

Summary
RdgB is a bacterial dNTPase with a strong in vitro preference for non-canonical DNA precursors dHapTP, dXTP and dITP that contain deaminated or aminogroup-modified purines. Utilization of these nucleotides by replisomes in rdgB mutants of E. coli produces modified DNA, on which EndoV-nicking near the base analogs initiates excision repair. Some EndoV-initiated excision events cause chromosomal fragmentation, which becomes inhibitory if recombinational repair is also inactivated (the rdgB recA co-inhibition). To reveal the sources and the identities of the non-canonical DNA precursors, intercepted by RdgB in E. coli, we characterized 17 suppressors of the rdgB recA co-inhibition. Ten suppressors affect genes of the RNA/DNA precursor metabolism, identifying the source of non-canonical DNA precursors. Comparing chromosomal fragmentation with the density of EndoV-recognized DNA modifications distinguishes three mechanisms of suppression: 1) reduction of the non-canonical dNTP production; 2) inhibition of the base analog excision from DNA; 3) enhancement of the cell tolerance to chromosomal fragmentation. The suppressor analysis suggests IMP as the key intermediate in the synthesis of the clastogenic DNA precursor, most likely dITP.
Keywords: rdgB, recA, dITP, clastogenesis, IMP, EndoV
Introduction
Rare non-canonical DNA precursors are used by DNA polymerases (Bessman et al., 1958), resulting in DNA containing base analogs, such as uracil, hypoxanthine, hydroxylaminopurine or 8-oxo-guanine, all subject to removal by excision repair. Excision repair goes through the obligate intermediate with a single-stranded interruption (Friedberg et al., 2006). It is proposed that, in growing cells, such single strand interruptions are occasionally encountered by replication forks, causing chromosomal fragmentation (Kouzminova & Kuzminov, 2006, Kuzminov, 2001). The cell employs recombinational repair of double-strand breaks, mediated by RecA, RecBCD and RuvABC in Escherichia coli (Kuzminov, 1999), to reassemble chromosomes fragmented as a result of base analog incorporation and subsequent excision (Bradshaw & Kuzminov, 2003, Burgis et al., 2003, Kouzminova & Kuzminov, 2004, Lukas & Kuzminov, 2006, Ting et al., 2008).
The non-canonical nucleotides, which contain deaminated and aminogroup-modified purine bases (hypoxanthine, xanthine and 6-N-hydroxylaminopurine (Fig. 1)), are either shown or suspected to incorporate during replication and increase the frequency of single strand repair interruptions in the chromosomal DNA (Abdul-Masih & Bessman, 1986, Bessman et al., 1958, Burgis et al., 2003, Myrnes et al., 1982, Piccirilli et al., 1990, Thomas et al., 1979). All three base analogs are actively removed from DNA in E. coli by excision repair initiated by endonuclease V (EndoV) (Burgis et al., 2003, He et al., 2000, Yao et al., 1994, Yao & Kow, 1995). To reduce the frequency of excision, the cell intercepts the corresponding deoxyribonucleoside triphosphates with the help of a specialized dNTPase, called RdgB in E. coli, HAM1 in yeast, and ITPA in humans (Bradshaw & Kuzminov, 2003, Burgis & Cunningham, 2007, Chung et al., 2002, Hwang et al., 1999, Kozmin et al., 1998, Lin et al., 2001, Noskov et al., 1996)). Although in E. coli, hydroxylaminopurine incorporation is primarily reduced by molybdoenzymes (Burgis et al., 2003, Kozmin et al., 2000, Kozmin & Schaaper, 2007), the RdgB enzyme of E. coli is highly specific towards non-canonical nucleotides containing not only hypoxanthine and xanthine, but also hydroxylaminopurine (Burgis & Cunningham, 2007, Chung et al., 2002), and the structural basis of this specificity has recently been determined (Savchenko et al., 2007). In the absence of the RdgB enzyme, E. coli cells accumulate EndoV-recognized modifications in its DNA (Bradshaw & Kuzminov, 2003, Budke & Kuzminov, 2006), supposedly leading to a higher density of EndoV nicking and elevated chromosomal fragmentation. Rec+ cells can repair the resulting double-strand breaks, but the recA mutants cannot, which results in the rdgB recA co-inhibition (Bradshaw & Kuzminov, 2003, Clyman & Cunningham, 1987, Lukas & Kuzminov, 2006). The poisoning of the double mutant is due to the nfi gene-coded EndoV, because the rdgB recA nfi triple mutant grows at the wild type rates and with no signs of chromosomal fragmentation (Bradshaw & Kuzminov, 2003). In addition to the complete suppression of the rdgB recA co-inhibition by the nfi inactivation, both strong and weak non-nfi suppressors could be also isolated that were suspected to act by reducing the incorporation of EndoV-recognized DNA modifications (Jill Bradshaw, Scott Horrell and A.K., unpublished).
Fig. 1. Deaminated or amino group-modified base analog toxicity and the expected types of suppressors of the rdgB recA co-inhibition.

Hap, 6-N-hydroxylaminopuriune; ba, base analog; nc-, non-canonical (for dNTP or dNMP). Stages are identified by the genes. Numbers in the stars denote the segment of the metabolic scheme where suppressors are possible: #1, decreased production of nc-dNTP; #2, increased alternative interception of nc-dNTP; #3, decreased excision of the base analog from DNA; #4, slowing down replication or accelerating the excision repair; #5, ameliorating the recombinational repair defect of the recA(Ts) mutant; #6, tolerance to double-strand DNA breaks.
In this work we systematically isolated and characterized these non-nfi suppressors of the rdgB recA co-inhibition with the purpose of revealing the sources and identities of the non-canonical DNA precursors, intercepted by RdgB. Since nucleotides with deaminated and aminogroup-modified purine bases can readily form in vitro as a result of chemical (Shapiro & Pohl, 1968) or enzymatic (Clement & Kunze, 1990) oxidation, they could similarly form in vivo as a result of metabolic oxidation. Alternatively, they could be spurious by-products of normal nucleotide metabolism. The enzymatic activities inactivated by the non-nfi suppressors of the rdgB recA co-lethality should distinguish between the two ideas. If these suppressors inactivate the genes involved in production or handling of strong oxidizers in the cell, this would argue in favor of metabolic oxidation as the source of the non-canonical DNA precursors, intercepted by RdgB. Alternatively, if the non-nfi suppressors inactivate various genes involved in the RNA and DNA precursor production, this would argue that the non-canonical DNA precursors are byproducts of the normal nucleotide metabolism.
Results
Rationale and expectations
The current knowledge of the nucleotide metabolism in E. coli, of the RdgB in vitro substrates, and of the EndoV-initiated excision (Burgis & Cunningham, 2007, Kow, 2002, Neuhard & Nygaard, 1987) can be assembled in a multistep model that explains the severe growth inhibition of the rdgB recA(Ts) mutants at the non-permissive temperatures as being ultimately the result of accumulation of unrepaired double-strand DNA breaks (Fig. 1). The model predicts several steps at which the rdgB recA growth inhibition could be suppressed: 1) inactivation of spurious oxidation/deamination of the canonical DNA precursors or inactivation of genes of normal nucleotide metabolism; 2) activation of alternative interceptors of the non-canonical dNTP; 3) inactivation of base analog excision from DNA; 4) decelerating replication or accelerating excision repair; 5) ameliorating the recA defect (we used a point mutation resulting in a temperature-sensitive RecA protein); 6) tolerance to clastogenesis. In addition, since the three base analogs use distinct set of enzymes to become nucleotides (Fig. 1, top), the identity of suppressors in the nucleotide metabolism may point to a particular base analog that is intercepted by RdgB in the dNTP form.
Suppressors of rdgB recA(Ts)
To isolate suppressors, we conducted insertional mutagenesis of the rdgB recA(Ts) double mutant and plated the mutagenized cells at the non-permissive temperature, analyzing the survivors. Most of the suppressors restored growth of the rdgB recA(Ts) double mutant at 42°C partially, while a few of them, like inactivations of hpt, suppressed completely, as judged by the close match between 30°C and 42°C titers (Fig. 2A). A total of 16 genes were inactivated two or more times, the nfi gene being the champion with 18 hits. Although some of the mutations that were isolated only once were likely to be real suppressors (especially those that belonged to metabolism of DNA or DNA precursors), we further characterized only mutants that were isolated more than once (Table 1). The complete list of isolated suppressors can be found in Table S4.
