

Abstract
Over the past decade, high-throughput studies have identified many novel transcripts. While their existence is undisputed, their coding potential and functionality have remained controversial. Recent computational approaches guided by ribosome profiling have indicated that translation is far more pervasive than anticipated and takes place on many transcripts previously assumed to be non-coding. Some of these newly discovered translated transcripts encode short, functional proteins that had been missed in prior screens. Other transcripts are translated, but it might be the process of translation rather than the resulting peptides that serve a function. Here, we review annotation studies in zebrafish to discuss the challenges of placing RNAs onto the continuum that ranges from functional protein-encoding mRNAs to potentially non-functional peptide-producing RNAs to non-coding RNAs. As highlighted by the discovery of the novel signaling peptide Apela/ELABELA/Toddler, accurate annotations can give rise to exciting opportunities to identify the functions of previously uncharacterized transcripts.
Keywords: Apela/ELABELA/Toddler, coding potential, gene annotation, ncRNAs, peptides, short ORFs, zebrafish
Introduction
Over the past decade, advances in high-throughput sequencing technologies have led to the identification of a large number of previously unknown transcripts [1-7]. While their existence is undisputed, their biological functions are largely unclear. Their lack of an obvious, long protein-coding open reading frame (ORF) and the lack of clear homologs in other organisms led to the initial assumption that most of these transcripts were non-coding, i.e. not translated into a protein. While one recent genome-wide coding prediction study supported the non-coding nature of the vast majority (~99%) of annotated lncRNAs [8], recent advances in assessing translation genome-wide have revealed much more widespread translational activities than anticipated. In particular, the development of ribosome profiling, a technique that allows the mapping of ribosome occupancy at sub-codon resolution [9-11], has revolutionized the genome-wide identification of translated sequences and revealed ribosome association with hundreds of predicted non-coding and un-annotated transcripts [12-19]. These transcripts have emerged as a previously unrecognized source of short proteins in essentially all organisms, ranging from bacteria [20, 21], yeast [9, 12, 18, 22] and plants [23-31] to flies [17, 32-37] and vertebrates [10, 11, 13-15, 38-44]. Here, we review the challenges and opportunities of annotating coding and non-coding transcripts. We use recent gene annotation studies in zebrafish to highlight widely applicable approaches and the lessons learned from these studies.
Challenges in gene annotation
Annotating typical protein coding genes is usually straightforward because their transcripts stand out by containing one long and often conserved ORF. This feature has been used in most classical, automated gene annotations, and correctly identifies the majority of known protein-coding ORFs. Many automated annotation algorithms have employed an ad hoc 100-amino-acid threshold on the length of protein-coding ORFs [1, 3, 45-47]. However, many proteins are smaller than 100 amino acids (e.g. several ribosomal proteins [48] and yeast mating factors [49]), and recent studies from plants [23, 31, 50-55], insects [33, 34, 37, 56-59] and fish [14, 39] (Box 1) show that uncharacterized yet functionally important small proteins had been missed using ORF threshold approaches [14, 15, 43-45, 60]. The small size of these proteins has also hampered their identification through homology searches or random mutagenesis screens.
Box 1 The case of Apela/ELABELA/Toddler – an embryonic peptide mistaken for a non-coding RNA.
The meandering path towards the eventual discovery of Apela as a novel embryonic signal that activates the GPCR APJ/Apelin receptor to promote mesendodermal migration highlights the challenges in accurately predicting the coding potential of unknown transcripts. Apela (also named Ende [94], ELABELA [39] and toddler [14]) was initially identified as an embryonically expressed transcript of unknown function in two independent screens [94, 95]. Detailed expression analyses demonstrated enrichment in endodermal tissues during mouse embryogenesis (hence its initial name “Ende” (Endoderm expressed)) [94]. However, the coding potential of this gene remained an open question. It was speculated that this transcript might be either non-coding or encode a potential peptide of 80 amino acids based on the longest ORF contained within the EST under investigation [94]. After its initial discovery as an embryonically expressed transcript, Apela was “rediscovered” several times in genome-wide annotation pipelines, resulting in opposing coding predictions even within the same model organism. It was predicted to be either a non-coding RNA in mESCs [96] and in zebrafish [68] or protein-coding in the same cell type and species [10, 13, 69]. The reasons for these discrepancies in coding predictions were the distinct computational [68, 69, 96] and experimental [10, 13] classification pipelines. In the case of Apela, either ribosome profiling-based identification of translated regions [10, 13] or classification guided by phylogenetic conservation [69] was sufficient to correctly annotate this gene as coding. Experimental studies eventually revealed that a different and shorter ORF than originally proposed [94] is translated, giving rise to a 58-aa (in mammals: 54-aa) peptide [14, 39]. The existence of this short peptide has by now been confirmed through several lines of evidence: antibody detection [39], mass spectrometry [14], Alkaline-Phosphatase and GFP fusions [14, 39], biological activity of an in vitro synthesized Apela peptide fragment [14], loss-of-function phenotypes by mutagenesis of the coding ORF [14, 39], and most compellingly by rescue of the mesendodermal mis-migration phenotypes in apela mutants by wild type but not by frame-shifted apela mRNA [14].
