

Abstract
Our previous study indicated that 8 patients from a family with a history of congenital heart disease had simple atrial septal defect (ASD) and carried the same mutation at codon 310 in the GATA4 gene. In the present study, to identify the functional defects caused by this mutation in an in vivo model, the transgene DNA constructs were microinjected into mice to generate a transgenic mouse model. The mice were genotyped using PCR and DNA sequencing. Protein expression was measured by western blot analysis. qPCR was used to determine the copy number of the transgenes. The heart tissue was fixed and sectioned by conventional procedures. The Vevo 2000 system was used to perform echocardiography on the mice. The expression of GATA4 target genes was measured using the real-time PCR system. The incidence of ASD in the heterozygous transgenic mice was found to be greater than that in the wild-type control mice (P<0.05). In addition, the expression of α-myosin heavy chain (α-MHC) in the heart tissues from the homozygous mice was lower than that in the heart tissues from their wild-type littermates (P<0.05). In conclusion, these results suggest that the introduction of GATA4 M310V negatively affects the normal expression of α-MHC. In accordance with previous findings on GATA4 mutation screening and in vitro experiments, this study confirms that GATA4 M310V mutation may lead to the development of the congenital heart defect, ASD.
Keywords: congenital heart disease, GATA4, atrial septal defect, transgenic mouse
Introduction
Congenital heart disease (CHD) is a multifactorial disease; genetic and environmental factors play a key role in the occurrence of this disease. It is considered that there are over 1,700 genes that are required for heart development. The genetically modified mouse is a good anatomical model for common cardiac malformations which is powerful experimental tool for understanding human CHD (1). The GATA4 gene is essential to normal heart development. GATA4 deficiency is embryonically lethal in mice, as this leads to severe ventricular developmental defects, resulting in cardiac malformation (2). GATA4 also regulates the expression of cardiac structural genes, including α-myosin heavy chain (α-MHC), cardiac troponin C (cTNC), atrial natriuretic factor (ANF), brain natriuretic peptide (BNF), A1 adenosine receptor (ADORA1), muscarinic acetylcholine receptor M2 (CHRM2, cholinergic receptor, muscarinic 2), and angiotensin II 1A receptors (AngII1a) (3,4). When GATA4 zinc finger structures and other cardiac-specific transcription factors [NK2 homeobox 5 (NKX2.5), T-box transcription factor (TBX5) and myocyte-specific enhancer factor 2 (MEF2C)] interact to form complexes, these complexes play a role in transcriptional regulation and regulate heart development (5–8).
The GATA4 gene is an important factor in the cardiac gene network and mutations in GATA4 have been confirmed to be associated with the occurrence of atrial septal defect (ASD). GATA4 has been used to distinguish patients with familial CHD from those with sporadic CHD (9–11). GATA4 transgenic mice have been used to as a model to study the pathogenesis of human CHD. GATA4 knockout is lethal to mouse embryos, which suffer severe cardiac malformation (2). Double heterozygous GATA4-TBX5 mice exhibit atrioventricular septal defects (12), and double heterozygous GATA4-GATA5 mice present with a phenotype of stenosis in the aorta and pulmonary artery (13). Heterozygous GATA4 mutation may result in ASD phenotypes, ventricular septal defect, endocardial cushion defect, right ventricular dysplasia and cardiomyopathy (14). GATA4 G295S transgenic mice generated by Misra et al presented with a normal abdominal cardiac morphology and cyclization pattern, but a weak ventricular myocardium and a single ventricle in homozygous mice, which died at the gestational age of 11.5 days; however, the heterozygous line propagated and one subset carried the phenotypes of semilunar valve stenosis and ASD (15). This indicated that the complete lack of GATA4 gene activity has a severe impact on cardiac development in mice, which cannot survive or reproduce. The homozygous animal model limits the functional study of GATA4 and to a certain extent cannot represent human disease. However, heterozygous transgenic mice have phenotypes that resemble, reasonably closely, human CHD phenotypes, which are caused by genetic mutations.
Our group discovered a representative family with simple ASD in 2007 (9). Since then, thorough individual examinations were performed (e.g., collection of medical history, physical examination and echocardiography) upon a total of 31 family members from 1st- to 3rd-level relatives. A total of 8 patients, including the probands, were diagnosed with simple ASD. Subsequently, an in-depth study on the mechanisms of GATA4 M310V mutation associated with familial ASD by our group revealed that GATA4 M310V mutation affects α-MHC promoter activity in in vitro experiments (16).
