

Abstract
Advanced materials that are highly biocompatible and easily modifiable with biomolecules are of great importance for bio-interfacing and the development of biodevices. Here, a biocompatible conducting polymer based nanocomposite was electrochemically synthesized through the electropolymerization of poly(3, 4-ethylene dioxythiophene) (PEDOT) in the presence of graphene oxide (GO) as the only dopant. GO contains many negatively charged carboxyl functional groups and is highly dispersible in aqueous solutions, enabling its facile incorporation and even distribution throughout the conducting polymer. PEDOT/GO films exhibited minimal cytotoxicity after 24 h and supported neuron growth with significantly longer neurites than a control PEDOT/PSS film, indicating that the PEDOT/GO film provides a positive growth signal to developing neurons. While some of the negatively charged functional carboxyl groups of GO “dope” the PEDOT, others are exposed freely on the surface of the nanocomposite allowing easy functionalization of the PEDOT/GO composite with biomolecules. Functional laminin peptide, RNIAEIIKDI (p20), was covalently bound to the surface of the PEDOT/GO film and maintained its bioactivity, as evidenced by an increased neurite outgrowth from neurons cultured on the functionalized composite surface. The ease of biomolecule functionalization of the PEDOT/GO nanocomposite, along with its low electrochemical impedance, minimal toxicity and permissiveness to neuron growth, underlines its potential as a material for widespread biosensing, neural interfacing and tissue engineering applications.
Introduction
With advances in the preparation and characterization of graphene and graphene oxide (GO), there has been exponentially growing interest in these materials because of their outstanding electrical, physical and chemical properties.1–3 The application of graphene4–9 and GO10–13 for interacting with biological systems has only been recently explored, though it has demonstrated great potential for fields such as biosensing, tissue engineering, and drug delivery.14, 15 To date, studies evaluating the biocompatibility of graphene and GO have been inconclusive, with some reports demonstrating severe dose-dependent toxicity,16, 17 while others indicate that graphene nanomaterials may enhance cell growth.12, 18 These conflicting results suggest that the biocompatibility of graphene and GO depends heavily on their specific chemical and physical states as well as their preparation methods, and further investigation is warranted.19, 20
Another class of conductive organic material, conducting polymers, has been extensively studied in biological and biomedical fields such as biosensors, neural tissue engineering and neural electrodes.21–25 In these applications, it is desired to immobilize biomolecules to the polymer in order to impart functionalities specific for interfacing with the biological systems. Such modification often requires the substrate material to have at least one derivatizable functional group, which many of the conducting polymers, such as polyethylenedioxythiophene (PEDOT), lack. In order to add functional groups to PEDOT, generally two strategies have been adopted. One is the direct addition of functional groups to the monomer 3,4-ethylenedioxythiophene (EDOT), followed by polymerization of the modified EDOT monomer.26–32 This method requires tedious synthesis and purification procedures for the modified EDOT monomers, and the added functional groups may pose electronic and steric limitations during polymerization.33 The other strategy is the copolymerization of EDOT with other monomers or molecules that possess functional groups.34–36 Although considerably simple, this method is still unsatisfactory because the existence of these molecules may impair the conductivity and stability of the resultant PEDOT. Another method of imparting bioactive function to PEDOT is to dope the polymer with the bioactive molecules directly. Peptides, drugs and proteins have been directly incorporated in PEDOT for neural interfacing or controlled drug delivery.25, 37–39 However, only negatively charged biomolecules can be used as dopants and most of them are poor dopants because of their weak charge and large size. Poor dopants lead to difficulty in electropolymerization and low conductivity of the resulting polymer. Furthermore, the biomolecules are entrapped throughout the film, limiting the exposure of the functional domain at the surface.
GO possesses many oxygen containing functional groups, such as carboxyl, hydroxyl and epoxide, rendering it hydrophilic and dispersible in aqueous solutions.40–42 This property, along with its abundance of negatively charged carboxyl groups, makes it an excellent dopant for the electropolymerization of conducting polymers. Additionally, GO has recently been shown to act as a promoter of neuronal growth and maturation, making it an interesting candidate as a neural interfacing material.43–45 Conducting polymer/GO nanocomposites have exhibited favorable electrical properties, energy storage and stability;46, 47 however, the performance of PEDOT/GO nanocomposites as biomaterials has yet to be substantially characterized. In this work, we report the straightforward electrochemical synthesis of a PEDOT material doped exclusively with GO and demonstrate its in vitro compatibility with neuronal cells. The GO sheets are partially entrapped by PEDOT on the surface of the nanocomposite and many of the carboxyl functional groups of GO on the surface are exposed freely, enabling biomolecule decoration on the PEDOT/GO film surface via carbodiimide conjugation. We achieve successful covalent immobilization of peptide RNIAEIIKDI (p20), the functional neurite outgrowth domain of extracellular matrix protein, laminin,48–51 and this immobilization procedure may be universally applied to bioactive proteins and peptides for a variety of bio-interfacing applications.