Fig. 2. Isolation of suppressors of the rdgB recA co-inhibition and quantification of their effect on growth of the double mutant.

A. Spotting serial dilutions of cultures grown at 28°C and subsequently incubating them at 43°C reveals the rdgB recA(Ts) synthetic inhibition, as well as its suppression. The strains are: WT, AB1157; rdgB recA, (JB30); rdgB recA deoB, (BB004); rdgB recA dusB, (BB020); rdgB recA hpt, (BB028); rdgB recA dgt, (BB036). B. Steps of the protocol used for quantification of growth inhibition in the double mutant and the extent of its suppression: 1) a saturated overnight culture grown at 28°C is diluted 10−5-fold; 2) 10 μl spots are incubated on an LB plate at 28°C or 42°C for 17 hours, 3) agar stubs with the spotted cells are cut out of the plate and resuspended in saline for 1 hour; 4) the cell suspensions are serially diluted, spotted by 10 μl onto LB agar and incubated at room temperature to determine the titer for each spot at the original temperatures. The titer of growth at 42°C divided by the titer of growth at 30°C (28°C) represents “growth” or its inhibition and is reported in “C”. C. Quantification of growth inhibition and its suppression by the protocol illustrated in “B”. The values are means of 4 to 20 independent measurements ± SE. D. Additional copies of the functional genes, identified by the most numerous suppressors, further decrease viability of the double rdgB recA mutant. pK80, low copy-number vector; pBB13, pK80 carrying nfi, deoB, deoD, hpt, purR, dgt, and tdk functional genes.
Table 1.
Suppressors of rdgB recA inviability that were further characterized.
| Insertion Suppressors | ||||
|---|---|---|---|---|
| #II* | Gene | Function of the protein | Original assignment** | Final assignment*** |
| 18 | nfi | Endonuclease V (deoxyinosine 3′-endonuclease) | 3 | 3 |
| 15 | dgt | dGTP triphosphohydrolase | 1 | 1 or 6 |
| 9 | tdk | Thymidine / deoxyuridine kinase | 1 | 1 or 6 |
| 5 | hpt | Hypoxanthine-guanine phosphoribosyltransferase | 1 | 1 |
| 3 | purR | PurR-Hypoxanthine transcriptional repressor | 1 | 1 or 6 |
| 3 | yecT | Hypothetical protein | ? | — (out) |
| 3 | yrdD | 180 AAs hypothetical protein, homology to DNA topoisomerases | ? (3 or 5) | 3 |
| 2 | deoB | Phosphopentomutase | 1 | 1 |
| 2 | deoD | Purine nucleoside phosphorylase | 1 | 1 |
| 2 | dusB | tRNA dihydrouridine synthase | ? (4, 5, 6) | 1, 2 or 4 |
| 2 | glnG | NtrC transcriptional dual regulator | ? (4, 5, 6) | — (out) |
| 2 | gmk | Deoxyguanylate kinase | 1 | 1 |
| 2 | hdfR | HdfR transcriptional repressor of flagellar master operon flhDC | ? (4, 5, 6) | 1, 2 or 4 |
| 2 | putA | Fused PutA transcriptional repressor / proline dehydrogenase | ? (4, 5, 6) | 3 |
| 2 | putP | Transmembrane proline transporter | ? (4, 5, 6) | — (out) |
| 2 | rpoC | RNA polymerase β′ subunit | ? (4, 5, 6) | 4 or 6 |
| Multicopy suppressors | ||||
| #II* | Gene | Function of the protein | ||
|
|
||||
| 29 | yjjX | ITPase/XTPase | 2 | 2 |
| 16 | purA | Subunit of adenylosuccinate synthetase | 1 | 1 |
| 7 | rnb | Ribonuclease II (tRNA processing, mRNA degradation) | ? (4, 5, 6) | 1, 2 or 4 |
| 1 | nepI | Inosine and guanosine ribonucleoside exporter | 1 | 3 |
To further characterize the inactivational suppressors that were isolated at least twice, we constructed precise deletions of the corresponding non-essential genes, with removal of the drug-resistance markers. In most cases the precise deletions suppressed as well as the original pRL27 inserts, but there were a few exceptions. In contrast to the weakly suppressing pRL27 insertions into putP, a complete deletion of the gene did not suppress, suggesting that either suppression required the parts of the proteins upstream of the inserts or that a neighboring gene was involved (likely putA, whose precise deletion did suppress). A precise deletion of yecT also did not suppress; however, we expected this result, as all three pRL27 “yecT” inserts were, actually, in the intergenic region between argS and yecT, upstream of yecT and co-directional with it. Yet, neither deleting the argS-yecT intergenic region, nor raising the yecT+ copy number by cloning it onto a multicopy plasmid (Table S3) suppressed the rdgB recA inhibition. The glnG suppression turned out to be an artefact of our calculation of the suppression as a ratio of 42°C titer to 30°C titer — the ΔglnG mutation did not increase the 42°C titer of the rdgB recA(Ts) mutant, but it did decrease its 30°C titer, which made it appear as a suppressor. The removal of putP, yecT and glnG mutants shortened the list of “suppressors to characterize” to 13. Finally, since the ΔrpoC::cat construct suppressed slightly better than the ΔrpoC construct from which the cat gene was removed, we show only results with the former in the major analyses, but with the latter in the epistatic analysis, where weaker individual phenotypes increase resolution.
Of the thirteen suppressor genes, two are involved in the DNA metabolism (nfi and, tentatively, yrdD), whereas seven can be classified as genes of the DNA precursor metabolism (dgt, tdk, hpt, purR, deoD, deoB, and gmk). We also isolated suppressors in genes that were hard to classify (dusB, hdfR, putA, and rpoC). Two of the genes on the list, gmk (Baba et al., 2006) and rpoC, are essential, with pRL27 inserts landing in the very end of the gene (rpoC) or just outside the termination codon (gmk). For mutations in rpoC, we deleted the part of the gene from the place of the earlier insert, which removed the last 138 bp of the gene. For gmk, we constructed an antidirectional cat insertion 44 bp downstream of the stop codon to mimic the antisense effect of pRL27 inserts. Both the partial rpoC deletion and the gmk antisensing construct still suppressed.
In addition to insertional suppressors, we also identified multicopy suppressors of the rdgB recA(Ts) co-inhibition. One of the most frequently isolated suppressors in this category was the purA+ gene, previously identified as an rdgB recA multicopy suppressor (Clyman & Cunningham, 1991). We also identified yjjX+, rnb+ and nepI+ as multicopy suppressors. Multicopy suppressors were confirmed by inactivating the suspected suppressor gene with a partial deletion (Table S3); none of these inactivated plasmids suppressed the rdgB recA(Ts) inhibition at 42°C. All 17 of the constructed mutants and multicopy suppressors were characterized with several assays that allowed us to sort them into three separate groups.
Growth rate of the suppressed strains
Although our original rdgB recA(Ts) double mutant almost fails to grow at 42°C, it is not truly co-lethal, but rather shows a severe synthetic inhibition. While suppression of synthetic lethality can be quantified simply by the colony forming unit titer of the suppressed strains, quantifying the degree of suppression of a co-inhibited combination requires a more sophisticated approach. We quantified the degree of growth inhibition in the double mutant at 42°C, as well as acceleration by each of our suppressor mutants in the ΔrdgB62 recA200(Ts) background by the assay that we originally developed for the pta recBC(Ts) co-inhibited strains (Shi et al., 2005). This is an integral assay for both viability and the rate of growth (Fig. 2B) that shows more than 100,000-fold difference between the wild type and the double mutant (Fig. 2C, the left two bars).