In the present study, in order to identify the site of GATA4 mutation capable of producing functional defects in an in vivo model, a GATA4 M310V same-point mutation was generated in transgenic mice. The mice were bred and screened, producing a stable transgenic mouse line. We found that the incidence of ASD in the heterozygous transgenic mice was higher than that in their wild-type control littermates (P<0.05). In addition, the peak pulmonary artery pressure (PPAP) and speed in the heterozygous mice were higher compared with their wild-type control littermates (P<0.05). The expression levels of GATA4 downstream target genes (α-MHC) in the homozygous mice were lower than those in their wild-type control littermates (P<0.05). To summarize, GATA4 M310V mutations were associated with a higher incidence of ASD-like cardiac malformation in heterozygous transgenic mice compared with the wild-type controls. Due to the changes in the expression of downstream target genes in the homozygous transgenic mice, it was inferred that the GATA4 M310V transgenic mouse model may be used to simulate ASD reasonably well. The effects of mutant GATA4 M310V were found to involve downstream genetic changes that may result in the development of CHD.
Materials and methods
Construction and breeding of GATA4 M310V transgenic mice
According to the human GATA4 encoding sequence, we performed gene synthesis, followed by site-specific mutagenesis and construct amplification in the pcDNA3.1-N-Myc-GATA4 (A928G) vector. The DNA constructs were then microinjected into mouse embryos to generate GATA4 M310V transgenic C57BL/6 mice. Each F0 transgene-positive transgenic mouse represented an individual transgenic line and was housed in a cage with wild-type mice (each cage contained 2 females and 1 male). All transgenic mice were genotyped by polymerase chain reaction (PCR) to screen for positive transgenic animals. The PCR products were verified by DNA sequencing. The animals were bred using the aforementioned method until the 4th generation. All animal experiments were carried out according to the approval of the Animal Studies Ethics Committee of the Peking University People’s Hospital, Beijing, China.
Evaluation of the genetic stability of the exogenous genes of GATA4 M310V transgenic mice
Fluorescence-based quantitative PCR (qPCR) was used to determine the copy number of transgenes in the mouse model and to further screen a stably inherited transgenic line in this study. The stably inherited transgenic mouse line was selected by qPCR based on the following criteria: the genomic DNA of wild-type mice was used as a control, the genomic DNA of transgenic mice was used as a test sample, and GATA4 and GCG were used as target fragments. Following confirmation of the sequence, the GATA4 M310V plasmid DNA and GCG plasmid DNA were 10-fold serially diluted with sterile distilled water to produce 109, 108, 107, 106, 105, 104 and 103 copy/μl standards. qPCR was used to amplify 2 sets of standards (the templates for GATA4 and GCG genes) and to generate a standard curve for amplification efficiency. The 2−ΔΔCt method (17) was used to measure the exogenous gene copy number of the transgenic mice. The formula was as follows: 2−ΔΔCt = 2−[(Ct GATA4 - Ct GCG)] in transgenic samples - [(Ct GATA4 - Ct GCG)] in wild-type samples.
Assessment of GATA4 protein expression
A total of 3 (4-week-old) F3 transgene-positive transgenic mice and their 3 wild-type littermates were selected. The protein expression of GATA4 in the heart tissue was measured by western blot analysis using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal reference. The rabbit polyclonal antibody to GATA4 (ab84593; Abcom, Cambridge, UK) which reacts with the human one was selected at the concentration of 1 μg/ml to assess GATA4 protein expression.
Comparative study of cardiac morphology and functions between transgenic and wild-type mice
The dead bodies of newborn heterozygous transgenic mice were collected [approximately one tenth of the heterozygous transgenic mice died shortly after birth (0–5 days after birth)]. Homozygous newborn mice and their wild-type littermates were euthanized by carbon dioxide inhalation. The animals were dissected by cutting open the chest to expose and remove the heart by cutting the root of the major arteries. The dissected hearts were rinsed in 0.01 mol/l phosphate buffer to remove the blood and were immediately placed in freshly prepared 4% neural buffered formalin. The geart tissue was then fixed for 18 h, followed by conventional dehydration, paraffin immersion and embedding procedures. The paraffin-embedded tissue was serially sectioned into 4-μm-thick slices and stained in hematoxylin and eosin (H&E) solution. The 8-week-old heterozygous transgenic mice and their wild-type littermates were selected and anesthetized with sodium pentobarbital (50 mg/kg body weight). The cardiac index was calculated and following imaging of the mice, their heart rates were carefully maintained at approximately 490 beats/min. An ultra-high resolution and real-time ultrasonic molecular imaging system (Vevo 2100; VisualSonics, Toronto, ON, Canada) was used to perform M-mode echocardiography, two-dimensional echocardiography and color Doppler echocardiography on the mice. Images of apical four-chamber view (A4C), parasternal long axis view of the left ventricle, and parasternal short axis view of the left ventricle (at the aortic valve level) were then collected. The basic data measurement and calculation of the heart and parameters of cardiac function were obtained using conventional methods. The experimental procedures were performed under double-blind conditions with regard to genotype. The Chi-square test and Fisher’s exact test were used to determine the difference in the incidence of ASD. The Student’s t-test was used to compare the independent sample results of echocardiography. P<0.05 represents a statistically significant difference.