Materials and Methods
Materials
Graphite powder was purchased from Bay Carbon Inc. (SP-1, Bay City). 3,4-Ethylenedioxythiophene (EDOT), poly(sodium-4-styrenesulfonate) (PSS, MW ~ 70,000), phosphate buffered saline (PBS, pH 7.4, 10 mM sodium phosphate and 0.9% NaCl), glutaraldehyde (25% in H2O), osmium tetroxide (OsO4, 4 wt.% in H2O), hexamethyldisilazane (HMDS), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and N-Hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich. The peptide RNIAEIIKDI (p20) was synthesized at the University of Pittsburgh Peptide Synthesis Facility. All other chemicals were of analytical grade, and Milli-Q water from a Millipore Q water purification system was used throughout.
Electrodeposition
GO was synthesized through the oxidization of graphite powder according to the modified Hummers method,{Hummers, 1958 #615}52, 53 and characterized using transmission electron spectroscopy (TEM) (JEOL JEM-2100F). PEDOT/GO films were electrodeposited onto platinum/iridium (Pt/Ir) microelectrodes (standard tip, diameter: 2–3 μm, MicroProbes, Gaithersburg, MD) for electrochemical characterization or gold sputtered plastic microscope coverslips (macroelectrode area: 0.38 cm2) for surface characterization and cell culture using a Gamry Potentiostat, FAS2/Femtostat (Gamry Instruments, Warminster, PA) with Gamry Framework software. A conventional three-electrode system with the Pt/Ir or gold electrode acting as the working electrode, a platinum foil as the counter electrode, and a silver/silver chloride (Ag/AgCl) reference electrode (CH Instruments, Austin, TX) was used. The PEDOT/GO was electropolymerized from an aqueous solution containing 0.02 M EDOT and 10 mg/mL GO. PEDOT/PSS films were synthesized from an electropolymerization solution containing 0.02 M EDOT and 0.1 M PSS. A constant potential of 1.0 V was applied to achieve a charge density of 200 nC total for microelectrodes or 100 mC/cm2 for macroelectrodes.
Modification of PEDOT/GO with p20
The peptide p20 was covalently immobilized on the surface of the PEDOT/GO coated electrodes through an amine reaction between carboxyl groups on the GO and amine groups on the peptide. The PEDOT/GO electrodes were incubated in a solution of 0.2 mg/ml p20, 0.2 M EDC and 0.2 M NHS in sterile H2O for 3 h at room temperature, and then thoroughly washed with sterile PBS to remove any free p20, EDC or NHS. In another set of samples, PEDOT/GO films were incubated with p20 in the absence of EDC/NHS as a control for physical adsorption. The amount of p20 on the surface of the covalently modified PEDOT/GO film was quantified using amino acid hydrolysis followed by high performance liquid chromatography as previously described.51
Electrochemical Impedance Spectroscopy
The electrochemical impedance spectroscopy (EIS) was measured with an Autolab potentiostat/galvanostat, PGSTAT128N (Metrohm Autolab) with Nova 1.8 software using a three-electrode system with a platinum foil counter electrode and Ag/AgCl reference electrode. The EIS was measured in PBS in the frequency range from 10 Hz to 100 kHz using an alternating current sinusoid of 20 mV in amplitude with the direct current potential set to 0 V.
PEDOT/GO Film Surface Analysis
The surface of PEDOT/GO films was characterized using Fourier transform infrared (FTIR) spectroscopy, scanning electron microscopy (SEM) and x-ray photoelectron spectroscopy (XPS). FTIR measurements were carried out using a Bruker Vertex 70 spectrometer equipped with a Hyperion 2000 microscope. A 20x attenuated total reflectance (ATR) objective was employed to record the spectra of deposited thin films. The ATR spectra were converted to transmittance spectra via the standard method within the spectrometer operation software package, OPUS 6.5.
The surface morphologies and microstructures of the PEDOT/GO films were examined with an XL30 SEM (FEI Company) operated at 10 kV. Samples with neurons growing on the surface were analyzed with the same SEM, but at a lower operating potential of 5 kV. Samples with cells were treated with 2.5% glutaraldehyde and 1% OsO4, both for one hour in sequence, followed by dehydration. The dehydration was performed by soaking the samples in 30% and 50% ethanol in PBS, 70% and 90% ethanol in water, and 100% ethanol in sequence for 15 min each, followed by immersion in HMDS for 15 min.