From this analysis, we can distinguish three classes of suppressors (Fig. 2C). The deletions that enhance growth of the double mutant almost to the wild type levels (10,000–100,000-fold), besides the nfi positive control, are deoD, hpt, deoB and hdfR. The multicopy suppressors yjjX+, rnb+ and purA+ are also strong suppressors. Suppressors with enhancement in the 1,000–10,000 range are the nepI+ multicopy, the gmk<—cat antisense construct, as well as ΔyrdD and ΔdusB. Finally, the mutations that confer less than 1,000-fold growth enhancement are deletions of tdk, rpoC (the very 3′-terminus), purR, dgt, and putA (Fig. 2C). The mere 3-fold growth enhancement of the double mutant by the weakest suppressor putA is an evidence of sensitivity of our suppressor analysis.
If the inactivation of a suppressor gene works by alleviating the burden of chromosomal fragmentation in rdgB mutants, then overproducing the corresponding gene should make the ΔrdgB62 recA200(Ts) cell sicker at elevated temperatures (supposedly via increased chromosomal fragmentation). We mildly overexpressed genes nfi, deoB, deoD, hpt, dgt, purR, and tdk, whose inactivation suppressed rdgB recA(Ts) most frequently, by cloning them with their native promoters on a low-copy number vector. Indeed, the rdgB recA(Ts) inhibition at 42°C in the presence of the resulting plasmid was 200-times stronger (Fig. 2D), verifying the nature of the most frequent suppressors. Also consistent with the deleterious effect of the inactivated genes, we were unable to introduce a high copy number plasmids containing the same genes into the rdgB recA(Ts) double mutant without some of the genes being inactivated. Originally, we hoped that additional copies of the most frequently inactivated genes would allow us to concentrate on isolation of less frequent suppressors. However, the plasmid made the double mutant so sick that, after its introduction, we could not isolate any suppressors at all, even if we lowered the incubation temperature to 40°C.
The “missing” suppressors
If one looks at the pathways of xanthine (X) and hypoxanthine (H) conversion into GMP and AMP (Seeger et al., 1995, Zalkin & Nygaard, 1996) (Fig. 3A), it is striking how the isolated deoD, hpt and purA+++ suppressors all point to IMP as the critical intermediate in the biosynthesis of clastogenic precursor, intercepted by RdgB. To make sure that we were not missing the “XMP” side of the story due to some technical reasons, we introduced the corresponding changes on the xanthine part of the scheme, ΔxapA, Δgpt and guaA+++ (together with guaB+++) (Fig. 3A) (Seeger et al., 1995, Zalkin & Nygaard, 1996), into the rdgB recA(Ts) double mutant and tested its growth at 42°C relative to 30°C. We found that ΔxapA and guaAB+++ had no effect on the growth inhibition, while both Δgpt and guaB+++ alone inhibited the double mutant even further (Fig. 3B). We conclude that XMP is not an intermediate in the biosynthesis of RdgB-intercepted clastogenic precursor.
Fig. 3. The “missing” suppressors and the effect of slow growth.

A. A scheme of xanthine (X) and hypoxanthine (H) conversion into nucleotides. R-1-P, ribose-1-phosphate; R-5-P, ribose-5-phosphate; PRPP, 5-phosphorybosyl-1-pyrophosphate. Isolated inactivational and overproductional suppressors on the “hypoxanthine side” are indicated by spiked and smooth ovals, correspondingly. The corresponding genes on the “xanthine side” (guaA, gpt and xapA) are in bold. B. The effect of “missing suppressors” on the degree of rdgB recA co-inhibition. The values are means of four-five independent measurements ± SE. C. Slowing the overall metabolism with chloramphenicol marginally increases growth of the double mutant. The values are means of four independent measurements ± SE (in most cases, masked by the size of the symbols). D. Slowing growth of wild type cells with thinner media marginally increases growth of the double mutant. 20 AA, all aminoacids added, according to the recipe of Neidhardt (Neidhardt et al., 1974); 6 AA, the six aminoacids that AB1157 requires (arginine, glutamine, histidine, leucine, proline, and threonine) are added. The values are means of three independent measurements ± SE.
Suppression and the slow growth
Although none of the isolated suppressors significantly slowed down growth of otherwise wild type cells, we still considered the trivial possibility that inhibition of the rate of growth of the double mutant could enhance its growth at 42°C by allowing more time for damage repair (Fig. 1, situation #4). Interestingly, slowing down cells with low concentrations of chloramphenicol (Fig. 3C) or in minimal media supplemented with fewer aminoacids or with glycerol as a carbon source (Fig. 3D) did enhance growth of the double mutant at 42°C. The maximal growth enhancement was 6-fold with chloramphenicol and up to 9-fold with a poorer media, so it could have explained the weakest suppressors we have isolated, putA, dgt and purR (compare with Fig. 2C). However, the same concentration of chloramphenicol or poor medium also inhibited growth of the wild type control up to 10,000-fold (Fig. 3CD), whereas such a dramatic inhibitory effect was never observed for any of our suppressor mutants. Besides, if it were simply slow growth that suppressed the rdgB recA coinhibition, we should have isolated all kinds of mutants in the central metabolism that make cells grow slowly. Instead, the three weakest suppressors were isolated repeatedly (Table 1). We conclude that slow growth alone cannot explain even the weakest suppressors of the rdgB recA co-inhibition that we have isolated.
Suppression of rdgB recA(Ts) is not due to suppression of recA(Ts) alone
Although the DNA repair defect of ΔrecA mutation cannot be suppressed, we were using a point mutation allele (recA200) which, theoretically, could have been partially suppressed by metabolic changes, explaining the enhanced growth of the rdgB recA(Ts) double mutant at 42°C. To address the possibility that some of the suppressors ameliorate the recombinational repair defect of the recA(Ts) mutation (Fig. 1, situation #5), we quantified the UV sensitivity of each suppressor in the recA200(Ts) background at 42°C. We found that all of the suppressors were at least as UV sensitive as recA200(Ts) alone at 42°C, with several mutants being marginally more UV-resistant, while some other mutants being somewhat more UV-sensitive (Fig. 4A). Although the difference in survival after this dose of UV between the most resistance and the most sensitive suppressors was almost 40-fold, the UV-resistance had no correlation with suppression of the growth defect (Fig. 4B), indicating that none of our suppressors acted to ameliorate the recA(Ts) defect.
Fig. 4. Testing the suppression of the recA defect.
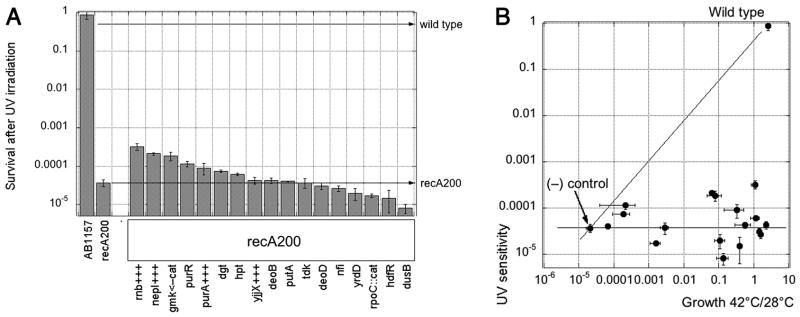
A. The effect of suppressors on UV-sensitivity of a single recA200 mutant at 42°C. Serial dilutions were spotted on LB agar plates and irradiated with a single dose of 12 J/m2. The data are means of four independent measurements ± SE. B. Suppression does not ameliorate the recA(Ts) defect. All the suppressors were represented by their growth at 42°C versus 28°C in the rdgB recA(Ts) double mutant (X axis), as well as by their survival of UV light in the recA(Ts) single mutant (Y axis). Wild type for both axes is AB1157; (−) control is rdgB recA(Ts) (JB30) for the X-axis, recA(Ts) (JC9941) for the Y-axis. Error bars are from the corresponding assays.