Assessment of GATA4 downstream target gene expression
Total RNA from the heart tissues of the homozygous transgenic mice and their wild-type littermates (control mice) was extracted using TRIzol® reagent (Life Technologies, Grand Island, NY, USA). Two micrograms of RNA were immediately reverse-transcribed into cDNA using the Promega Reverse Transcription kit (Promega, Madison, WI, USA). The expression levels of the α-MHC, cTNC, GATA6, NKX2.5 and TBX5 genes were measured using the Applied Biosystems StepOnePlus™ real-time PCR system using the KAPA SYBR® FAST qPCR kit Master Mix (2X) Universal (Kapa Biosystems, Worburn, MA, USA). The primer sequences are listed in Table I. The relative average gene expression in the cardiac tissues of the transgenic mice and their wild-type littermates was calculated using GAPDH as an internal reference and the 2−ΔΔCt formula. Each sample was evaluated in triplicate on a qPCR plate. SPSS 13.0 statistical software (SPSS, Inc., Chicago, IL, USA) was used to perform non-parametric tests to screen 2 independent samples. P-values <0.05 were considered indicative of statistically significant differences.
Table I.
Primer sequences.
| Gene name | Primer sequences |
|---|---|
| GATA4 (PCR) | F: CCTTCGACAGCCCGGTCCT R: TGCACAGATAGTGACCCGTCCC |
| GATA4 (sequencing) | F: CTGTGCCAACTGCCAGACC R GTGCCCGTAGTGAGATGACAG |
| GATA4 (copy no.) | F: GGCCTCTCCTGTGCCAACTGC R CTTCATGTAGAGGCCGCAGGCA |
| GCG | F: AACATTGCCAAACGTCATGATG R: GCCTTCCTCGGCCTTTCA |
| GAPDH | F: CATCACTGCCACCCAGAAGACTG R: ATGCCAGTGAGCTTCCCGTTCAG |
| α-MHC | F: GCTGGAAGATGAGTGCTCAGAG R: CCAGCCATCTCCTCTGTTAGGT |
| cTNC | F: GATGGTTCGGTGCATGAAGGAC R: CTTCCGTAATGGTCTCACCTGTG |
| GATA6 | F: ATGCGGTCTCTACAGCAAGATGA R: CGCCATAAGGTAGTGGTTGTGG |
| TBX5 | F: CCAAAGACAGGTCTTGCGATTCG R: TTCTCCTCCCTGCCTTGGTGAT |
| NKX2.5 | F: GGTCTCAATGCCTATGGCTAC R: GCCAAAGTTCACGAAGTTGCT |
α-MHC, α-myosin heavy chain; cTNC, cardiac troponin C; NKX2.5, NK2 homeobox 5; TBX5, T-box transcription factor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; F, forward; R, reverse.
Results
Establishment and breeding of GATA4 M310V transgenic mice
All transgenic mice were genotyped using PCR to screen for positive transgenic animals (Fig. 1A). Genomic DNA was verified by sequencing (Fig. 1B). The animals were bred using the aforementioned method (as described in the Materials and methods) to the 4th generation. A stably inherited transgenic line was screened. The positive heterozygous transgenic mice were inbred to generate homozygous transgenic mice.
Figure 1.

(A) Agarose gel image illustrating the genotyping of the transgenic mouse model. The size of the PCR product was 168 bp. The arrow indicates the 200 bp size of the molecular marker. Lanes 1,3,5 represent the genomic DNA products of 3 positive transgenic mice after genotyping and PCR amplification. Lane 2 represents the corresponding region in the wild-type littermate that had no amplified PCR fragment. Lane 4 represents the negative result of water. This animal was used as a negative control in this experiment. (B) Direct DNA sequencing to confirm the presence of mutation. The arrow indicates the corresponding methionine to valine amino acid substitution of GATA4 M310V point mutation.
Evaluation of genetic stability of exogenous genes in GATA4 M310V transgenic mice
The GATA4 M310V application and melting curves are illustrated in Fig. 2C and D, and the GATA4 M310V standard curve is illustrated in Fig. 2F. No sign peaks were found in the melting curve and no primer dimers or other non-specific amplification were observed. The data obtained by PCR amplification were therefore considered reliable. The correlation coefficient of the GCG standard curve coefficient was R2=0.999 with 101% amplification efficiency, and the correlation coefficient of the GATA4 standard curve was R2=0.988, with 97% amplification efficiency. The 2−ΔΔCt method of qPCR revealed that the amplification efficiency between the reference and target genes was very close, ranging from 90–110%.