XPS analysis of PEDOT/GO films after treatment with p20 in the presence or absence of EDC/NHS was performed with a K-Alpha XPS system (Thermo Scientific) equipped with a monochromated Al Kα source (1486.68 eV). High resolution scans of the C1s and N1s regions were taken at two locations on each sample.
Primary Neuron Culture
PEDOT/GO coated macroelectrodes were fixed to the surface of 24-well culture plates with Kwik-Sil (World Precision Instruments) and sterilized with exposure to UV light for 15 min. Following sterilization, the polymer surfaces were washed with sterile PBS. Cortical tissue was isolated from E18 Sprague-Dawley rat embryos and treated with 0.025% Trypsin in a digestion buffer containing 137 mM NaCl, 5 mM KCl, 7 mM Na2HPO4, and 25 mM HEPES. Neurons were dissociated with gentle tritruation and maintained in Neurobasal medium (Invitrogen, 21103-049) supplemented with B27 (Invitrogen, 17504-044), GlutaMax (Invitrogen, 35050-061) and Antibiotic-Antimycotic (Invitrogen 15240-062). For neuron growth assays, cells were seeded on PEDOT/GO and PEDOT/PSS surfaces at a density of 100k cells per electrode and grown for 3 days. For neuron viability and death assays, polymer samples were cut to fit into 96-well plates and seeded with neurons at a density of 10k per well. For the cell cultures intended to assess the p20 functionalization on PEDOT/GO films, similar procedures were followed. In order to measure the neurite length easily by preventing the formation of very long and interconnected neurites, neurons were seeded on the PEDOT/GO surfaces at a density of 100k cells per electrode and grown for only 24 h before fixation and immunocytochemical analysis.
Immunofluorescence Staining and Quantification
Neurons growing on the polymer surfaces were fixed in 4% paraformaldehyde in PBS for 15 min and washed several times with PBS. The cells were immersed in a blocking buffer (5% goat serum/0.2% triton-X in PBS) for 20 min followed by incubation in mouse monoclonal antibody against β-III-tubulin (TuJ1, 1:1000, Sigma) for 1 h. After washing in PBS, the cells were incubated in goat anti-mouse Alexa Fluor 488 (1:1000, Invitrogen) secondary antibody for 1 h, washed in PBS and counterstained for nuclei using Hoechst 33342 (Invitrogen).
TuJ1-immunoreactive cells were imaged using a fluorescence microscope. For each experimental group, 10 random 10x images were collected from each sample (n=3). Neuron density was quantified by counting the number of TuJ1-immunoreactive cells that extended at least one neurite that measured longer than the width of the cell body. Neurite analysis was performed using the NeuronJ plugin for ImageJ (http://rsbweb.nih.gov/ij/). Neurites extending from each TuJ1+ cell body were traced and measured, and the average neurite length was calculated.
Neuron Viability and Toxicity Assay
The viability of neurons growing on the PEDOT/GO composite and PEDOT/PSS films, as indicated by their mitochondrial activity, was assessed with the MTT Cell Proliferation Assay Kit (Molecular Probes). The ratio of absorbance signal at 570 nm to 630 nm (reference wavelength) was used to assess metabolic activity. All polymer samples were normalized to a blank containing the polymer sample with no cells, and compared to a positive control containing cells growing on the tissue culture polystyrene (TCP) well surface.
Percentage of cell death was assessed using the propidium iodide (PI) assay. PI fluoresces after binding to the nuclear material of dead cells, while the plasma membrane of healthy cells excludes the dye. Polymer samples were prepared and neuron culture performed as in the MTT assay. Fluorescence was evaluated in a spectrometer with an excitation at 530 nm and emission at 618 nm. Polymer samples were normalized to controls containing the same polymer with 100% dead cells, and compared to cells growing on the TCP control surface.
Statistical Analysis
All statistical analyses were carried out in SPSS software. Student’s t-tests were utilized for comparisons of two experimental groups and one-way analysis of variance (ANOVA) tests followed by Bonferroni’s post hoc analysis were utilized for comparisons of more than two experimental groups. Statistical significance was considered for p < 0.05 (*) and p < 0.01 (**). All data is presented as the mean (±SEM).
Results and Discussion
Synthesis and Characterization of PEDOT/GO Film
GO was synthesized using the modified Hummers method and its microsheet morphology was confirmed with transmission electron microscopy (TEM) (Supplementary Figure 1). For PEDOT/GO film synthesis, electropolymerization of EDOT was carried out in aqueous solution containing only EDOT and GO. No additional electrolyte was used in order to avoid the involvement of any dopant other than GO. In the presence of the negatively charged GO, EDOT could be successfully electropolymerized on the electrode surface, indicating that GO, itself, acts to sufficiently dope the polymer film. To maintain a conductive polymerization solution, a GO concentration of 10 mg/ml was utilized. Because solutions containing lower amounts of GO resulted in slower or less charge passage during the polymerization reaction, a high concentration of GO was selected to ensure adequate film growth. The resulting film is uniform, and the incorporated GO created a network-like surface morphology (Figure 1).