Epistatic analyis of weak suppressors
To reveal how many independent pathways the weak suppressors represent, we ran epistatic analysis by combining the putA, rpoC, dgt, purR and tdk mutations pairwise in the rdgB recA background and comparing the resulting enhancement of growth with the one by the corresponding single mutants, as well as with the expected products of single mutant’s effects (Fig. 5). To our surprise, we found no clear epistasis between any two of the mutations, indicating that all the five protiens operate in independent pathways. The only possible (partial) epistasis is shown by the dgt tdk combination, which would make sense, as both tdk (codes for thymidine kinase) and dgt (codes for dGTP hydrolase) belong to the nucleotide metabolism. The effects of the other nine double mutant combinations were of two types: either synergistic (exceeding the multiplicative expectation) or multiplicative. Interestingly, all combinations of purR with other defects, with the exception of tdk, lead to the strongly synergistic effect on viability (Fig. 5A), suggesting independent pathways that strongly expand if the other of the pair is mutated. The three pairwise combinations between putA, rpoC and dgt produced either minor synergistic effects or just additive (multiplicative) effect (Fig. 5B), suggesting independent and weakly- or non-expanding pathways. Finally, all four combinations with tdk were strictly additive or even mildly epistatic, suggesting independent non-expanding pathways (Fig. 5C). Thus, epistatic analysis failed to reveal strong grouping among the five weak suppressors, with the exception of dgt and tdk.
Fig. 5. Epistatic analysis of the weak suppressors.
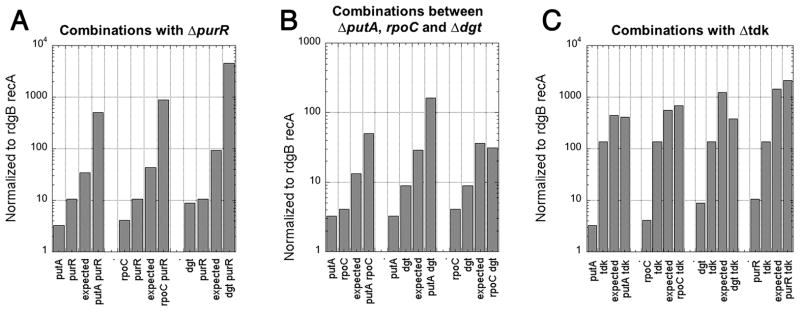
Suppressor mutations were combined pairwise in the rdgB recA(Ts) background, and the effect of their combination on the viability of the strain at 42°C was compared to the “expected” multiplicative effect of the double mutation (product of the effects of the corresponding single mutations). In each group of bars, the effects of single mutations and the calculated effect of the double mutation are normalized to the viability of the original rdgB recA(Ts) mutant and then compared with the observed effect of the double mutation. The double mutant values are averages of four independent measurements. Error bars are not shown because the averages are normalized. A. Three out of four possible combinations with purR show strong synergy — the observed effect is more than 10-fold higher that the multiplicative one. B. The pairwise combinations between putA, rpoC and dgt tend to be either weakly synergistic (less than 10-fold higher than the multiplicative effect) or additive (= multiplicative: the effect of the double mutant roughly equals the product of the effects of the single mutants). C. All four combinations with tdk show either additivity or weak epistasis.
Chromosomal fragmentation
A major detectable consequence of the rdgB mutation is increased chromosomal fragmentation due to double-strand DNA breaks (DSBs) (Kouzminova et al., 2004, Lukas & Kuzminov, 2006). These DSBs are detected in the double rdgB recBC(Ts) mutant as linear subchromosomal fragments that migrate into the lanes during pulsed-field gel electrophoresis, while intact circular chromosomes remain in the plugs (Fig. 6A) (Bradshaw & Kuzminov, 2003, Kouzminova et al., 2004). Chromosomal fragmentation is about twice as high at 37°C in the rdgB recBC(Ts) double mutant compared to recBC(Ts) alone, over the wild type level (Fig. 6B). This fragmentation is a consequence of increased EndoV-initiated removal of modified bases from the rdgB mutant, since at 37°C, the nfi rdgB62 recBC(Ts) triple mutant has the same level of chromosomal fragmentation as the recBC(Ts) single mutant (Fig. 6B). If chromosomal fragmentation is the killer of the rdgB recA mutants, then the only expected suppressors are those that decrease fragmentation commensurate with the degree of their suppression. If chromosomal fragmentation is not the proximal poison, then there would be suppressors that did not decrease fragmentation (suppression by tolerance to double-strand breaks).
Fig. 6. The effect of suppressors on chromosomal fragmentation in the rdgB recBC double mutant.

A. A representative PFGE showing chromosomal fragmentation for each control strain and four suppressors. CZ, compression zone. Intact chromosomes remain in the wells at the origin of the gel, while linear sub-chromosomal fragments migrate into the lanes. B. Quantification of chromosomal fragmentation. Values are means of 6–17 independent measurements ± SE. The wild type (AB1157) and dut recBC(Ts) (AK107) strains are (−) and (+) controls for fragmentation (Kouzminova et al., 2004). C. Plotting growth (X) versus fragmentation (Y) reveals two groups of suppressors. All the suppressors were represented by their growth at 42°C versus 28°C in the rdgB recA(Ts) double mutant (X axis), as well as by their fragmentation in the rdgB recBC(Ts) double mutant (Y axis). Wild type is AB1157 for the X-axis, recBC(Ts) (SK129) for the Y-axis; (−) control is rdgB recA(Ts) (JB30) for the X-axis, rdgB recBC(Ts) (JB36) for the Y-axis. Error bars are from the corresponding assays.
We tested all isolated suppressors in the chromosomal fragmentation assay and found that they belong to three categories depending on the extent of suppression (Fig. 6B). Mutations in yrdD, putA and gmk<—cat, as well as overproduction of purA+, yjjX+, nepI+ and rnb+ reduced fragmentation to the level around the recBC(Ts) single mutant, behaving like the nfi suppressors. Mutations in deoD, hpt, dusB and hdfR reduced fragmentation to intermediate levels. Finally, mutations in deoB, purR tdk, rpoC::cat and dgt did not change the fragmentation level (Fig. 6B). Plotting growth of the suppressed mutants versus their chromosomal fragmentation reveals two groups of suppressors (Fig. 6C), which coincide with the expectations (see above). In the group that we call “tolerance to DSBs” chromosomal fragmentation stays at the level of the negative control. In the group “prevention of DSBs” the degree of suppression correlates with the decrease in chromosomal fragmentation, apparently reflecting the corresponding gene’s contribution to the production of clastogenic DNA precursors that become EndoV-recognized DNA modifications when incorporated into DNA, or to their subsequent repair.
Density of EndoV-recognized DNA modifications
To identify suppressors that reduce the amount of the non-canonical DNA precursors that RdgB intercepts, we measured the density of endonuclease V-recognized DNA modifications in supercoiled plasmid DNA extracted from suppressor mutant strains in the ΔrdgB62 background using an in vitro assay. EndoV initiates excision repair of hypoxanthine, xanthine (He et al., 2000, Yao & Kow, 1997) and, presumably, HAP (Burgis et al., 2003), by introducing a nick near the modified nucleotide. The supercoiled plasmid species were separated from relaxed plasmid by agarose gel electrophoresis, and the relative amounts of each were quantified after hybridization (Fig. 7A). The resulting relaxation is taken as measurement of the density of EndoV-recognized modifications in DNA, isolated from a particular strain.
Fig. 7. The effect of suppressors on the density of EndoV-recognized DNA modifications.

A. Assay for EndoV-sensitivity of a supercoiled plasmid (the conversion between supercoiled and relaxed circular forms in a 22.5 kbp plasmid). B. Quantification of EndoV-sensitivity. The density of EndoV-recognized DNA modifications in each suppressor in the rdgB background is shown at the percentage of a 22.5 kbp plasmid that is nicked after being treated with EndoV. Values are means of 4–16 independent measurements ± SE. The wild type (AB1157) is the background, the rdgB single mutant (JB29) shows the elevated levels of EndoV-recognized DNA modifications, while the rdgB nfi double mutant (BB112) shows further elevation due to the additional excision repair defect. C. Plotting growth (X) versus EndoV-sensitivity (Y) reveals two groups of suppressors. All the suppressors were represented by their growth at 42°C versus 28°C in the rdgB recA(Ts) double mutant (X axis), as well as by EndoV-sensitivity of their DNA in the rdgB single mutant (Y axis). Wild type is AB1157 for both axes; (−) control is rdgB recA(Ts) (JB30) for the X-axis, rdgB (JB29) for the Y-axis. Error bars are from the corresponding assays.