Figure 2.

(A and D) Amplification curves; (C and F) standard curves; (B and E) melting curves of 102, 103, 104, 105, 106, 107 and 108 copies of the mutant GATA4 M310V plasmid and single copy GCG gene.
Protein expression of GATA4
To assess the changes in GATA4 protein expression in the hearts of F3 transgene-positive transgenic mice, western blot analysis was used to analyze the protein expression levels of GATA4. The results revealed that the protein expression level of GATA4 was higher in the hearts of the transgenic mice than in those of the wild-type mice (P<0.05; Fig. 3).
Figure 3.

Representative result of western blot analysis. GATA4 protein expression in wild-type and F3 transgene-positive transgenic mouse hearts. In transgenic mouse hearts, the protein expression of GATA4 was increased (P<0.05). TG, transgenic; WT, wild-type.
Cardiac phenotype of heterozygous transgenic mice Observation of cardiac structure in newborn mice
Approximately one tenth of the heterozygous transgenic mice died shortly after birth (0–5 days after birth). To observe the cardiac structural defects of the GATA4 M310V heterozygous transgenic mice, the dead newborn pups were collected and their hearts were dissected and subjected to conventional histological analysis. H&E staining of the serial heart sections revealed that, among the 15 newborn heterozygous mice, 9 mice (60%) had the ASD phenotype in their hearts (Fig. 4A) and 6 mice (40%) had no obvious structural defect. A total of 20 newborn wild-type littermates were selected as the controls and only 1 mouse (5%) had the ASD phenotype in its heart. The remaining mice had no obvious structural defects (Fig. 4B). The details of the transgenic and wild-type mice regarding cardiac structure are presented in Table II, suggesting that GATA4 M310V produces the phenotype of ASD in the heart.
Figure 4.

Hematoxylin and eosin (H&E) staining of coronal heart sections in the newborn mice. (A) Representative coronal heart autopsy section of neonatal heterozygous transgenic mouse. It shows an incomplete atrial septum with connective blood cells between the left and the right atria, but no obvious morphological abnormality of the myocardial cells. (B) Representative coronal heart sections of the wild-type littermates. No obvious abnormalities in cardiac tissue structure and myocardial cell morphology were observed. (A) Newborn heart of GATA4 M310V transgenic mouse. (B) Newborn heart of wild-type control mouse. LA, left atrium; RA, right atrium; LV, left ventricle; RV, right ventricle.
Table II.
Results obtained from the heart sections on the incidence of ASD between the transgenic mice and the wild-type mice.
| Heart section results | Mouse group
|
|
|---|---|---|
| GATA4 M310V heterozygous transgenic mice (n=15) | Wild-type mice (n=20) | |
| ASD | 60% (9)a | 5% (1) |
| Normal | 40% (6) | 95% (19) |
The Chi-square test was used to analyze the 4-fold table. Fisher’s exact test was used to determine the difference in the incidence of ASD between the transgenic and wild-type mice.
P<0.05 represents a statistically significant difference between 2 groups. ASD, atrial septal defect.
Echocardiography for the diagnosis and assessment of cardiac structure and function in the mice
To access the differences in cardiac structure and function between the transgenic mice and their wild-type littermates, 10 transgenic and 6 wild-type mice (8-weeks old) were selected for M-mode echocardiography, two-dimensional echocardiography and color Doppler echocardiography using the Vevo 2100 ultra-high resolution and real-time ultrasonic molecular imaging system. A4C was selected when using color Doppler echocardiography to assess atrial septal integrity. Horizontal sections of the parasternal long axis view of the left ventricle and the parasternal short axis view of the left ventricle (at the aortic valve level) were selected under color Doppler echocardiography and M-mode echocardiography to indicate the aortic and pulmonary artery flow velocities. Among the heterozygous transgenic mice, 8 out of the 10 heterozygous transgenic mice presented with intermittent blood flow occurring between the left and right atria; however, only 1 out of the 6 wild-type littermates presented with the similar symptom (significant difference between groups, P<0.05) (Table III and Fig. 5. The results of color Doppler echocardiography on assessing aortic and pulmonary artery peak velocities and pressure revealed that 6 out of the 10 heterozygous transgenic mice exhibited mild pulmonary stenosis, whereas no abnormality was observed in their wild-type littermates. No statistically significant difference was observed in left ventricular function, left ventricular size and left ventricular wall thickness between the heterozygous transgenic mice and their wild-type littermates (Table IV).
Table III.