The translational landscape is a continuum
Attempts to distinguish between genes that encode non-coding RNAs versus mRNAs that generate functional proteins are further complicated by recently emerging evidence for the pervasive nature of translation [9, 10, 12-18, 43, 44]. This situation is analogous to the concept of pervasive transcription [5, 7], where the majority of the genome is transcribed, but many of the resulting RNAs may be non-functional. Similarly, the phenomenon of pervasive translation implies that not all newly identified peptides are functional. In fact, a large fraction of translation products appears to be highly unstable and degraded within minutes after production [61]. Thus, translation of ORFs could in some cases be neutral, or alternatively, it could be the translation process itself rather than the resultant peptide that serves a function (Fig. 1). Precedent for regulatory functions of translated ORFs whose generated proteins do not have a function comes from studies of ORFs that control the expression of downstream coding ORFs (e.g. GCN4 upstream ORF (uORF) - [62, 63]), balance protein-isoform levels (e.g. C/EBP ORFs - [64, 65]) or regulate mRNA stability via nonsense mediated RNA decay (NMD) (e.g. CPA1 uORF [66], [18, 67]). The translational landscape is thus emerging as a continuum ranging from mRNAs that encode functional proteins to non-coding RNAs, with a transition zone of RNAs that produce peptides that might not be functional (see Figure 1). The big challenge for the future will be to unravel the biological significance of the translational continuum: Which of the short peptides are functional? To which extent does translation serve regulatory functions, and to which extent is it just neutral? Future experimental studies will be required to confirm or adjust the predicted position of each ORF on the translational landscape.
Figure 1. A continuum from protein-coding to non-coding RNAs.
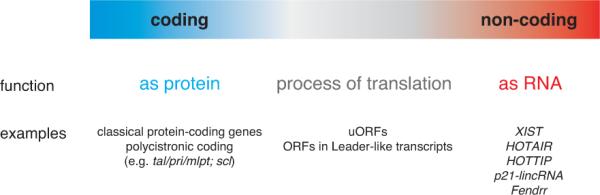
Protein-coding transcripts (blue) and non-coding RNAs (ncRNAs, red) are at either end of the spectrum of translation. The transition zone in between is populated by transcripts with translated ORFs whose peptide products might not be functional. Illustrated here are only transcripts that are functional or potentially functional (non-functional transcripts are not included).
Lessons from transcript annotation studies in zebrafish
The annotation of the zebrafish genome has moved at a rapid pace [13-15, 68-73] and offers interesting case studies for distinguishing coding from non-coding RNAs. Five studies in zebrafish have specifically investigated the coding potential of transcripts, focusing either on identifying non-coding transcripts [68, 69], testing and revising previous non-coding RNA predictions [13] or identifying uncharacterized protein-coding genes [14, 15] (see Box 2 for details, Fig. 2).
Box 2 Studies in zebrafish.
1) Identifying non-coding transcripts
Ulitsky and Shkumatava et al., 2011 [68]: This study predicted 567 putative lncRNA genes, including conserved lncRNAs such as megamind and cyrano. Previously un-annotated intergenic loci were defined by the boundaries of transcription units provided by H3K4me3 chromatin marks outlining the transcriptional start sites, and 3P-Seq defining the polyadenylation sites. The requirement of such “start-end” signatures increased the confidence in gene calls as compared to purely RNA-Seq-based transcript assemblies, which can contain truncated and lowly expressed transcripts. Thus, single exon genes were included in the prediction. Distinction between coding and non-coding transcripts relied mainly on a computational tool called Coding Potential Calculator (CPC [74]; see main text).
Pauli and Valen et al., 2012 [69]: This study predicted 1,133 putative lncRNAs in 859 loci. RNA-Seq-based transcript assemblies were used as a starting point for identifying multi-exonic long non-coding RNAs (lncRNAs) expressed during the first five days of zebrafish embryogenesis. Even low-expressed lncRNAs could be identified because of the depth of the RNA-Seq libraries. The identification pipeline aimed at discovering lncRNAs at high confidence, and thus excluded transcripts that (1) were identified at only one developmental stage or by only Cufflinks [97] or Scripture [98], (2) had a high repeat content, (3) were single exons, and (4) contained ORFs longer than 300nts. At the heart of distinguishing coding and non-coding transcripts was the phylogenetic assessment of the codon substitution frequency (PhyloCSF [75]; see main text).
2) Revising non-coding predictions with ribosome profiling
Chew et al., 2013 [13]: This study developed the Translated ORF Classifier (TOC), a ribosome profiling based classifier, to distinguish bona-fide coding ORFs from ORFs within 5’UTRs (leaders) and 3’UTRs (trailers). TOC employs four metrics to assess the coding potential of each ORF within a transcript: (1) ‘Translational Efficiency’ (TE) measures the level of translation of a specific ORF (density of RPFs) in relation to the expression level of the transcript and has been used in prior studies to evaluate translation [9, 10]; (2) ‘Fraction Length’ (FL) is defined as the fraction of the transcript covered by the ORF; (3) ‘Inside versus Outside’ (IO) builds on the observation that the ribosome-protected fraction of bases in a protein coding ORF is much higher than the fraction of bases protected outside of the ORF; and (4) the ‘Disengagement Score’ (DS) captures the efficiency of ribosome release after encountering a stop codon – a feature characteristic of ribosomes translating canonical proteins resulting in few reads over the 3’ UTR. The combination of these four metrics resulted in a significantly higher discriminatory power than TE alone.
3) Identifying translated and protein-coding transcripts
Pauli et al., 2014 [14]: This study expanded the search for novel protein-coding genes within the zebrafish genome and predicted 700 previously non-annotated protein-coding transcripts in 399 loci, including 26 loci encoding 28 putative secreted peptides. To screen for confidently translated, un-annotated protein-coding transcripts, ORFs were extracted from transcripts that had neither been annotated as coding in the major gene sets in zebrafish (Ensembl and RefSeq) nor been aligned from other organisms (xenoRefSeq). Each of these ORFs was assessed for its coding potential by an improved version of TOC [13] that included a fifth feature, ‘Cover’ (number of bases within an ORF that contain single-nucleotide mapped RPFs). Apart from peptides that had not been described before in any other organism or that appear to be newly discovered paralogs of known peptides, the resultant manually curated list of 700 protein-coding transcripts also contains peptides with annotated homologs in other species (see Box 3). 134/700 transcripts are predicted to encode short (< 100aa) proteins (95 non-redundant small ORFs (smORFs)), one of which is the 58-aa long signal Apela/ELABELA/Toddler (Box 1).