This EndoV-sensitivity assay has demonstrated previously that rdgB mutants accumulate modified bases in their DNA (Bradshaw & Kuzminov, 2003, Budke & Kuzminov, 2006). Suppressor mutations that reduce the levels of modified nucleotides in the DNA precursor pools should reduce the amount of EndoV-recognized modifications in the DNA of rdgB mutants. On the other hand, suppressors that interfere with removal of the modified bases from DNA should have at least as many or even more EndoV-recognized DNA modifications as a single rdgB mutant, as for example observed for the nfi suppressors (Bradshaw & Kuzminov, 2003, Budke & Kuzminov, 2006). Both types of suppressor mutations are expected to have greater viability than the rdgB recA double mutant and reduced chromosomal fragmentation compared to the rdgB62 recBC double mutant. Finally, suppressors that speed up the rate of DNA repair in lieu of increased EndoV activity are also possible, — these should have extremely low densities of EndoV-recognized DNA modifications.
The EndoV in vitro assay was used to quantify the amount of modified bases in plasmid DNA extracted from the selected suppressors. Mutations in hpt, deoB, dusB, hdfR, anti-sensing of gmk, as well as high copy number of purA+, yjjX+ and rnb+ decrease the levels of EndoV-recognized DNA modifications and, therefore, must lower the amount of non-canonical nucleotides in the DNA precursor pool, indicating that the corresponding enzymes participate in their generation or, in case of overproduction, in their utilization/hydrolysis (Fig. 7B). Mutations in deoD, purR, dgt and tdk do not change or slightly increase the level of EndoV-sensitivity of DNA from that of the rdgB single mutant (Fig. 7B). Finally, in addition to the nfi suppressor, which accumulates more EndoV-recognized DNA modifications, deletions of putA, rpoC and yrdD, as well as increased copy number of nepI+, elevate the amount of EndoV-recognized DNA modifications, suggesting that these suppressors decrease the modified base excision.
The two groups of suppressors are clearly separated by the “growth-versus-EndoV-sensitivity” analysis (Fig. 7C). In the “Reduction in EndoV-sensitivity” group, made mostly of stronger suppressors, the degree of suppression correlates with decrease in EndoV-sensitivity. In the “Reduction in excision” group, represented by both strong and weak suppressors, the degree of suppression correlates with increase in EndoV-sensitivity.
Discussion
RdgB is a bacterial dNTPase with a strong in vitro preference for dHapTP, dXTP and dITP (Bradshaw & Kuzminov, 2003, Burgis & Cunningham, 2007, Chung et al., 2002). In rgdB mutants, these non-canonical DNA precursors with deaminated or aminogroup-modified purines are utilized by DNA polymerases, while excision of the corresponding base analogs from DNA is initiated by EndoV-nicking (Burgis et al., 2003, He et al., 2000, Yao et al., 1994, Yao & Kow, 1995). This excision repair, by still unknown mechanisms, is translated into chromosomal fragmentation, which becomes inhibitory in recombinational repair mutants, so that rdgB recA(Ts) double mutants cannot grow at 42°C (Bradshaw & Kuzminov, 2003, Clyman & Cunningham, 1987). To reveal the sources and identities of the non-canonical DNA precursors, intercepted by RdgB in E. coli, we systematically isolated and characterized suppressors of the rdgB recA(Ts) co-inhibition at 42°C by insertional mutagenesis and also by overproduction. Although we found a total of 38 different loci affected by suppressing inserts or multicopy clones, we limited our analysis to the 17 mutations or multicopy clones that were isolated more than once. We first quantified the growth enhancement to classify the suppressors as strong, intermediate or weak (Fig. 2). We then tested the trivial possibilities that some suppressors are working through general inhibition of the cellular metabolism or through amelioration of the defect of the recA(Ts) point mutation and found no evidence of either (Fig. 3 and 4). We quantified the effect of suppressors on the chromosomal fragmentation due to the rdgB defect and found that in most suppressors the improved growth correlates with the alleviation of chromosomal fragmentation (Fig. 6). Finally, we quantified EndoV-sensitivity of plasmid DNA, isolated from suppressed rdgB mutant cells and found that half of the suppressors reduce this sensitivity, while the other half has either no effect or increases it (Fig. 7). Plotting the chromosomal fragmentation values against those of EndoV-sensitivity for the same mutants reveals three distinct groups of suppressors (Fig. 8A): 1) those that suppress via reduction of the nc-dNTP production (reduce both fragmentation and EndoV-sensitivity); 2) those that suppress by inhibiting or eliminating excision of the base analogs from DNA (reduce fragmentation, while increasing EndoV-sensitivity); 3) those that, apparently, increase the tolerance of the cells to fragmented chromosomes (no change or increase in fragmentation or EndoV-sensitivity).
Fig. 8. Summary of suppressors and the identity of the RdgB-intercepted non-canonical dNTP, revealed by a tentative pathways of its biosynthesis.

A. Summary: plotting fragmentation (X) versus EndoV-sensitivity (Y) reveals three groups of suppressors. This is the third possible pairwise comparison, of the types presented in Fig. 4C and 5C. All the suppressors are represented by their fragmentation in the rdgB recBC(Ts) double mutant (X axis), as well as by EndoV-sensitivity of their DNA in the rdgB single mutant (Y axis). Wild type is recBC(Ts) (SK129) for the X-axis, AB1157 for the Y-axis; (−) control is rdgB recBC(Ts) (JB36) for the X-axis, rdgB (JB29) for the Y-axis. Error bars are from the corresponding assays. B. A scheme of the nucleotide metabolism to explain our suppressor results. R-1-P, ribose-1-phosphate; R-5-P, ribose-5-phosphate; PRPP, 5-phosphorybosyl-1-pyrophosphate; Hx, hypoxanthine. Inactivational and overproductional suppressors are indicated by spiked and smooth ovals, correspondingly. The only hypothetical step (Gmk-catalyzed IMP—>IDP conversion) is shown by a broken arrow.
Suppressors that reduce both clastogenesis and EndoV-recognized DNA modifications must do so via decreasing the intracellular concentrations of non-canonical dNTPs. These are deletions of deoB, deoD, hpt, dusB, hdfR, antisensing of the essential gmk, as well as overproduction of yjjX+, purA+ and rnb+. Their known enzymatic specificities allow one to distinguish between the three known in vitro substrates of RdgB (Fig. 1). Perhaps the least clear suppressors in this group are inactivation of hdfR, which codes for a transcription regulator, dusB, which codes for tRNA dihydrouridine synthase, and overproduction of rnb+, coding for ribonuclease II (RNase II), one of the two main exonucleases involved in mRNA decay. The three activities have no direct connection to the purine nucleotide metabolism and probably act by regulating gene expression of real suppressors via repression, translation or mRNA stability.
Other suppressors of the first group are more informative (Fig. 8B). The DeoB protein is phosphopentomutase that catalyzes interconversion between ribose-1-phosphate and ribose-5-phosphate, implicating nucleotide salvage pathways, while the DeoD protein is purine phosphorylase I that uses ribose-1-phosphate to convert a hypoxanthine, guanine or adenine base into the corresponding nucleoside (Zalkin & Nygaard, 1996), implicating purine nucleotide metabolism. Since inactivation of the analogous enzyme that makes xanthosine (XapA, purine nucleoside phosphorylase II (Hammer-Jespersen et al., 1980)) does not suppress the rdgB recA co-inhibition (Fig. 3B), deoD suppressors point to hypoxanthine as the purine, intercepted in the dNTP form by RdgB. Interestingly, the Hpt protein is hypoxanthine phosphoribosyltransferase, which converts hypoxanthine and, less efficiently, guanine into the corresponding NMPs (Zalkin & Nygaard, 1996). Again, IMP emerges as the key intermediate, as mutations in the enzymes that make AMP from adenine (apt) or XMP from xanthine (gpt) were not isolated as suppressors, and inactivation of gpt actually further inhibits rdgB recA (Fig. 3B). IMP is an intermediate in the synthesis of both AMP (via PurA adenylosuccinate synthetase) and XMP (via GuaB IMP dehydrogenase), while XMP is similarly converted into GMP by GuaA GMP synthetase (Fig. 3A) (Zalkin & Nygaard, 1996). GuaAB overproduction has no strong effect on the rdgB recA co-inhibition, while PurA overproduction is one of the strongest suppressors of it (Clyman & Cunningham, 1991) (Fig. 8A), again pointing to IMP as the critical intermediate in the synthesis of the clastogenic DNA precursor. Finally, Gmk protein is guanylate kinase, an essential enzyme catalyzing GMP—>GDP reaction, and IMP is a known secondary substrate of this enzyme, while XMP is not (Oeschger & Bessman, 1966). Once IDP is formed by Gmk, there is nothing that can prevent its further conversion into dITP (Fig. 8B). Finally, multiple isolations of yjjX+ overproduction as a strong suppressor actually do not distinguish between dITP versus dXTP as the intracellular target of RdgB, since YjjX protein hydrolyses ITP and XTP equally well (Zheng et al., 2005). However, the overall results of our suppressor analysis indicate dITP as the main in vivo target of RdgB, with IMP being the key intermediate in its biosynthesis (Fig. 8B).