The echocardiography results about the incidence of ASD in transgenic and the wild-type mice.
| Echocardiography | Mouse group
|
|
|---|---|---|
| GATA4 M310V heterozygous transgenic mice (n=10) | Wild-type mice (n=6) | |
| ASD | 80% (8)a | 17% (1) |
| Normal | 20% (2) | 83% (5) |
The Chi-square test was used to analyze the 4-fold table. Fisher’s exact test was used to determine the difference in the incidence of ASD between the transgenic and wild-type mice.
P<0.05 represents a statistically significant difference between 2 groups. ASD, atrial septal defect.
Figure 5.

Echocardiography showing an atrial septal defect (ASD) in the heterozygous transgenic mice: (A–D) color Doppler echocardiographic images; and (E–F) pulsed Doppler echocardiographic images. (A) An apical four chamber view of the heterozygous mouse heart, displaying the blood circulation between the atria. (B) An apical four chamber view of the wild-type mouse heart, with no obvious abnormality. (C and D) Images of pulmonary valve peak velocity of the heterozygous transgenic mouse and its wild-type littermate, respectively. (E and F) The pulsed Doppler echocardiographic images across the pulmonary valves of the heterozygous transgenic mouse and its wild-type littermate, respectively. (G and H) The pulsed Doppler echocardiographic images across the aortic valves of the heterozygous transgenic mouse (G) and its wild-type littermate (H). RV, right ventricle; LV, left ventricle; RA, right atrium; LA, left atrium; AO, aorta.
Table IV.
Echocardiographic measurements.
| Measurement | Wild-type mice (n=6) | Transgenic mice (n=9) |
|---|---|---|
| Aortic peak velocity (AV Peak Vel; mm/sec) | 788.47±72.71 | 919.40±195.79 |
| Aortic (AV) peak pressure (mmHg) | 2.43±0.53 | 3.51±1.51 |
| Pulmonary artery peak velocity (PV Peak Vel; mm/sec) | −640.20±32.89a | −879.44±213.58 |
| Pulmonary (PV) artery peak pressure (mmHg) | 1.66±0.16a | 3.13±1.63 |
| End-diastolic left ventricular anterior wall thickness (LVAW; day, mm) | 0.80±0.07 | 0.89±0.11 |
| End-systolic left ventricular anterior wall thickness (LVAW; sec, mm) | 1.29±0.23 | 1.45±0.16 |
| End-diastolic left ventricular diameter (LVID; day, mm) | 3.30±0.48 | 3.39±0.28 |
| End-systolic left ventricular diameter (LVID; sec, mm) | 2.17±0.66 | 2.04±0.24 |
| End-diastolic left ventricular posterior wall thickness (LVPW; day, mm) | 0.71±0.09 | 0.80±0.13 |
| End-systolic left ventricular posterior wall thickness (LVPW; sec, mm) | 1.16±0.18 | 1.28±0.17 |
| Ejection fraction (EF, %) | 64.17±17.23 | 71.05±7.28 |
| Fractional shortening (FS, %) | 35.29±12.13 | 39.72±5.79 |
| Left ventricular end-diastolic volume (LV Vol; day, μl) | 45.45±14.94 | 47.49±9.34 |
| Left ventricular end-systolic volume (LV Vol; sec, μl) | 17.75±13.17 | 13.68±4.09 |
| Heart rate (HR, bpm) | 491.00±7.75 | 487.67±6.02 |
Results are represented as the means ± standard deviation. The Student’s t-test was used to compare the independent samples between 2groups.
P<0.05 represents a statistically significant difference between 2 groups. Heart rate was maintained at approximately 490 beats/min. Pulmonary artery peak velocity and pressure in the transgenic mice were higher than those in their wild-type littermates (P<0.05).
Expression of GATA4 target genes
To assess the changes in GATA4 transcript target gene expression in the hearts of teh homozygous transgenic mice, RNA was extracted from the hearts of newborn homozygous mice and qPCR was used to analyze the expression levels of the α-MHC, cTNC, GATA6, NKX2.5 and TBX5 genes. The results revealed that expression levels of α-MHC, as a GATA4 downstream target gene, was lower in the hearts of the homozygous transgenic mice than in those of the wild-type mice (P<0.05; Fig. 6), suggesting that the introduction of GATA4 M310V inhibits the normal expression of this downstream gene. However, the expression level of cTNC, as a GATA4 downstream target gene, was higher in the hearts of the homozygous transgenic mice than in those of the wild-type mice (P<0.05), in accordance with the increased protein expression of GATA4. There was no change in the expression levels of other GATA4 downstream genes (NKX2.5 and GATA6) and GATA4 coactivating genes (TBX5). It was thus inferred that the introduction of the GATA4 M310V exogenous gene suppressed the expression of the GATA4 downstream gene, α-MHC, which may subsequently cause the abnormal development of atrial septum in the hearts of mice.