Bazzini et al., 2014 [15]: This study used a ribosome-profiling based strategy similar to [13] and [14] to identify translated ORFs, focusing specifically on short (20-100aa) protein products. ORFscore (see main text) was used to quantify the coding potential of each ORF within a given transcript. Applying ORFscore to 2,450 previously predicted non-coding RNAs and un-annotated transcripts [68, 69, 73] identified 303 putative protein-coding transcripts, including 214 transcripts in 190 loci predicted to encode small polypeptides (20–100 aa). A small fraction of these peptides (6/190) were confirmed by mass-spectrometry. This study also introduced a second computational tool, the so-called micro peptide detection pipeline (micPDP). MicPDP uses PhyloCSF [75] to identify conserved peptides. Of the 63 micPDP-identified conserved zebrafish smORFs, 23 were also found by ORFscore. A complete overlap between ORFscore- and micPDP-predicted peptides is not expected, since these two approaches are complementary and tailored towards active translation and conservation, respectively. The resultant list includes a large number of translated ORFs (170 loci) derived from repetitive element-containing transposons.
Figure 2. Overview of zebrafish transcript annotation pipelines.

Outline of five zebrafish transcript annotation pipelines with their input data, strategies of classification and output data. Two pipelines focused on identifying non-coding transcripts [68, 69], one on testing and revising previous non-coding RNA predictions [13] and two on identifying uncharacterized protein-coding genes [14, 15]. Single asterisk (*): an ORF threshold < 30aa was used for transcripts mapping to genomic regions without alignments [69]. Double asterisk (**): 435 lncRNAs from [68] with sense overlapping transcripts in the Embryonic Transcriptome from [69]. smORF, short ORFs encoding a peptide < 100aa. For details see main text and Box 2.
Distinguishing coding from non-coding transcripts by computational analysesy
Two studies used computational tools to predict hundreds of putative lncRNAs expressed during zebrafish embryogenesis [68, 69]. Both studies were based on the genome-wide identification of transcription units followed by computational filtering pipelines aimed at distinguishing coding from non-coding sequences. While Ulitsky & Shkumatova et al. [68] employed the Coding Potential Calculator (CPC [74]; see later), Pauli & Valen et al. [69] used a combination of PhyloCSF (phylogenetic codon substitution frequency [75]; see later), Blast and an ORF cutoff. The unexpectedly small overlap between the two predicted lncRNA sets (29 putative lncRNAs (~5%)) exemplifies the pitfalls of employing purely computational filtering pipelines.
While the implementation of computational approaches is relatively straightforward, optimizing thresholds to obtain both high specificity and high sensitivity is challenging. For example, CPC is a support vector machine classifier that uses six sequence features to discriminate between coding and non-coding transcripts [74]. Three features score the extent and quality of the longest in-frame ORF within a transcript (size, coverage and integrity (start-stop)); the remaining three features use BLASTX to assess the likelihood that putative homologous protein sequences are present in other organisms (number of hits, significance of hits, frame distribution of hits). CPC has been shown to reliably distinguish coding from non-coding sequences in other studies [74], but it was not a stringent predictor for non-coding transcripts in Ulitsky & Shkumatova et al. [68]; subsequent analyses using ribosome profiling suggested that 20-43% of the predicted putative lncRNAs are indistinguishable from protein-coding mRNAs [13-15].
PhyloCSF is a conservation-based method relying on the observation that proteins are primarily conserved at the amino acid level rather than the nucleotide level [75]. The fact that multiple codons can code for the same amino acid allows some nucleotide substitutions to occur (synonymous changes), but restricts substitutions that would disrupt the amino acid sequence (non-synonymous changes). This feature is the basis of a number of methods -- which have progressively become more sophisticated -- from pairwise sequence comparisons to modeling the phylogenetic relationship between multiple species [15, 75-80]. PhyloCSF outperforms several other similar methods and can identify unknown protein-coding ORFs encoding peptides as short as 13aa [75, 81]. The combination of PhyloCSF and BLAST in Pauli et al. resulted in a low number (<10%) of false-positives (protein-coding genes that were incorrectly predicted to be non-coding) [69] as revealed by ribosome profiling based re-classification [13-15]. However, some conserved lncRNAs (e.g. megamind and cyrano) were filtered out, because they scored high with PhyloCSF.
Non-coding RNA annotations can be improved by ribosome profiling
Because of the limitations of computational pipelines in discriminating coding potential, a subsequent study revisited the lncRNA predictions from [68, 69] experimentally by ribosome profiling [13]. Single-nucleotide mapped ribosome protected fragments were used to develop the so-called Translated ORF Classifier (TOC) (Box 2), which can distinguish bona-fide coding ORFs from ORFs within 5’UTRs (leaders) and 3’UTRs (trailers). TOC also revealed that many of the translated ORFs in “non-coding” RNAs are more similar to uORFs than to canonical proteins (Figure 2). This discovery introduced the concept of “leader-like transcripts” [13] (see also [82]). Leader-like transcripts are characterized by ribosome profiles that resemble the uORF-containing 5’ leaders of classical protein-coding genes. 5’ leaders can have one or several translated small ORFs, generating products as short as a single amino acid (ATG-stop). Although ribosome profiling experiments and mass-spectrometric peptide detection indicate that peptides can be generated from leader-like transcripts and uORFs, the peptide sequence is rarely conserved, and the generated peptides are generally thought to be non-functional [10, 12, 13, 15, 16, 40, 42-44, 83-85].