Suppressors of the second group could potentially provide insights into the mechanism of EndoV-initiated excision of DNA-hypoxanthine. While the EndoV incision step of DNA-hypoxanthine removal is thoroughly characterized in vitro (Kow, 2002), the subsequent steps of this so-called “alternative excision repair” are still a mystery, which did not even yield to the powerful screen for RdgB-dependent mutants (Lukas & Kuzminov, 2006). This time there is a candidate for a potential EndoV helper in this reaction, the 180 aa YrdD protein with unknown functions and a limited homology to topoisomerases (it is too small to be a functional topoisomerase itself). In the fragmentation-vs-EndoV-sensitivity comparison, yrdD is the closest mutant to the nfi mutant (Fig. 8A), as both show dramatically reduced chromosomal fragmentation and at the same time increase in EndoV-sensitivity, which we now can interpret as accumulation of DNA-hypoxanthine. Another interesting suppressor that clusters with nfi is nepI+ overproduction; the NepI protein is an inosine/guanosine exporter (Gronskiy et al., 2005). How this pumping of inosine out of the cell creates an EndoV-like phenotype is at present unclear. If the protein could pump inosine into the cell, the high intracellular concentration of inosine could directly inhibit the DNA-hypoxanthine-incision activity of EndoV (similar to free uracil inhibition of uracil-DNA-glycosylase (Lindahl et al., 1977)), essentially making NepI-overproducing strains nfi phenocopies. Another suppressor that behaves like nepI+ high copy number, inactivation of putA, has no known links to DNA metabolism.
The last group of suppressor, clustering above the negative control in the fragmentation-EndoV-sensitivity comparison in Fig. 8A are tdk, purR, dgt and rpoC. Possible suppression mechanisms for the representatives of this group are problematic, like for putA above, partly because they are all weak suppressors. Since tdk, dgt and purR belong to the nucleotide metabolism, it is tempting to place them together with deoBD, hpt, gmk and purA+++ (especially purR, which is a purA regulator) but they reduce neither fragmentation, nor EndoV-sensitivity (at least within the limits of detection of our procedures), so apparently relieve the rdgB recA synthetic inhibition by some other means. A generic explanation of action for this group is via increased “tolerance” to chromosomal fragmentation, yet specific mechanisms of such a tolerance are unclear at this point. Existence of such tolerance in the face of the significant level of chromosomal fragmentation, also observed for the rfaQGP suppressors of the seqA recA co-lethality (Rotman et al., 2009), suggests heterogeneity of double-strand breaks, specifically a mixture of the predominant/nonlethal type and a minor/lethal type. The suppressors of the third group may specifically target the minor/lethal type of double-strand breaks (possibly increasing the predominant and less lethal type in parallel).
Overall, our systematic suppressor analysis of the rdgB recA synthetic inhibition revealed IMP as the key intermediate in the synthesis of the clastogenic DNA precursor, most likely dITP. At least half of the isolated suppressors affect various genes involved in the RNA/DNA precursor production, demonstrating that the non-canonical DNA precursors are byproducts of the normal nucleotide metabolism. These findings highlight the importance of the nucleotide pool sanitizing activity of RdgB in the avoidance of chromosomal fragmentation due to the subsequent EndoV-initiated excision. The details of this excision repair reaction to remove DNA-hypoxanthine still remain a mystery.
Experimental Procedures
Strains and media
The bacterial strains used were derivatives of Escherichia coli AB1157 and MG1655 (Table S1). Deletion mutants were constructed by the deletion-replacement method of Datsenko and Wanner (Datsenko & Wanner, 2000) or by P1 transduction of deletion-replacement alleles from the Keio collection (Baba et al., 2006). Drug resistance markers used during the construction of these strains were removed using the plasmid pCP20, which was then cured by passaging the strain at 37 °C (Datsenko & Wanner, 2000). The relevant genotypes of each newly-constructed strain were verified by PCR for deleted genes or, for recA and recBC(Ts) alleles, by the characteristic UV-sensitivities. For routine culturing, cells were grown in LB liquid medium (10 g peptone, 5 g yeast extract, 5 g NaCl per 1 L, pH 7.4 with 250 μl 4M NaOH) or on LB plates (15 g agar per 1 L LB broth). Antibiotics for maintaining plasmids and selecting transductants were used at the following concentrations: 30–100 μg/ml spectinomycin, 10 μg/ml chloramphenicol, 50 μg/ml kanamycin, and 30–100 μg/ml ampicillin.
Insertion suppressor screen
The plasmid pRL27 (Larsen et al., 2002) was used for insertional mutagenesis. The strain JB30 was electroporated with 10 ng of pRL27, the transformants were plated on LB medium with 10 μg/ml kanamycin, and the plates were incubated at 43°C overnight. Potential suppressors of the rdgB recA synthetic lethality appeared as colonies of various sizes on the transformation plate. These colonies were re-streaked on LB supplemented with 50 μg/ml kanamycin and incubated at 43°C. To verify that suppression was due to insertion of the transposable element from pRL27, rather than due to a spontaneous mutation, each potential suppressor was backcrossed by P1 transduction into the original rdgB recA(Ts) strain and verified for suppression by spot-testing at 28°C and 42°C. The genes disrupted by insertion were identified by sequencing outwards from the insertion element, as before (Bradshaw & Kuzminov, 2003, Kouzminova & Kuzminov, 2004, Kouzminova et al., 2004). For confirmed suppressors, we constructed in-frame deletions of the suppressing genes to verify that suppression was due to gene inactivation.
Plasmids
The plasmid pBB11 was constructed by cloning functional alleles of the deoBD, dgt, hpt, purR, nfi, and tdk genes, generated by PCR from genomic DNA of AB1157, one at a time into the multiple cloning site of pMTL22 (Chambers et al., 1988) using restriction sites engineered into the PCR primers. Each cloned gene was individually shown to be functional by inhibiting growth at 42°C of its respective ΔrdgB62 recA200(Ts) + suppressor gene deletion triple mutant. The assembled multigene cassette was then cloned into the low copy number pK80 plasmid (Kuzminov & Stahl, 1997) using the XhoI and BamHI sites, giving pBB13. Primers and plasmid intermediates used during construction are given in Table S2 and S3.
Multicopy suppressor screen
Libraries of chromosomal DNA fragments from AB1157 ΔrdgB62 were generated by digestion with EcoRI, HindIII, MluI, BssHII or SalI to completion and cloning into the corresponding sites on pMTL22 (Chambers et al., 1988). The plasmid library was transformed by electroporation into AB1157 ΔrdgB62 recA200(Ts) and plated on LB + 100 μg/ml ampicillin at 42°C to select for suppressors. To verify that suppression was due to increased copy number of a multicopy suppressor gene, rather than due to a spontaneous suppressor, candidate suppressing plasmids were isolated and re-introduced back into AB1157 ΔrdgB62 recA200(Ts). Confirmed multicopy suppressors were identified by sequencing. For multicopy suppressor plasmids containing more than one functional gene, the gene responsible for suppression was identified by subcloning.
Growth inhibition assay
A fresh saturated LB culture of the strain to be tested was serially diluted in saline (1% NaCl). 10 μl of the 10−5 dilution was spotted onto duplicate LB plates, which were incubated for 17 hours either at 28°C or at 42°C, after which the spots were cut out of the agar plate and resuspended in 1.0 ml of saline by shaking for one hour. The suspension was serially diluted in saline and titered on LB agar plates incubated at room temperature. Growth inhibition was calculated as the 42°C titer divided by the 28°C titer.