Figure 6.
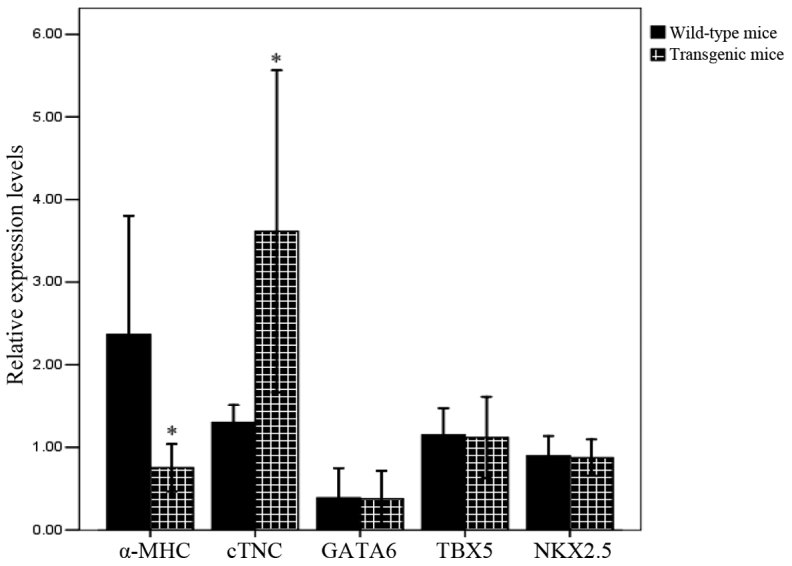
Gene expression in the heart tissue of homozygous transgenic mice. qPCR results demonstrated that the expression of the α-myosin heavy chain (α-MHC) gene in the hearts of homozygous transgenic mice was significantly lower than that in the hearts of their wild-type littermates (*P<0.05), while the expression of the cTNC gene in the hearts of homozygous transgenic mice was significantly higher than that in the hearts of their wild-type littermates (*P<0.05). However, no significant difference was observed in the expression of GATA6, TBX5 or NKX2.5 between the 2 groups.
Discussion
The findings of the present study are as follows: i) the GATA4 gene plays an important role in the development of ASD; ii) the GATA4 downstream gene, α-MHC, is associate with GATA4 function. The altered expression of this gene affects the normal cardiac atrial septal formation during development. This is associated with the incidence of ASD. The conclusions of this study were based on the following: i) in a previous study of ours on GATA4 mutation screening among patients with CHD revealed that 8 patients with simple ASD from a Chinese family with a history of CHD demonstrated the point mutation of GATA4 M310V (9). ii) In another previous study of ours, in vitro experiments revealed that GATA4 mutation affected its dose-positive correlation with α-MHC activity (16). iii) GATA4 M310V transgenic mice developed for the purpose of this study revealed ASD phenotypes. iv) GATA4 downstream gene (α-MHC) expression in the hearts of GATA4 M310V transgenic mice was significantly lower compared with the wild-type littermates (P<0.05). In short, it was here inferred that GATA4 M310V mutation may result in some functional defects, which suppress the normal expression of GATA4 downstream genes and affect the normal development of the cardiac septum, resulting in the development of ASD.
ASD, in which blood flows between the left and right atria of the heart, is a very common form of CHD. However, the incidence of ASD and its etiology remain unclear. GATA4 gene mutations have been demonstrated in many human families and are associated with ASD and pulmonary stenosis (9,11,18). In a previous study by our group, we presented a representative family with familial ASD (9).
The GATA4 gene is required for normal heart development (2). In addition, the GATA4 gene is a regulatory factor for heart development and dose-related adjustment (19). Different degrees of GATA4 gene defect have been found to produce ASD in a mouse model, producing a variety of heart malformations (14). In this study, a GATA4 M310V transgenic mouse model presenting with ASD phenotypes was developed. Echocardiography and histological examination were used on the cardiac sections to determine the differences in cardiac structure between the heterozygous transgenic mice and their wild-type littermates. The heterozygous transgenic mice carried the same ASD phenotypes as the 8 patients with simple ASD from a previously tested family with CHD. It was here inferred that these specific phenotypes of simple ASD are triggered by GATA4 gene mutation during heart formation, resulting in partially functional defects in the heart.