Ribosome profiling identifies uncharacterized protein-coding genes
In addition to the “leader-like” transcripts containing uORF-like ORFs, ribosome profiling also revealed hundreds of uncharacterized, bona-fide proteins in un-annotated or non-coding transcripts [14, 15]. Building on the study by Chew et al. [13], Pauli et al. [14] used TOC and adopted a broad annotation strategy to identify ~400 un-annotated protein-coding genes in zebrafish (Fig. 2). Bazzini et al. [15] focused on identifying short peptides (<100 aa) and employed two approaches, a translation-independent, PhyloCSF-based method (micPDP) and a translation- dependent metric called “ORFscore” (see Fig. 2). ORFscore is based on the property of translating ribosomes to translocate in discrete one-codon/three-nucleotide steps. This feature, known as “phasing”, results in an unequal distribution of RPFs over the three nucleotides of each codon [9, 86, 87], and has previously been used for detecting frame-shifts in protein-coding ORFs [87]. ORFscore identified 303 putative protein-coding transcripts, including 190 loci predicted to encode small polypeptides (20–100 aa). MicPDP predicted 63 conserved zebrafish smORFs, 40 of which had not been found by ORFscore. Interestingly, about half of the predicted smORFs from Bazzini et al. [15] are derived from repetitive element-containing transposons.
The studies in zebrafish highlight the challenges in transcript annotation: classification schemes are diverse and difficult to directly compare and most methods tend to be biased towards capturing specific subgroups of transcripts. For example, a pipeline that works well for finding conserved lncRNAs like megamind and cyrano [68] might not be efficient in filtering out protein-coding genes [13-15]. Conversely, a pipeline that effectively filters out the majority of protein-coding transcripts might also miss conserved lncRNAs [69]. Moreover, in the absence of other lncRNA-specific features, lncRNA identification remains solely based on the lack of protein-coding features.
Best practices in transcript annotation
Based on the studies in zebrafish, the most powerful classification can be achieved by combining tools from two domains: experimental evidence of translation and evolutionary conservation (Fig. 3). Both of these domains come with shortcomings: experimental evidence relies on high-quality data and the putative peptide being translated at the time of sample collection; sequence conservation can only identify conserved ORFs, and relies either on sufficient genome or transcriptome alignment data or annotation data from other organisms. Because these two approaches are complementary, results can be aggregated into a common prediction, which improves overall transcript classification.
Figure 3. Levels of gene annotation.
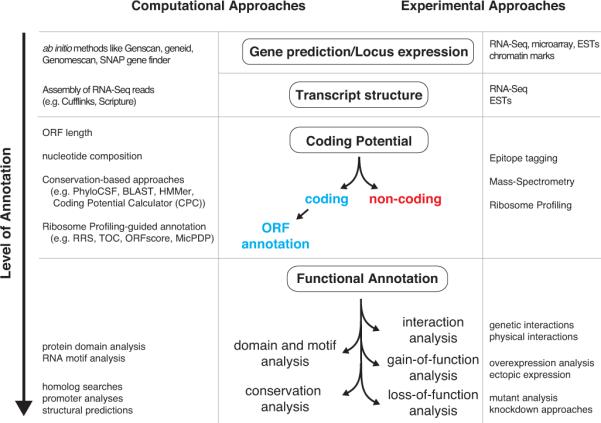
At the most basic level the presence of a gene is indicated by evidence of expression of the locus or by computational prediction of its locus (top). The next level of annotation is the determination of a transcript's exon-intron structure, which is usually followed by the prediction of its coding potential (middle). The ultimate level of annotation is reached by discovering a gene's function (bottom). The computational methods (left) and experimental approaches (right) that can be used to reach each level of gene annotation are outlined.
Predicting coding potential by computational approaches
In the absence of genome-wide experimental data, coding predictions can be made computationally. One strategy that has proven effective is the sequence alignment-based phylogenetic assessment of codon substitution frequencies (PhyloCSF) [75] (see above). Even though its predictive power scales with the quality of the alignments, PhyloCSF correctly predicted Apela/ELABELA/Toddler as a protein-coding transcript in zebrafish [69] (Box 1), which lacks high-quality alignments over the majority of genomic sequences. However, if alignments are of poor quality or missing, PhyloCSF will not perform well in classifying novel transcripts by phylogenetic measures. Coding predictions should therefore be complemented by approaches that are independent of whole genome alignments. These are either aimed at identifying homologous proteins in other species (i.e. BLAST [88]) or use sequence features inherent to protein-coding ORFs such as sequence bias, codon composition or ORF length (e.g. CPAT (Coding-Potential Assessment Tool [89]). Finally, ORF size thresholds, while inherently problematic, can increase the stringency of classification pipelines (e.g. [69]).
All computational methods have the limitation that they rely on the fulfillment of certain conditions, ranging from the availability of well-aligned genomes for multiple species to conservation or sequence bias of the peptide sequence. These conditions might not always be met, and in cases where they are, they might not be exclusive to proteins. These concerns will become less relevant as genome and transcriptome annotations increase in number and quality. Several projects (e.g. the Drosophila modENCODE project [90]) are directed towards sequencing multiple related organisms to obtain comparable datasets that will facilitate evolutionary comparisons for predicting functional elements. Filling these “phylogenetic discovery gaps” will increase the sensitivity in the search for short peptides.