UV sensitivity assay
A fresh saturated culture of the strain to be tested was subcultured 1:50 – 1:100 into 2 ml of LB medium and incubated at 28°C with agitation until it reached OD600 0.3 – 0.6, then shifted to 42°C and incubated with agitation for 30 minutes, and then serially diluted ten-fold to 10-6 in saline pre-warmed to 42°C. 10 μl of each dilution were spotted onto duplicate LB plates pre-warmed to 42°C, the spots were allowed to dry in a 42 °C incubator for 15 minutes, and one duplicate plate was given 9–12 J/m2 UV while the other was kept unirradiated. The plates were incubated for another hour at 42 °C and then shifted to 28°C overnight. UV survival was determined as the titer of the irradiated culture divided by the titer of the unirradiated culture.
EndoV treatment
Plasmid DNA for in vitro EndoV treatment was isolated from growing LB cultures at OD600 0.7 (to maximize the density of the modified nucleotides in DNA) by the alkaline lysis method (Birnboim, 1983). To reduce spurious EndoV nicking of DNA and to maximize deoxyinosine-specific activity of the enzyme, we carried out the reaction using 2 mM MgCl2 at an acidic pH (Yao & Kow, 1997). 50–100 ng of plasmid DNA was treated for 15 min at 37°C with 1.0 mU Endo V (Trevigen) per ng DNA in either 15 or 100 μl reaction containing 10 mM PIPES pH 6.4, 50 mM NaCl, and 2 mM MgCl2. Reactions were stopped either by addition of EDTA pH 8.0 to 94 mM or by addition of SDS to 1% followed by ethanol precipitation. The reactions were run on a 1.1% agarose TAE gel for 20 hours at 2.35 V•cm−1, and transferred to a positively charged nylon membrane (Amersham) by capillary transfer. The relative amounts of supercoiled and relaxed circular pK96 were quantified after hybridization to a radiolabeled pK96 probe, phosphorimaging and measuring the signal intensity of each band using the ImageQuant or Multigauge 3 (Fuji) software.
Quantitation of chromosomal fragmentation
Fresh overnight cultures of strains to be tested were diluted into fresh LB medium containing 10 μCi 32P-orthophosphate (MP Biomedicals) such that they would grow to OD600 0.3 after one hour at 22°C followed by four hours at 37°C. 0.5 ml of each culture were pelleted by centrifugation and resuspended in 60 μl TE. After 2 minutes of incubation at 37°C, 5 μl of 5 mg/ml proteinase K and 60 μl of 1X lysis buffer (0.2% sarcosyl, 10 mM Tris HCl pH 8.0, 5 mM EDTA) containing 1.5% agarose heated to 70°C were added and the suspension was transferred to a plug mold. The solidified plugs were transferred to a tube containing 1 ml of 1X lysis buffer and incubated for 24 hours at 60°C, then run on a 1.0% agarose (Bio-Rad Pulsed Field Certified) 0.5X TBE gel using a Gene Navigator (Pharmacia Biotech) CHEF pulsed-field apparatus set to 180 V using the following switching times: 90 sec for 7 hours, 115 sec for 7 hours, and 125 sec for 8 hours. Following electrophoresis, the gel was dried, exposed overnight to a phosphorimaging screen, and the relative amounts of radioactivity in the lanes of the gel vs. the plugs were quantified using the ImageQuant software. The percent fragmentation for each sample was calculated as the amount of 32P signal in the lane divided by the total 32P signal in the well plus lane.
Supplementary Material
Acknowledgments
We thank Jill Bradshaw and Scott Horrell for help at the initial stages of this project. This work was supported by grant # RSG-05-135-01-GMC from the American Cancer Society and by grant # GM 073115 from the National Institutes of Health.
References
- Abdul-Masih MT, Bessman MJ. Biochemical studies on the mutagen, 6-N-hydroxylaminopurine. Synthesis of the deoxynucleoside triphosphate and its incorporation into DNA in vitro. J Biol Chem. 1986;261:2020–2026. [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:Article number: 2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessman MJ, Lehman IR, Adler J, Zimmerman SB, Simms ES, Kornberg A. Enzymatic synthesis of deoxyribonucleic acid. III. The incorporation of pyrimidine and purine analogues into deoxyribonucleic acid. Proc Natl Acad Sci USA. 1958;44:633–640. doi: 10.1073/pnas.44.7.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnboim HC. A rapid alkaline extraction method for the isolation of plasmid DNA. Methods Enzymol. 1983;100:243–255. doi: 10.1016/0076-6879(83)00059-2. [DOI] [PubMed] [Google Scholar]
- Bradshaw JS, Kuzminov A. RdgB acts to avoid chromosome fragmentation in Escherichia coli. Mol Microbiol. 2003;48:1711–1725. doi: 10.1046/j.1365-2958.2003.03540.x. [DOI] [PubMed] [Google Scholar]
- Budke B, Kuzminov A. Hypoxanthine incorporation is nonmutagenic in Escherichia coli. J Bacteriol. 2006;188:6553–6560. doi: 10.1128/JB.00447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgis NE, Brucker JJ, Cunningham RP. Repair system for noncanonical purines in Escherichia coli. J Bacteriol. 2003;185:3101–3110. doi: 10.1128/JB.185.10.3101-3110.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgis NE, Cunningham RP. Substrate specificity of RdgB protein, a deoxyribonucleoside triphosphate pyrophosphohydrolase. J Biol Chem. 2007;282:3531–3538. doi: 10.1074/jbc.M608708200. [DOI] [PubMed] [Google Scholar]
- Chambers SP, Prior SE, Barstow DA, Minton NP. The pMTL nic− cloning vectors. I. Improved pUC polylinker regions to facilitate the use of sonicated DNA for nucleotide sequencing. Gene. 1988;68:139–149. doi: 10.1016/0378-1119(88)90606-3. [DOI] [PubMed] [Google Scholar]
- Chung JH, Park HY, Lee JH, Jang Y. Identification of the dITP- and XTP-hydrolyzing protein from Escherichia coli. J Biochem Mol Biol. 2002;35:403–408. doi: 10.5483/bmbrep.2002.35.4.403. [DOI] [PubMed] [Google Scholar]
- Clement B, Kunze T. Hepatic microsomal N-hydroxylation of adenine to 6-N-hydroxylaminopurine. Biochem Pharmacol. 1990;39:925–933. doi: 10.1016/0006-2952(90)90209-4. [DOI] [PubMed] [Google Scholar]
- Clyman J, Cunningham RP. Escherichia coli K-12 mutants in which viability is dependent on recA function. J Bacteriol. 1987;169:4203–4210. doi: 10.1128/jb.169.9.4203-4210.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyman J, Cunningham RP. Suppression of the defects in rdgB mutants of Escherichia coli K-12 by the cloned purA gene. J Bacteriol. 1991;173:1360–1362. doi: 10.1128/jb.173.3.1360-1362.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, D.C: 2006. p. 1118. [Google Scholar]
- Gronskiy SV, Zakataeva NP, Vitushkina MV, Ptitsyn LR, Altman IB, Novikova AE, Livshits VA. The yicM (nepI) gene of Escherichia coli encodes a major facilitator superfamily protein involved in efflux of purine ribonucleosides. FEMS Microbiol Lett. 2005;250:39–47. doi: 10.1016/j.femsle.2005.06.051. [DOI] [PubMed] [Google Scholar]
- Hammer-Jespersen K, Buxton RS, Hansen TD. A second purine nucleoside phosphorylase in Escherichia coli K–12. II. Properties of xanthosine phosphorylase and its induction by xanthosine. Mol Gen Genet. 1980;179:341–348. doi: 10.1007/BF00425462. [DOI] [PubMed] [Google Scholar]
- He B, Qing H, Kow YW. Deoxyxanthosine in DNA is repaired by Escherichia coli endonuclease V. Mutat Res. 2000;459:109–114. doi: 10.1016/s0921-8777(99)00063-4. [DOI] [PubMed] [Google Scholar]