GATA4 is involved in the regulation of the expression of cardiac structural genes, including α-MHC, cTNC and ANF (3,4). It has been found that the conserved GATA sites are in the GATA4 regulatory region of many cardiac promoters and GATA4 and potently activate their promoters (20). α-MHC is the major contractile protein in the heart. Two putative GATA-binding sites are in the proximal enhancer of the α-MHC gene which suggests that GATA4 regulates its expression (21). Both MEF2C and GATA4 activate the gene expression of ANF and α-MHC (22). The cTNC gene has been used to examine the molecular mechanisms that regulate cardiac muscle-specific transcription. It has been proven that GATA4 binds specifically to the CEF-1 site of the cTNC enhancer (23). GATA6, another member of the GATA family, is also critical to cardiac development and disorders. It has been reported that GATA4 and GATA6 act cooperatively in the heart (24). NKX2.5 is a restrictive transcription factor of the heart and is involved in normal cardiac development and the function of the cardiac conduction system (25–27). NKX2.5 regulates transcription and often interacts with other transcriptional factors, including GATA4, MEF2C (25,28,29) and Hand2 (30). The TBX5 gene plays a key role in cardiac morphology and the conduction system. Model mice with a missing copy of the T-box transcription factor, TBX, have ASD and sporadic ventricular septal defect (VSD) (31).
In a previous in vitro study published by our group, we demonstrated that following the introduction of the GATA4 expression vector using α-MHC promoter, the increased concentration of GATA4 expression vector also enhanced its promoter activity (16) (P<0.05). However, as we demonstrated in the present study, the increased concentration of the mutant GATA4 expression vector reduced the activity of the α-MHC promoter. qPCR revealed that the expression of the GATA4 downstream target gene, α-MHC, in the heart tissues of the homozygous mice was lower than that in the heart tiussues of the wild-type control mice (P<0.05), suggesting that the introduction of GATA4 M310V inhibits the normal expression of this downstream gene. However, the expression level of cTNC, as a GATA4 downstream target gene, were higher in the hearts of the homozygous transgenic mice than in those of the wild-type mice (P<0.05), in accordance with the increased protein expression of GATA4. However, the expression of GATA6, NKX2.5 and TBX5 was not altered. It was thus inferred that the GATA4 M310V point mutation may result in the amino acid substitution of methionine to valine in a corresponding position, leading to functional defects in GATA4. This change also caused the suppression of downstream gene α-MHC expression. The α-MHC gene may be associated with the ASD phenotype of GATA4 M310V transgenic mice.
This study focused on an important familial mutation of the GATA4 gene found in a clinical study to further establish the findings of basic research. In this study, a GATA4 M310V transgenic mouse model of ASD phenotypes was developed. Compared with the heart autopsies of embryonically lethal mice and hearts from newborn GATA4 knockout mice, this transgenic mouse line had a longer survival time and ensured the stable inheritance of the exogenous gene through the transgenic line. This mutant transgenic mouse model may be suitable for research into human CHD. This mouse model may facilitate the study of the genetic and environmental modifications that cause cardiac malformations. It may also help the in-depth understanding of the mechanisms underlying septal defects. This mouse model demonstrated a specific gene mutation which may lead to the functional defects in the animals. Studies on the potential function of GATA4 genes are warranted to determine the effect and changes associated with GATA4 mutation.
References
- 1.Bentham J, Bhattacharya S. Genetic mechanisms controlling cardiovascular development. Ann NY Acad Sci. 2008;1123:10–19. doi: 10.1196/annals.1420.003. [DOI] [PubMed] [Google Scholar]
- 2.Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- 3.Brunskill EW, Witte DP, Yutzey KE, Potter SS. Novel cell lines promote the discovery of genes involved in early heart development. Dev Biol. 2001;235:507–520. doi: 10.1006/dbio.2001.0313. [DOI] [PubMed] [Google Scholar]
- 4.Temsah R, Nemer M. GATA factors and transcriptional regulation of cardiac natriuretic peptide genes. Regul Pept. 2005;128:177–185. doi: 10.1016/j.regpep.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 5.Sarkozy A, Conti E, Neri C, D’Agostino R, Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A, Dallapiccola B. Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet. 2005;42:e16. doi: 10.1136/jmg.2004.026740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garg V, Kathiriya IS, Barnes R, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 7.Christoforou N, Chellappan M, Adler AF, Kirkton RD, Wu T, Addis RC, Bursac N, Leong KW. Transcription factors MYOCD, SRF, Mesp1 and SMARCD3 enhance the cardio-inducing effect of GATA4, TBX5, and MEF2C during direct cellular reprogramming. PLoS One. 2013;8:e63577. doi: 10.1371/journal.pone.0063577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–3942. doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Han ZQ, Yan WD, Tang CZ, Xie JY, Chen H, Hu DY. A novel mutation in GATA4 gene associated with dominant inherited familial atrial septal defect. J Thorac Cardiovasc Surg. 2010;140:684–687. doi: 10.1016/j.jtcvs.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Xiang R, Fan LL, Huang H, Cao BB, Li XP, Peng DQ, Xia K. A novel mutation of GATA4 (K319E) is responsible for familial atrial septal defect and pulmonary valve stenosis. Gene. 2014;534:320–323. doi: 10.1016/j.gene.2013.10.028. [DOI] [PubMed] [Google Scholar]