Predicting coding potential based on experimental evidence of translation
Ribosome profiling is currently the ‘state of the art’ experimental approach to assess translation genome-wide. Due to its high-throughput nature it needs to be performed and analyzed with caution because not all sequencing reads originate from a translating ribosome. The predominant contaminant in sequenced ribosome profiling data is ribosomal RNAs, but other ncRNAs such as tRNAs, miRNAs and snoRNAs can result in sequenced reads. Perhaps even more confounding, structured parts of RNA, RNA degradation products and RNA protected by non-ribosomal proteins could give rise to noise on both mRNAs and non-coding RNAs [11, 82]. Differences in sample preparation can also give rise to dissimilarities of ribosome profiles over certain regions and thus result in distinct gene classifications. For example, several ORFs that are classified as protein-coding based on ribosome profiling experiments performed according to one protocol [15] appear to lack ribosome-protected fragments using an alternative protocol [13, 14] and vice versa.
Combining experimental and computational approaches
In light of these experimental limitations, the best results for transcript classification are obtained by combining ribosome profiling data with computational methods specifically targeted at distinguishing ‘real’ translation from noise. Building on the evidence for translation that the ribosome profiling assay provides, these analyses outperform those that are merely based on genomic and meta-genomic features by providing a relatively unbiased view of translated regions. Using specifically developed algorithms that can handle the noise inherent to ribosome profiling assays, the likelihood of each ORF's translational capabilities can be assessed. Several methods have been developed that capture features of translating protein-coding ORFs, including context of the translated ORF within the transcript (TOC [13, 14] and RRS [82]) and phasing of ribosome protected fragments (ORFscore [15]). The newest addition to these methods is the fragment length organization similarity score (FLOSS) [16]. This method takes advantage of the observation that sequenced reads not originating from actively translating ribosomes tend to have a different read-length distribution. Comparison of read lengths within a candidate ORF to read lengths in canonical proteins assesses the likelihood of an ORF to be coding. Together, these features allow the distinction of bona-fide protein-coding ORFs from translated uORFs/leader-like ORFs, untranslated ORFs and – at least in theory - reads originating from non-ribosomal sources.
Candidate translated ORFs can be confirmed by several approaches. Gene-by-gene approaches include the epitope-tagging of putative proteins and mass-spectrometric (MS) validation (e.g. [14, 15]). MS has become a powerful technology also for large-scale and de-novo identification of unknown peptide products [40, 42-44]. It therefore complements ribosome profiling. One caveat of MS-based peptide prediction is that lack of MS-evidence should not be taken as evidence against a protein being produced, since MS has an inherent bias towards detecting certain peptides but not others, and some proteins might be extremely short-lived [61].
Prospects
Function of pervasive translation
The presence of ribosomes on ORFs that do not share the features of canonical protein-coding ORFs poses a number of questions: Are these peptide products functional or simply translational noise? Or does the act of translation itself have a function? The process of translation might serve regulatory roles, e.g. by destabilizing the transcript by triggering non-sense mediated decay (NMD) or – in case of uORFs - by modulating translation of a downstream protein-coding ORF. Pervasive translation might also provide a pool of neutral gene products that selection can act upon. In analogy to the proposed idea of de-novo gene birth as a function for pervasive transcription [91], pervasive translation might generate a peptide that provides a selective advantage under certain conditions. Evolutionarily younger protein-coding genes might have non-coding homologs in syntenic positions in other organisms [92]. This process could be viewed as reverse pseudogenization, in which a protein-coding gene loses its coding function and for which the XIST RNA provides a prominent example [93]. The uncertainty as to the function of this novel population of translated ORFs raises ample opportunities for future studies. What is the extent of small ORFs encoding functional peptides, and what are their functions? What is the extent of regulatory versus neutral translation? To which extent can we computationally predict whether a translated ORF is likely to function as a peptide? While computational algorithms can greatly improve gene annotations, ultimately, the questions regarding functionality of coding or non-coding gene products will need to be addressed by functional studies (see Box 1 and 2).
Challenges of gene annotation
The case of Apela/ELABELA/Toddler demonstrates that important peptides are yet to be discovered and that carefully guided computational classification based on experimental data can find these hidden gems (Box 1). Annotation is best viewed as a gradual process (Fig. 3) where every piece of evidence adds to the final picture, culminating in the eventual identification of the gene's function(s). Currently, the field has many tools for gene annotation, but frequent mis-classifications indicate that there is ample room for improvement. A particular challenge for future tools will be to integrate information from a variety of both experimental and computational sources to predict the coding potential of uncharacterized transcripts. Independent lines of evidence will increase the power of predictions and facilitate rapid annotation of the complete set of genes in new organisms. Functional characterization, carried out on a gene-by-gene basis, is still the final bottleneck. However, identifying a gene's function is what really matters, and every step in the gene annotation pipeline contributes a piece of information towards this ultimate goal.
Box 3 Levels of gene annotation.