Figure 1.

SEM images of the electrodeposited PEDOT/GO film illustrating the rough, network-like morphology of the surface. The film was electropolymerized at 1.0 V for 600 s in 0.02 M EDOT solution containing 10 mg/mL GO. Scale bar in (a) is 5 μm. Scale bar in (b) is 1 μm.
FTIR analysis of the synthesized GO sheets and the PEDOT/GO films verified successful incorporation of GO into the film (Figure 2). Pure GO exhibits peaks at 3396 cm−1, 1726 cm−1, 1404 cm−1, 1283 cm−1, and 1058 cm−1 that represent carboxylic O-H stretching and vibration, carboxylic C=O stretching and vibration, O-H deformation, epoxy C-O stretching and vibration, and alkoxy C-O stretching and vibration, respectively.47, 54 The spectrum of the electrodeposited PEDOT/GO nanocomposite contains the characteristic peaks for C=O stretching and vibration of carboxyl groups at 1744 cm−1 and O-H deformation at 1410 cm−1. The PEDOT polymer does not contain either carboxyl or hydroxyl functional groups, so these must be attributed to GO, the sole dopant in the polymerization solution, indicating that the GO sheets have been successfully incorporated into the polymer film. Notably, the presence of the carboxylic carbonyl peak indicates that the film contains carboxylic acid functional groups provided by the GO sheets that can be utilized for biomolecule immobilization with carbodiimide crosslinking. The carboxylic O-H stretching and vibration band that should be apparent around 3400 cm−1 is absent in the PEDOT/GO spectrum, and is likely obscured by the tail of the ~ 1 eV bipolaron absorption band, a typical attribute of conductive polymers.55
Figure 2.
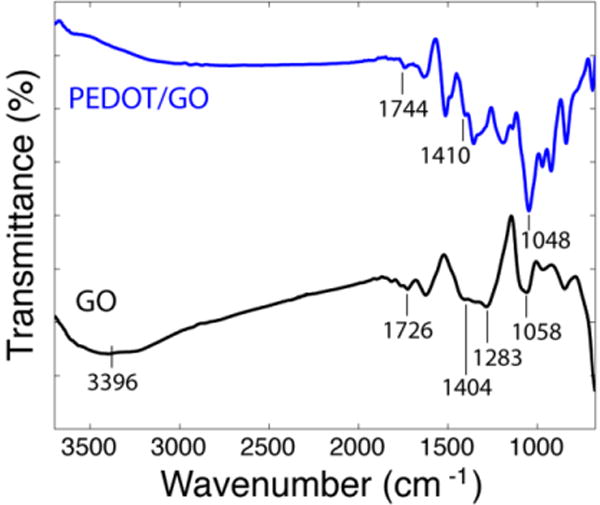
FTIR spectra of GO sheets synthesized using the modified Hummers method (black curve) and electropolymerized PEDOT film doped with GO sheets (blue curve).
Cytotoxicity of PEDOT/GO Nanocomposite
Although PEDOT doped with various molecules, such as heparin,56 poly(styrene sulfonate), (PSS)57 and adhesive peptides58 has been shown to be non-cytotoxic, and soluble GO has demonstrated inconsistent toxicity effects,12, 17, 20 the biocompatibility of GO incorporated in conducting polymers has not previously been studied. To explore the cytocompatibility of the GO doped conducting polymer film, the viability and death of neurons growing on the PEDOT/GO surface after 24 h were evaluated with the MTT viability and propidium iodide (PI) exclusion assays and compared to PEDOT films containing the commonly and extensively studied dopant PSS. To isolate the effects of the polymer surface directly on the cell viability/death, the surfaces were not coated with laminin, an extracellular matrix protein widely used to promote neuron attachment and growth on various surfaces.59, 60 There was no significant difference in viability between the PEDOT/GO and PEDOT/PSS films, with each group exhibiting greater than 96% of the metabolic activity of neurons growing on a control TCP surface (Figure 3a). Neurons growing on the PEDOT/GO surface did not undergo a higher percentage of death than the cells on the PEDOT/PSS surface (GO: 12.79 ± 5.0; PSS: 20.61 ± 3.78, Figure 3b).
Figure 3.