- 11.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 12.Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, Garg V. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol. 2009;326:368–377. doi: 10.1016/j.ydbio.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laforest B, Nemer M. GATA5 interacts with GATA4 and GATA6 in outflow tract development. Dev Biol. 2011;358:368–378. doi: 10.1016/j.ydbio.2011.07.037. [DOI] [PubMed] [Google Scholar]
- 14.Rajagopal SK, Ma Q, Obler D, et al. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol. 2007;43:677–685. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Misra C, Sachan N, McNally CR, Koenig SN, Nichols HA, Guggilam A, Lucchesi PA, Pu WT, Srivastava D, Garg V. Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet. 2012;8:e1002690. doi: 10.1371/journal.pgen.1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Y, Han Z, Han H, Chen Y. Mechanism involved in the familial atrial septal defects caused by GATA-4 M310V single gene mutation. China Circ J. 2013;3:211–214. In Chinese. [Google Scholar]
- 17.Ballester M, Castelló A, Ibáñez E, Sánchez A, Folch JM. Real-time quantitative PCR-based system for determining transgene copy number in transgenic animals. Biotechniques. 2004;37:610–613. doi: 10.2144/04374ST06. [DOI] [PubMed] [Google Scholar]
- 18.Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat. 2006;27:293–294. doi: 10.1002/humu.9410. [DOI] [PubMed] [Google Scholar]
- 19.Zeisberg EM, Ma Q, Juraszek AL, Moses K, Schwartz RJ, Izumo S, Pu WT. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J Clin Invest. 2005;115:1522–1531. doi: 10.1172/JCI23769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pu WT, Ishiwata T, Juraszek AL, Ma Q, Izumo S. GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Dev Biol. 2004;275:235–244. doi: 10.1016/j.ydbio.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 21.McBride K, Nemer M. Regulation of the ANF and BNP promoters by GATA factors: Lessons learned for cardiac transcription. Can J Physiol Pharmacol. 2001;79:673–681. doi: 10.1139/y01-037. [DOI] [PubMed] [Google Scholar]
- 22.Molkentin JD, Kalvakolanu DV, Markham BE. Transcription factor GATA-4 regulates cardiac muscle-specific expression of the alpha-myosin heavy-chain gene. Mol Cell Biol. 1994;14:4947–4957. doi: 10.1128/mcb.14.7.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morin S, Charron F, Robitaille L, Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000;19:2046–2055. doi: 10.1093/emboj/19.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ip HS, Wilson DB, Heikinheimo M, Tang Z, Ting CN, Simon MC, Leiden JM, Parmacek MS. The GATA-4 transcription factor transactivates the cardiac muscle-specific troponin C promoter-enhancer in nonmuscle cells. Mol Cell Biol. 1994;14:7517–7526. doi: 10.1128/mcb.14.11.7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317:614–619. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jay PY, Harris BS, Maguire CT, Buerger A, Wakimoto H, Tanaka M, Kupershmidt S, Roden DM, Schultheiss TM, O’Brien TX, Gourdie RG, Berul CI, Izumo S. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest. 2004;113:1130–1137. doi: 10.1172/JCI19846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terada R, Warren S, Lu JT, Chien KR, Wessels A, Kasahara H. Ablation of Nkx2-5 at mid-embryonic stage results in premature lethality and cardiac malformation. Cardiovasc Res. 2011;91:289–299. doi: 10.1093/cvr/cvr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda M, Briggs LE, Wakimoto H, Marks MH, Warren SA, Lu JT, Weinberg EO, Robertson KD, Chien KR, Kasahara H. Slow progressive conduction and contraction defects in loss of Nkx2-5 mice after cardiomyocyte terminal differentiation. Lab Invest. 2009;89:983–993. doi: 10.1038/labinvest.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlesinger J, Schueler M, Grunert M, Fischer JJ, Zhang Q, Krueger T, Lange M, Tönjes M, Dunkel I, Sperling SR. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 2011;7:e1001313. doi: 10.1371/journal.pgen.1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamagishi H, Yamagishi C, Nakagawa O, Harvey RP, Olson EN, Srivastava D. The combinatorial activities of Nkx2.5 and dHAND are essential for cardiac ventricle formation. Dev Biol. 2001;239:190–203. doi: 10.1006/dbio.2001.0417. [DOI] [PubMed] [Google Scholar]
- 31.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–721. doi: 10.1016/S0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]