Annotation of a gene is a continuous process that usually progresses through several iterations of computational predictions and experiments (Fig. 3), culminating in the determination of its function. These different levels of gene annotation make it difficult and diffuse to define the exact time of gene ‘discovery’, and raise the question at which point a transcript should be considered ‘annotated’. Discovery claims in gene identification and annotation are further complicated by the frequent disagreements in coding predictions between classification pipelines. Many transcripts are predicted as coding by one algorithm, but non-coding by another, and often only functional data can resolve discrepancies in gene annotations. A good example for a gene with contradicting annotations that have finally been resolved by zebrafish knockout studies is the recently confirmed protein-coding gene apela/elabela/toddler [14, 39] (see Box 1). Another factor that can confound discovery claims is the ‘hidden layer’ of gene annotations. Gene annotations are constantly changing, but updates can take a long time to be incorporated into public databases. For example, Ensembl's RNA-Seq-based protein-coding predictions for zebrafish [73] have not yet been incorporated into the Ensembl zebrafish genome build that is by default visible on the Ensembl website. Further complications for discovery claims arise from variable degrees of genome annotations across organisms. What is ‘novel’ (non-annotated or mis-annotated) in one organism might have been annotated in a different organism. Homology-based search algorithms like BLAST are often used to uncover potential homologs in other species, but the interpretation of BLAST-based search results requires caution. First, BLAST does not distinguish between orthology and paralogy. Thus, a BLAST hit of a known, functionally characterized gene could suggest that the gene in question is either an ortholog or a new paralog of the known gene. Distinguishing between these two possibilities requires further analyses such as reciprocal BLAST/BLAT searches. Paralogy is indicated if there is a gene in the same organism that has higher similarity to the BLAST hit than the gene in question. Second, BLAST searches usually include a category of genes that has only been predicted to be coding based on computational algorithms (i.e. BLAST hits like ‘predicted protein’, ‘hypothetical protein’) and might therefore be of limited trustworthiness (see main text). For example, amongst the 28 putative signaling peptides that had not been previously annotated as coding in zebrafish [14], 23 have BLAST hits in other organisms. However, only 6 peptides appear to be orthologs to annotated genes (e.g. otospiralin-like). The remaining hits are uncharacterized (6) or predicted (6) genes and novel paralogs of known genes (5) (e.g. new paralog of interferon 1, new paralog of cocaine- and amphetamine-regulated transcript (CART)). Apela falls into the class of 5 peptides that had no annotated or predicted homologs in other organisms. However, it could be claimed to have been “discovered” at least three times, by 1) the detection of its transcript [94], 2) the discovery of its coding potential and translated ORF [10, 13, 69] and 3) the characterization of its function [14, 39].
Acknowledgements
We thank Sean Eddy, Nicholas Ingolia, Antonio Giraldez and Ariel Bazzini, Igor Ulitsky, Guo-Liang Chew and other members of the Schier lab for helpful comments on the manuscript and discussions. This research was supported by funding from the National Institutes of Health (NIH) (A.P., A.F.S.) and the Human Frontier Science Program (HFSP) (A.P., E.V.).
References
- 1.Okazaki Y, Furuno M, Kasukawa T, Adachi J, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–73. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 2.Bertone P, Stolc V, Royce TE, Rozowsky JS, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–6. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 3.Carninci P, Kasukawa T, Katayama S, Gough J, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 4.Kapranov P, Cheng J, Dike S, Nix DA, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 5.ENCODE Project Consortium. Birney E, Stamatoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, et al. The reality of pervasive transcription. Plos Biol. 2011;9:e1000625. doi: 10.1371/journal.pbio.1000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bánfai B, Jia H, Khatun J, Wood E, et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 2012;22:1646–57. doi: 10.1101/gr.134767.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15:205–13. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- 12.Brar GA, Yassour M, Friedman N, Regev A, et al. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–7. doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chew G-L, Pauli A, Rinn JL, Regev A, et al. Ribosome profiling reveals resemblance between long non-coding RNAs and 5' leaders of coding RNAs. Development. 2013;140:2828–34. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pauli A, Norris ML, Valen E, Chew G-L, et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343:1248636. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014;33:981–93. doi: 10.1002/embj.201488411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014;8:1365–79. doi: 10.1016/j.celrep.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aspden JL, Eyre-Walker YC, Philips RJ, Amin U, et al. Extensive translation of small ORFs revealed by Poly-Ribo-Seq. Elife. 2014;3:e03528. doi: 10.7554/eLife.03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith JE, Alvarez-Dominguez JR, Kline N, Huynh NJ, et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep. 2014;7:1858–66. doi: 10.1016/j.celrep.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruiz-Orera J, Messeguer X, Subirana JA, Albà M. Long non-coding RNAs as a source of new peptides. arXiv preprint arXiv:1405. 2014;4174:1–40. doi: 10.7554/eLife.03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samayoa J, Yildiz FH, Karplus K. Identification of prokaryotic small proteins using a comparative genomic approach. Bioinformatics. 2011;27:1765–71. doi: 10.1093/bioinformatics/btr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hobbs EC, Fontaine F, Yin X, Storz G. An expanding universe of small proteins. Curr Opin Microbiol. 2011;14:167–73. doi: 10.1016/j.mib.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kastenmayer JP, Ni L, Chu A, Kitchen LE, et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Res. 2006;16:365–73. doi: 10.1101/gr.4355406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butenko MA, Patterson SE, Grini PE, Stenvik G-E, et al. Inflorescence deficient in abscission controls floral organ abscission in Arabidopsis and identifies a novel family of putative ligands in plants. Plant Cell. 2003;15:2296–307. doi: 10.1105/tpc.014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham MA, Silverstein KAT, Cannon SB, VandenBosch KA. Computational identification and characterization of novel genes from legumes. Plant Physiol. 