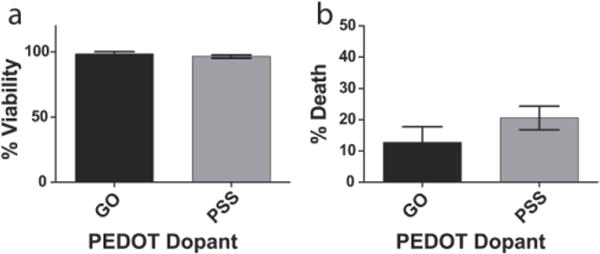
(a) Viability and (b) death of neurons growing on PEDOT films doped with GO or PSS at 24 h. PEDOT/GO films perform similarly to control PEDOT/PSS films, exhibiting no loss of viability and minimal cell death. Error bar represents SEM (n=5).
The mechanism of soluble GO cytotoxicity shown in previous reports remains unclear, but multiple processes have been suggested, including uptake into the cell or adsorption onto the cellular membrane and consequent apoptosis or death, disruption of membrane integrity and cellular exchange, interference with cell adhesion, or induction of oxidative stress16, 17, 61, 62. The absence of significant cytotoxicity caused by PEDOT/GO films in the current study may arise from the entrapment of the GO sheets within the film, hindering their ability to diffuse within the culture media and interact freely with the neurons. Cells growing on the surface of the film are largely contacting the PEDOT polymer, which has demonstrated biocompatibility with neuronal cells.57, 63 The minimal toxicity of PEDOT/GO films indicates that the nanocomposite has potential as a neural interfacing material. However, long-term toxicity studies must be performed to determine the full cytocompatibility of the material.
Neuron Growth on the PEDOT/GO Nanocomposite
To evaluate the neural biocompatibility of the PEDOT/GO composite, films were electrochemically deposited on gold sputtered coverslips, and the resulting PEDOT/GO coated coverslips were used as substrates to grow primary neuron cultures. SEM imaging revealed that neurons exhibited healthy growth on the surface of nanocomposite films in the absence of laminin treatment (Figure 4). Cells spread and flattened on the film and extended long, highly branched neurites that interconnected with other neurons, demonstrating that the surface supported neural attachment and maturation. Due to the specific network-like microstructure of the PEDOT/GO film, some of the smaller processes of neurons intimately grew along or around the partially exposed GO ridges on the surface of the film, potentially using the film morphology as a guidance cue for neurite outgrowth. Representative fluorescent images show neuron attachment and growth on PEDOT/GO and PEDOT/PSS films after 3 d in culture (Figure 5a, b). The neurons grew on the surface of the PEDOT/GO film at a density comparable to that of PEDOT/PSS, indicating that the GO is not specifically contributing any obstruction to the attachment of cells (Figure 5c). While previous GO biocompatibility studies have indicated that GO initiates downregulation of adhesion proteins, such as laminin, fibronectin, and focal adhesion kinase-1, leading to a decrease in cellular adhesion,17 our data suggest that GO entrapped in the polymer matrix may not have such adverse effects on neuron attachment. This agrees with a proposed mechanism for decreased cell adhesion that attributes altered gene expression to the activation of intracellular pathways after GO nanoparticles adhere to the cell membrane.17 GO sheets embedded in the PEDOT polymer matrix may be restricted from interacting with the cell membrane in a way that would initiate changes in gene expression, rendering the PEDOT/GO film a favorable surface for cell attachment and growth.
Figure 4.
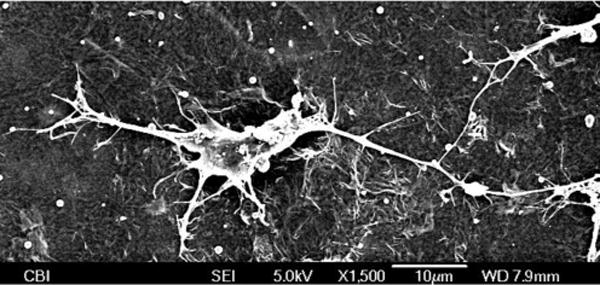
SEM image of a neuron growing on the PEDOT/GO surface at 1 d. The cell exhibits extensive neurite branching and forms contacts with other cells, demonstrating the biocompatibility of the PEDOT/GO film.
Figure 5.