- Hwang KY, Chung JH, Kim S-H, Han YS, Cho Y. Structure-based identification of a novel NTPase from Methanococcus jannaschii. Nature Struct Biol. 1999;6:691–696. doi: 10.1038/10745. [DOI] [PubMed] [Google Scholar]
- Kouzminova EA, Kuzminov A. Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol Microbiol. 2004;51:1279–1295. doi: 10.1111/j.1365-2958.2003.03924.x. [DOI] [PubMed] [Google Scholar]
- Kouzminova EA, Kuzminov A. Fragmentation of replicating chromosomes triggered by uracil in DNA. J Mol Biol. 2006;355:20–33. doi: 10.1016/j.jmb.2005.10.044. [DOI] [PubMed] [Google Scholar]
- Kouzminova EA, Rotman E, Macomber L, Zhang J, Kuzminov A. RecA-dependent mutants in E. coli reveal strategies to avoid replication fork failure. Proc Natl Acad Sci USA. 2004;101:16262–16267. doi: 10.1073/pnas.0405943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kow YW. Repair of deaminated bases in DNA. Free Radic Biol Med. 2002;33:886–893. doi: 10.1016/s0891-5849(02)00902-4. [DOI] [PubMed] [Google Scholar]
- Kozmin SG, Pavlov YI, Dunn RL, Schaaper RM. Hypersensitivity of Escherichia coli Δ(uvrB-bio) mutants to 6-hydroxylaminopurine and other base analogs is due to a defect in molybdenum cofactor biosynthesis. J Bacteriol. 2000;182:3361–3367. doi: 10.1128/jb.182.12.3361-3367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozmin SG, Schaaper RM. Molybdenum cofactor-dependent resistance to N-hydroxylated base analogs in Escherichia coli is independent of MobA function. Mutat Res. 2007;619:9–15. doi: 10.1016/j.mrfmmm.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozmin SG, Schaaper RM, Shcherbakova PV, Kulikov VN, Noskov VN, Guetsova ML, Alenin VV, Rogozin IB, Makarova KS, Pavlov YI. Multiple antimutagenesis mechanisms affect mutagenic activity and specificity of the base analog 6-N-hydroxylaminopurine in bacteria and yeast. Mutat Res. 1998;402:41–50. doi: 10.1016/s0027-5107(97)00280-7. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci USA. 2001;98:8241–8246. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A, Stahl FW. Stability of linear DNA in recA mutant Escherichia coli cells reflects ongoing chromosomal DNA degradation. J Bacteriol. 1997;179:880–888. doi: 10.1128/jb.179.3.880-888.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen RA, Wilson MM, Guss AM, Metcalf WW. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch Microbiol. 2002;178:193–201. doi: 10.1007/s00203-002-0442-2. [DOI] [PubMed] [Google Scholar]
- Lin S, McLennan AG, Ying K, Wang Z, Gu S, Jin H, Wu C, Liu W, Yuan Y, Tang R, Xie Y, Mao Y. Cloning, expression, and characterization of a human inosine triphosphate pyrophosphatase encoded by the ITPA gene. J Biol Chem. 2001;276:18695–18701. doi: 10.1074/jbc.M011084200. [DOI] [PubMed] [Google Scholar]
- Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B. DNA N-glycosidases. Properties of uracil-DNA glycosidase from Escherichia coli. J Biol Chem. 1977;252:3286–3294. [PubMed] [Google Scholar]
- Lukas L, Kuzminov A. Chromosomal fragmentation is the major consequence of the rdgB defect in Escherichia coli. Genetics. 2006;172:1359–1362. doi: 10.1534/genetics.105.051144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrnes B, Guddal PH, Krokan H. Metabolism of dITP in HeLa cell extracts, incorporation into DNA by isolated nuclei and release of hypoxanthine from DNA by a hypoxanthine-DNA glycosylase activity. Nucleic Acids Res. 1982;10:3693–3701. doi: 10.1093/nar/10.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhard J, Nygaard P. Purines and pyrimidines Escherichia coli and Salmonella typhimurium. In: Neidhardt FC, editor. Cellular and molecular biology. Washington, D.C: American Society for Microbiology; 1987. pp. 445–473. [Google Scholar]
- Noskov VN, Staak K, Shcherbakova PV, Kozmin SG, Negishi K, Ono B-C, Hayatsu H, Pavlov YI. HAM1, the gene controlling 6-N-hydroxylaminopurine sensitivity and mutagenesis in the yeast. Saccharomyces cerevisiae Yeast. 1996;12:17–29. doi: 10.1002/(SICI)1097-0061(199601)12:1%3C17::AID-YEA875%3E3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Oeschger MP, Bessman MJ. Purification and properties of guanylate kinase from Escherichia coli. J Biol Chem. 1966;241:5452–5460. [PubMed] [Google Scholar]
- Piccirilli JA, Krauch T, Moroney SE, Benner SA. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature. 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- Rotman E, Bratcher P, Kuzminov A. Reduced lipopolysaccharide phosphorylation in Escherichia coli lowers the elevated ori/ter ratio in seqA mutants. Mol Microbiol. 2009;72:1273–1292. doi: 10.1111/j.1365-2958.2009.06725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savchenko A, Proudfoot M, Skarina T, Singer A, Litvinova O, Sanishvili R, Brown G, Chirgadze N, Yakunin AF. Molecular basis of the antimutagenic activity of the house-cleaning inosine triphosphate pyrophosphatase RdgB from Escherichia coli. J Mol Biol. 2007;374:1091–1103. doi: 10.1016/j.jmb.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Seeger C, Poulsen C, Dandanell G. Identification and characterization of genes (xapA, xapB, and xapR) involved in xanthosine catabolism in Escherichia coli. J Bacteriol. 1995;177:5506–5516. doi: 10.1128/jb.177.19.5506-5516.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro R, Pohl SH. The reaction of ribonucleosides with nitrous acid. Side products and kinetics. Biochemistry. 1968;7:448–455. doi: 10.1021/bi00841a057. [DOI] [PubMed] [Google Scholar]
- Shi IY, Stansbury J, Kuzminov A. Defect in Acetyl-CoA<—>Acetate Pathway Poisons Recombinational Repair-deficient mutants of Escherichia coli. J Bacteriol. 2005;187:1266–1275. doi: 10.1128/JB.187.4.1266-1275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas KR, Manlapaz-Ramos P, Lundquist R, Olivera BM. Formation of Okazaki pieces at the Escherichia coli replication fork in vitro. Cold Spring Harbor Symp Quant Biol. 1979;43:231–237. doi: 10.1101/sqb.1979.043.01.028. [DOI] [PubMed] [Google Scholar]
- Ting H, Kouzminova EA, Kuzminov A. Synthetic lethality with the dut defect in Escherichia coli reveals layers of DNA damage of increasing complexity due to uracil incorporation. J Bacteriol. 2008;190:5841–5854. doi: 10.1128/JB.00711-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Hatahet Z, Melamede RJ, Kow YW. Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3′ endonuclease, from Escherichia coli. J Biol Chem. 1994;269:16260–16268. [PubMed] [Google Scholar]
- Yao M, Kow YW. Interaction of deoxyinosine 3′-endonuclease from Escherichia coli with DNA containing deoxyinosine. J Biol Chem. 1995;270:28609–28616. doi: 10.1074/jbc.270.48.28609. [DOI] [PubMed] [Google Scholar]
- Yao M, Kow YW. Further characterization of Escherichia coli endonuclease V. Mechanism of recognition for deoxyinosine, deoxyuridine, and base mismatches in DNA. J Biol Chem. 1997;272:30774–30779. doi: 10.1074/jbc.272.49.30774. [DOI] [PubMed] [Google Scholar]
- Zalkin H, Nygaard P. Biosynthesis of Purine Nucleotides Escherichia coli and Salmonella. In: Neidhardt FC, editor. Cellular and Molecular Biology. Washington, D.C: ASM Press; 1996. pp. 561–579. [Google Scholar]
- Zheng J, V, Singh K, Jia Z. Identification of an ITPase/XTPase in Escherichia coli by structural and biochemical analysis. Structure. 2005;13:1511–1520. doi: 10.1016/j.str.2005.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.