2004;135:1179–97. doi: 10.1104/pp.104.037531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silverstein KAT, Graham MA, Paape TD, VandenBosch KA. Genome organization of more than 300 defensin-like genes in Arabidopsis. Plant Physiol. 2005;138:600–10. doi: 10.1104/pp.105.060079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lease KA, Walker JC. The Arabidopsis unannotated secreted peptide database, a resource for plant peptidomics. Plant Physiol. 2006;142:831–8. doi: 10.1104/pp.106.086041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanada K, Zhang X, Borevitz JO, Li W-H, et al. A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Genome Res. 2007;17:632–40. doi: 10.1101/gr.5836207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanada K, Higuchi-Takeuchi M, Okamoto M, Yoshizumi T, et al. Small open reading frames associated with morphogenesis are hidden in plant genomes. Proc Natl Acad Sci USA. 2013;110:2395–400. doi: 10.1073/pnas.1213958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou P, Silverstein KA, Gao L, Walton JD, et al. Detecting small plant peptides using SPADA (Small Peptide Alignment Discovery Application). BMC Bioinformatics. 2013;14:335. doi: 10.1186/1471-2105-14-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haruta M, Sabat G, Stecker K, Minkoff BB, et al. A peptide hormone and its receptor protein kinase regulate plant cell expansion. Science. 2014;343:408–11. doi: 10.1126/science.1244454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costa LM, Marshall E, Tesfaye M, Silverstein KAT, et al. Central cell-derived peptides regulate early embryo patterning in flowering plants. Science. 2014;344:168–72. doi: 10.1126/science.1243005. [DOI] [PubMed] [Google Scholar]
- 32.Liu F, Baggerman G, D'Hertog W, Verleyen P, et al. In silico identification of new secretory peptide genes in Drosophila melanogaster. Mol Cell Proteomics. 2006;5:510–22. doi: 10.1074/mcp.M400114-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Kondo T, Hashimoto Y, Kato K, Inagaki S, et al. Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol. 2007;9:660–5. doi: 10.1038/ncb1595. [DOI] [PubMed] [Google Scholar]
- 34.Kondo T, Plaza S, Zanet J, Benrabah E, et al. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329:336–9. doi: 10.1126/science.1188158. [DOI] [PubMed] [Google Scholar]
- 35.Stark A, Lin MF, Kheradpour P, Pedersen JS, et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–32. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ladoukakis E, Pereira V, Magny EG, Eyre-Walker A, et al. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 2011;12:R118. doi: 10.1186/gb-2011-12-11-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magny EG, Pueyo JI, Pearl FMG, Cespedes MA, et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 2013;341:1116–20. doi: 10.1126/science.1238802. [DOI] [PubMed] [Google Scholar]
- 38.Crowe ML, Wang X-Q, Rothnagel JA. Evidence for conservation and selection of upstream open reading frames suggests probable encoding of bioactive peptides. BMC Genomics. 2006;7:16. doi: 10.1186/1471-2164-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell. 2013;27:672–80. doi: 10.1016/j.devcel.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slavoff SA, Heo J, Budnik BA, Hanakahi LA, et al. A human short open reading frame (sORF)-encoded polypeptide that stimulates DNA end joining. J Biol Chem. 2014;289:10950–7. doi: 10.1074/jbc.C113.533968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma J, Ward CC, Jungreis I, Slavoff SA, et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 2014;13:1757–65. doi: 10.1021/pr401280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim M-S, Pinto SM, Getnet D, Nirujogi RS, et al. A draft map of the human proteome. Nature. 2014;509:575–81. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilhelm M, Schlegl J, Hahne H, Gholami AM, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–7. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 45.Frith MC, Forrest AR, Nourbakhsh E, Pang KC, et al. The abundance of short proteins in the mammalian proteome. PLoS Genet. 2006;2:e52. doi: 10.1371/journal.pgen.0020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frith MC, Bailey TL, Kasukawa T, Mignone F, et al. Discrimination of non-protein-coding transcripts from protein-coding mRNA. RNA Biol. 2006;3:40–8. doi: 10.4161/rna.3.1.2789. [DOI] [PubMed] [Google Scholar]
- 47.Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput Biol. 2008;4:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshihama M. The human ribosomal protein genes: Sequencing and comparative analysis of 73 genes. Genome Res. 2002;12:379–90. doi: 10.1101/gr.214202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen P, Sapperstein SK, Choi JD, Michaelis S. Biogenesis of the Saccharomyces cerevisiae mating pheromone a-factor. J Cell Biol. 1997;136:251–69. doi: 10.1083/jcb.136.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katsir L, Davies KA, Bergmann DC, Laux T. Peptide signaling in plant development. Curr Biol. 2011;21:R356–64. doi: 10.1016/j.cub.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stenvik G-E, Butenko MA, Urbanowicz BR, Rose JKC, et al. Overexpression of INFLORESCENCE DEFICIENT IN ABSCISSION activates cell separation in vestigial abscission zones in Arabidopsis. Plant Cell. 2006;18:1467–76. doi: 10.1105/tpc.106.042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stenvik G-E, Tandstad NM, Guo Y, Shi C-L, et al. The EPIP peptide of INFLORESCENCE DEFICIENT IN ABSCISSION is sufficient to induce abscission in arabidopsis through the receptor-like kinases HAESA and HAESA-LIKE2. Plant Cell. 2008;20:1805–17. doi: 10.1105/tpc.108.059139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho SK, Larue CT, Chevalier D, Wang H, et al. Regulation of floral organ abscission in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2008;105:15629–34. doi: 10.1073/pnas.0805539105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gutierrez-Marcos JF, Costa LM, Biderre-Petit C, Khbaya B, et al. maternally expressed gene1 Is a novel maize endosperm transfer cell-specific gene with a maternal parent-of-origin pattern of expression. Plant Cell. 2004;16:1288–301. doi: 10.1105/tpc.019778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costa LM, Yuan J, Rouster J, Paul W, et al. Maternal control of nutrient allocation in plant seeds by genomic imprinting. Curr Biol. 2012;22:160–5. doi: 10.1016/j.cub.2011.11.059. [DOI] [PubMed] [Google Scholar]
- 56.Hanyu-Nakamura K, Sonobe-Nojima H, Tanigawa A, Lasko P, et al. Drosophila Pgc protein inhibits P-TEFb recruitment to chromatin in primordial germ cells. Nature. 2008;451:730–3. doi: 10.1038/nature06498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Timinszky G, Bortfeld M, Ladurner AG. Repression of RNA polymerase II transcription by a Drosophila oligopeptide. PLoS One. 2008;3:e2506. doi: 10.1371/journal.pone.0002506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Savard J, Marques-Souza H, Aranda M, Tautz D. A segmentation gene in tribolium produces a polycistronic mRNA that codes for multiple conserved peptides. Cell. 2006;126:559–69. doi: 10.1016/j.cell.2006.05.053. [DOI] [PubMed] [Google Scholar]