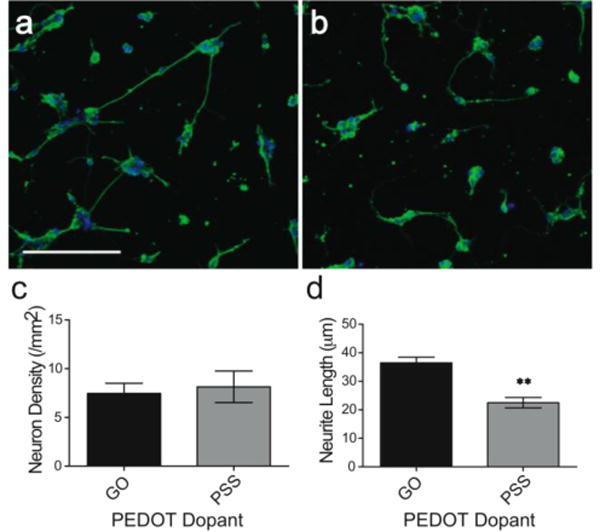
Neuron growth on PEDOT surfaces doped with GO and PSS at 3 d. Representative 20× fluorescent images of β-III-tubulin immunofluorescent reactivity (green) of neurons growing on (a) GO and (b) PSS doped PEDOT. Blue color is Hoechst nuclear counterstain. Scale bar represents 100 μm. (c) Neuron density and (d) average neurite length (± SEM, n=3) of cells growing on the polymer surfaces. GO doped PEDOT films support neurons with longer neurites extensions than PSS doped PEDOT films (** p < 0.01).
Neurons growing on the PEDOT/GO film exhibited significantly longer neurites than cells growing on the PEDOT/PSS film (Figure 5d, GO: 36.4 ± 2.0 μm; PSS: 22.5 ± 1.8 μm, p < 0.01). This finding is supported by previous work demonstrating that pure GO surfaces promote neurite outgrowth in hippocampal neurons,44 and soluble GO can enhance neurite outgrowth in SH-SY5Y neuroblastoma cells by potentially shuttling adsorbed proteins into the cell body during uptake.45 Although the GO from the PEDOT/GO film is likely not being taken into the neuron cell body due to its entrapment within the polymer matrix, its ability to strongly physically adsorb proteins, a consequence of the huge surface area of its single-layer carbon structure, may attract components of the cell media to the surface of the polymer film, enhancing growth cone outgrowth. Additionally, neurons have been shown to be extremely responsive to a variety of topographical cues, and in particular, surface roughness has been shown to promote neurite extension.61, 62 The rough, network-like surface morphology of the PEDOT/GO film (Figures 1 and 4), compared to the smooth and featureless surface of PEDOT/PSS at the same scale (previously reported in [50]) may contribute to the longer neurite outgrowth in PEDOT/GO as compared to PEDOT/PSS. Regardless of the mechanism, the desirable effect on neurite outgrowth demonstrates that PEDOT/GO films are an amenable material for supporting neuronal growth and maturation, and may be useful substrates for neural tissue interfacing applications.
Bioconjugation of PEDOT/GO Films with p20 Peptide
The GO sheets on the top layer of the PEDOT/GO films are partially embedded, as demonstrated by the network-like morphology of the film (Figure 1), and the exposed portions of the GO, rich in carboxyl groups (Figure 2), provide the PEDOT/GO films with many free functional groups. Utilizing carbodiimide conjugation to modify these functional groups, we demonstrate a novel method of biomolecule patterning on conducting polymer films. A laminin fragment peptide, p20, which is reported to promote neurite outgrowth,49, 50 was conjugated to the electrodeposited film. The peptide was covalently attached to the PEDOT/GO film through the formation of amide bonds between the carboxyl groups on the surface of PEDOT/GO and the amine groups of the p20, with the assistance of crosslinkers EDC and NHS. The presence of p20 on the film after carbodiimide modification was verified by hydrolysis and amino acid quantification (5.37 pmol-mm-1).
XPS analysis of the PEDOT/GO film evaluated the surface chemistry of the film after p20 immobilization with EDC/NHS (Figure 6). The deconvoluted C1s region (Figure 6a) consists of 4 peaks in addition to the main C-C peak located at 284.8 eV, including a C-O/C-S peak at 285.6 eV, an epoxy C-O-C peak at 286.9 eV, a N-C=O peak at 288.2 eV and an O-C=O peak at 288.8 eV.63, 64 The PEDOT contributes to the C-S and C-O-C peaks, the GO sheets contribute to the C-O, C-O-C, and O-C=O peaks, and the peptide contributes to the O-C=O and N-C=O peaks. Analysis of the C1s region of the PEDOT/GO film treated with p20 in the absence of EDC/NHS resulted in a similar deconvolution. During the amide bond formation in the presence of EDC/NHS, a carboxylic acid provided by the GO reacts with an amine on the peptide, resulting in a net gain of one amide bond and a net loss of one carboxylic acid bond. However, since both the GO and peptide contain carboxylic acids, a comparison of the ratio of amide to carboxylic acid between the experimental groups cannot be used to verify the formation of covalent amide bonds between the peptide and the film with the addition of EDC/NHS. The carboxylic acid signal of the GO sheets is likely variable across the film depending on the proportion of GO exposed to the surface versus embedded within the polymer matrix, so the ratio of amide to carboxyl will not reflect the amount of covalently attached peptide. A more appropriate method of evaluating the amide formation is to monitor the ratio of amine to amide bonds. During the covalent reaction, one amine in the peptide p20 reacts with a carboxylic acid group to form an amide bond, so there will be more amide and less amine after the covalent treatment, as compared to the physical adsorption treatment. A high-resolution scan of the N1s region of the film treated with p20 and EDC/NHS revealed a peak centered at 388.9 eV, corresponding to the nitrogen in the peptide (Figure 6b). Deconvolution of the N1s peak resulted in a C-N (amine) peak at 399.7 eV, a N-C=O (amide) peak at 400.3 eV, and a protonated amine peak at 401.8 eV.63, 65 The amide/amine ratio is 0.58, compared to 0.19 in the absence of EDC/NHS crosslinking, indicating that the EDC/NHS treatment produced covalent linkages between the peptide and the PEDOT/GO film.
