K-J, Lin DW, Miura M, Zhu R-Y, Gong W, Wasa M, Yu J-Q. J Am Chem Soc. 2014;136:8138. doi: 10.1021/ja504196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desimoni G, Faita G, Quadrelli P. Chem Rev. 2003;103:3119. doi: 10.1021/cr020004h. [DOI] [PubMed] [Google Scholar]
- 15.(a) Reddy BVS, Reddy LR, Corey EJ. Org Lett. 2006;8:3391. doi: 10.1021/ol061389j. [DOI] [PubMed] [Google Scholar]; (b) Feng Y, Chen G. Angew Chem Int Ed. 2010;49:958. doi: 10.1002/anie.200905134. [DOI] [PubMed] [Google Scholar]; (c) Tran LD, Daugulis O. Angew Chem, Int Ed. 2012;51:5188. doi: 10.1002/anie.201200731. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang Q, Chen K, Rao W, Zhang Y, Chen F-J, Shi B-F. Angew Chem, Int Ed. 2013;52:13588. doi: 10.1002/anie.201306625. [DOI] [PubMed] [Google Scholar]
- 16.(a) Wang B, Nack WA, He G, Zhang S-Y, Chen G. Chem Sci. 2014;5:3952. [Google Scholar]; (b) Chen K, Zhang S-Q, Xu J-W, Hu F, Shi B-F. Chem Comm. 2014;50:13924. doi: 10.1039/c4cc06652a. [DOI] [PubMed] [Google Scholar]
- 17.He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu J-Q. Science. 2014;343:1216. doi: 10.1126/science.1249198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dang Y, Qu S, Nelson JW, Pham HD, Wang ZX, Wang X. J Am Chem Soc. doi: 10.1021/ja512374g. [DOI] [Google Scholar]
- 19.Garbay-Jaureguiberry C, Ficheux D, Roques BP. Int J Pept Prot Res. 1992;39:523. doi: 10.1111/j.1399-3011.1992.tb00283.x. [DOI] [PubMed] [Google Scholar]
- 20.Kim MH, Lai JH, Hangauer DG. Int J Pept Prot Res. 1994;44:457. [PubMed] [Google Scholar]
- 21.Tang W, Zhang X. Chem Rev. 2003;103:3029. doi: 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]
- 22.Zhang N, Yang R, Zhang-Negrerie D, Du Y, Zhao K. J Org Chem. 2013;78:8705. doi: 10.1021/jo401435v. [DOI] [PubMed] [Google Scholar]
- 23.Glover SA, Mo G. J Chem Soc, Perkin Trans. 2002;2:1728. [Google Scholar]
- 24.Molinaro C, Scott JP, Shevlin M, Wise C, Ménard A, Gibb A, Junker EM, Lieberman D. J Am Chem Soc. 2015;137:999. doi: 10.1021/ja511872a. [DOI] [PubMed] [Google Scholar]
- 25.Amano Y, Inoue K, Nishiyama S. Synlett. 2008;134 [Google Scholar]
- 26.Birch AM, Kenny PW, Oikonomakos NG, Otterbein L, Schofield P, Whittamore PRO, Whalley DP. Bioorg Med Chem Lett. 2007;17:394. doi: 10.1016/j.bmcl.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 27.McClure DE, Lumma PK, Arison BH, Jones JH, Baldwin JJ. J Org Chem. 1983;48:2675. [Google Scholar]
- 28.Li M, Xia L. Chem Biol Drug Des. 2007;70:461. doi: 10.1111/j.1747-0285.2007.00581.x. [DOI] [PubMed] [Google Scholar]
- 29.Ortwine DF, Malone TC, Bigge CF, Drummond JT, Humblet C, Johnson G, Pinter GW. J Med Chem. 1992;35:1345. doi: 10.1021/jm00086a004. [DOI] [PubMed] [Google Scholar]
- 30.Stoddart LA, Vernall AJ, Denman JL, Briddon SJ, Kellam B, Hill SJ. Chem Biol. 2012;19:1105. doi: 10.1016/j.chembiol.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mei T-S, Leow D, Xiao H, Laforteza BN, Yu J-Q. Org Lett. 2013;15:3058. doi: 10.1021/ol401246u. [DOI] [PubMed] [Google Scholar]
- 32.(a) Davis AL, Choun OHP, Cook DE, McCord TJ. J Med Chem. 1964;7:632. doi: 10.1021/jm00335a014. [DOI] [PubMed] [Google Scholar]; (b) Davis AL, Chambers WH, Kelley DH, Fell DA, Haynes JR, Hulme KL, Gage LG, McCord TJ. J Med Chem. 1975;18:752. doi: 10.1021/jm00241a022. [DOI] [PubMed] [Google Scholar]
- 33.Cornejo A, Fraile JM, García JI, Gil MJ, Martínez-Merino V, Mayoral JA, Pires E, Villalba I. Synlett. 2005:2321. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.