- 59.Galindo MI, Pueyo JI, Fouix S, Bishop SA, et al. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007;5:e106. doi: 10.1371/journal.pbio.0050106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Basrai MA, Hieter P, Boeke JD. Small open reading frames: beautiful needles in the haystack. Genome Res. 1997;7:768–71. doi: 10.1101/gr.7.8.768. [DOI] [PubMed] [Google Scholar]
- 61.Baboo S, Cook PR. “Dark matter” worlds of unstable RNA and protein. Nucleus. 2014;5:281–6. doi: 10.4161/nucl.29577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mueller PP, Hinnebusch AG. Multiple upstream AUG codons mediate translational control of GCN4. Cell. 1986;45:201–7. doi: 10.1016/0092-8674(86)90384-3. [DOI] [PubMed] [Google Scholar]
- 63.Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–50. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 64.Calkhoven CF, Müller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000;14:1920–32. [PMC free article] [PubMed] [Google Scholar]
- 65.Wethmar K, Bégay V, Smink JJ, Zaragoza K, et al. C/EBPbetaDeltauORF mice--a genetic model for uORF-mediated translational control in mammals. Genes Dev. 2010;24:15–20. doi: 10.1101/gad.557910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaba A, Jacobson A, Sachs MS. Ribosome occupancy of the yeast CPA1 upstream open reading frame termination codon modulates nonsense-mediated mRNA decay. Mol Cell. 2005;20:449–60. doi: 10.1016/j.molcel.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 67.Tani H, Torimura M, Akimitsu N. The RNA degradation pathway regulates the function of GAS5 a non-coding RNA in mammalian cells. PLoS One. 2013;8:e55684. doi: 10.1371/journal.pone.0055684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ulitsky I, Shkumatava A, Jan CH, Sive H, et al. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–50. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pauli A, Valen E, Lin MF, Garber M, et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012;22:577–91. doi: 10.1101/gr.133009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, et al. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012;22:2054–66. doi: 10.1101/gr.139733.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aanes H, Winata CL, Lin CH, Chen JP, et al. Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res. 2011;21:1328–38. doi: 10.1101/gr.116012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aanes H, Østrup O, Andersen IS, Moen LF, et al. Differential transcript isoform usage pre- and post-zygotic genome activation in zebrafish. BMC Genomics. 2013;14:331. doi: 10.1186/1471-2164-14-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Howe K, Clark MD, Torroja CF, Torrance J, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kong L, Zhang Y, Ye Z-Q, Liu X-Q, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–9. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–82. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- 77.Yang Z, Nielsen R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol. 1998;46:409–18. doi: 10.1007/pl00006320. [DOI] [PubMed] [Google Scholar]
- 78.Ochman H. Distinguishing the ORFs from the ELFs: short bacterial genes and the annotation of genomes. Trends Genet. 2002;18:335–7. doi: 10.1016/s0168-9525(02)02668-9. [DOI] [PubMed] [Google Scholar]
- 79.Nekrutenko A, Makova KD, Li W-H. he K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome Res. 2002;12:198–202. doi: 10.1101/gr.200901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hanada K, Akiyama K, Sakurai T, Toyoda T, et al. sORF finder: a program package to identify small open reading frames with high coding potential. Bioinformatics. 2010;26:399–400. doi: 10.1093/bioinformatics/btp688. [DOI] [PubMed] [Google Scholar]
- 81.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–46. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guttman M, Russell P, Ingolia NT, Weissman JS, et al. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–51. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fritsch C, Herrmann A, Nothnagel M, Szafranski K, et al. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 2012;22:2208–18. doi: 10.1101/gr.139568.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arribere JA, Gilbert WV. Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res. 2013;23:977–87. doi: 10.1101/gr.150342.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Juntawong P, Girke T, Bazin J, Bailey-Serres J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc Natl Acad Sci USA. 2014;111:E203–12. doi: 10.1073/pnas.1317811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Michel AM, Choudhury KR, Firth AE, Ingolia NT, et al. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res. 2012;22:2219–29. doi: 10.1101/gr.133249.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Altschul SF, Gish W, Miller W, Myers EW, et al. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 89.Wang L, Park HJ, Dasari S, Wang S, et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.modENCODE Consortium. Roy S, Ernst J, Kharchenko PV, et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–97. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carvunis A-R, Rolland T, Wapinski I, Calderwood MA, et al. Proto-genes and de novo gene birth. Nature. 2012;487:370–4. doi: 10.1038/nature11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xie C, Zhang YE, Chen J-Y, Liu C-J, et al. Hominoid-specific de novo protein-coding genes originating from long non-coding RNAs. PLoS Genet. 2012;8:e1002942. doi: 10.1371/journal.pgen.1002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duret L, Chureau C, Samain S, Weissenbach J, et al. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science. 2006;312:1653–5. doi: 10.1126/science.1126316. [DOI] [PubMed] [Google Scholar]
- 94.Hassan AS, Hou J, Wei W, Hoodless PA. Expression of two novel transcripts in the mouse definitive endoderm. Gene Expr Patterns. 2010;10:127–34. doi: 10.1016/j.gep.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miura T, Luo Y, Khrebtukova I, Brandenberger R, et al. Monitoring early differentiation events in human embryonic stem cells by massively parallel signature sequencing and expressed sequence tag scan. Stem Cells Dev. 2004;13:694–715. doi: 10.1089/scd.2004.13.694. [DOI] [PubMed] [Google Scholar]
- 96.Guttman M, Donaghey J, Carey BW, Garber M, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trapnell C, Williams BA, Pertea G, Mortazavi A, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guttman M, Garber M, Levin JZ, Donaghey J, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–10. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]