Figure 6.
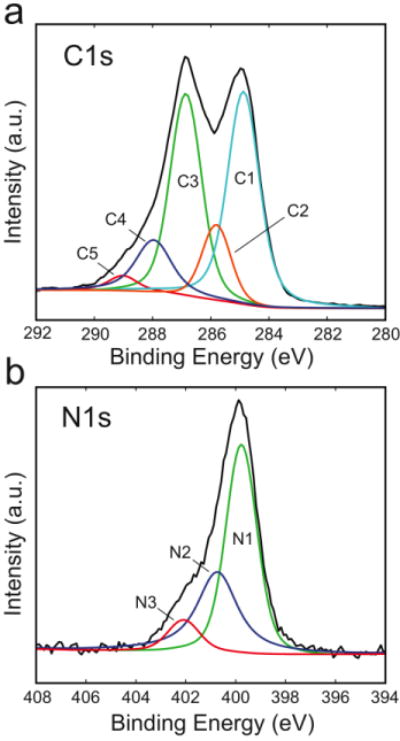
High resolution XPS spectra of the PEDOT/GO surface after treatment with p20 in conjugation with EDC/NHS. Deconvoluted peaks of the (a) C1s region (C1: C-C; C2: C-O/C-S; C3: C-O-C; C4: N-C=O; C5: O-C=O) and (b) N1s region (N1: C-N; N2: N-C=O; N3: protonated amine).
The electrical properties of the electrodeposited PEDOT/GO films before and after p20 immobilization were studied using EIS. As shown in Figure 7a, coating the electrode with the PEDOT/GO film resulted in decreased impedance across all frequencies measured. This significant impedance decrease may be attributed to an increase in the effective surface area of the electrode due to the network-like surface microstructure of the nanocomposite polymer film.66 Longer deposition times resulted in a progressive decrease in impedance, demonstrating the film properties can be tuned as desired by controlling deposition parameters (Supplementary Figure 2). At 1 kHz, a frequency relevant to single unit neural recording, the impedance is decreased by an order of magnitude after the PEDOT/GO deposition, indicating that the film may be a beneficial microelectrode coating to improve the recording and stimulation capability of neural electrodes.67 The Nyquist plot of the impedance (Figure 7b) demonstrates that the bare metal has mostly capacitive behavior, as indicated by its steep linear curve. The electrodes coated with PEDOT/GO films exhibit a knee that separates capacitive behavior at low frequencies and diffusive behavior, characterized by a more gradual slope, at higher frequencies.47, 68 The emergence of diffusion-dominated behavior may be attributed to the creation of a diffusion barrier by the conducting polymer film. After immobilization of p20 at the surface of the polymer film, the impedance increases slightly, a possible result of the creation of a nonconductive peptide layer at the electrode surface; however, the impedance remains significantly lower than that of the bare metal electrode.
Figure 7.
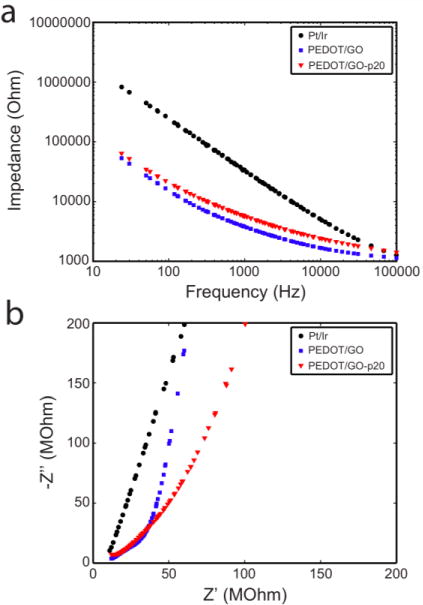
(a) Bode and (b) Nyquist plots of the electrochemical impedance behavior of platinum iridium microwires uncoated (black circles), coated with PEDOT/GO (blue squares) and coated with PEDOT/GO covalently modified with p20 (red triangles).
The bioactivity of the immobilized p20 was assessed with primary neuron culture on the functionalized PEDOT/GO films. After 24 h in culture, neuron attachment and average neurite length were quantified and compared among PEDOT/GO films unmodified with peptide (bare), and films modified with p20 via physical adsorption or covalent immobilization. Representative fluorescent images illustrating β-III-tubulin immunoreactivity and neurite outgrowth on each film are shown in Figure 8a–c. While there are no differences in the density of neurons attached to each film (Figure 8d), the average neurite length (Figure 8e) of the neurons grown on the PEDOT/GO films covalently modified with p20 is significantly longer than that on the other two films (bare: 14.29 ± 0.63 μm; adsorption: 14.59 ± 1.72 μm; covalent immobilization: 20.48 ± 1.45 μm, p < 0.05). This observation can be ascribed to the effect of p20, which is the neurite outgrowth domain of laminin protein, and has been shown to enhance neurite outgrowth when incorporated into conducting polymer films as a dopant.50, 51 There was no discernable effect of p20 when physically adsorbed on the PEDOT/GO film (Figure 8). It is possible that the peptide does not retain its bioactivity, potentially due to conformational changes as a consequence of the physical adsorption onto the film that may obstruct laminin receptors on the neurons from binding to the peptide. Covalent anchoring of p20 to the PEDOT/GO film leaves most of the peptide free to interact with the cell, preserving the bioactivity of the peptide. It is also possible that the physically adsorbed peptide desorbs over the course of the cell culture experiment, resulting in less neurite outgrowth. The covalently conjugated p20 is very stable and continues to support neurite outgrowth after presoaking in PBS at 37°C for 3 d prior to neuron culture (data not shown). This simple method of functionalizing GO doped PEDOT films with biomolecules and its superior effectiveness over traditional biomolecule adsorption clearly demonstrates the potential of the nanocomposite as a bio-interfacing material.
Figure 8.
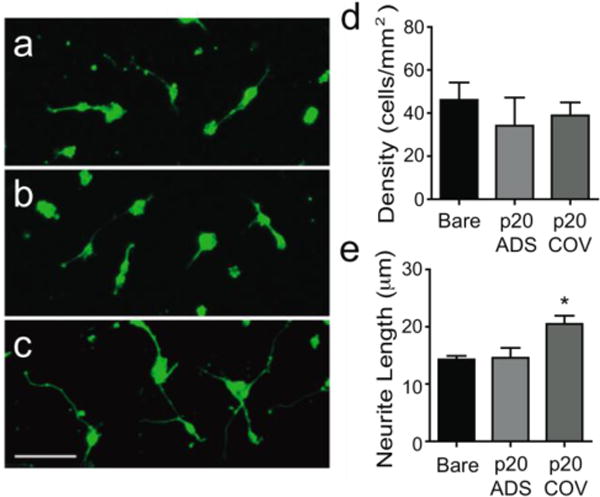
Neuron attachment and neurite outgrowth on PEDOT/GO surfaces modified with p20 peptide at 24 h. Representative 20x fluorescent images of β-III-tubulin immunofluorescent reactivity of neurons cultured on (a) bare, (b) physically adsorbed p20 (p20 ADS) and (c) covalently immobilized p20 (p20 COV) PEDOT/GO surfaces. Scale bar represents 50 μm. (d) Neuron density (± SEM, n=3) growing on the p20 modified PEDOT/GO surfaces. Modification with p20 did not result in a change in cell density. (e) Average neurite length (± SEM, n=3) of neurons growing on the p20 modified PEDOT/GO surfaces (* p < 0.05). Covalent immobilization, but not physical adsorption of p20 on the film surface enhanced neurite outgrowth.
Conclusions
We have successfully prepared a conducting polymer PEDOT material doped solely with GO. The electrodeposited PEDOT/GO films showed good conductivity, and they can significantly lower the impedance of the coated electrodes. The PEDOT/GO films possess a network-like surface structure due to the presence of partially embedded GO sheets, and they supported the growth of neurons with minimal toxicity. Most interestingly, the partially exposed GO pieces on the surface of the PEDOT/GO films are rich in free carboxyl groups, which offer the PEDOT/GO films active functional groups for surface modification. Functional laminin peptide, p20, was successfully bioconjugated to the surface of the PEDOT/GO film through a simple crosslinking reaction that may be universally applied to a multitude of biomolecules. It is expected that this biocompatible PEDOT/GO material with excellent modifiability will find important biological and biomedical applications, such as neural interfacing and biosensing.
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation Grant 0748001, 0729869 and DGE-0549352, National Institute of Health R01NS062019, the Department of Defence TATRC grant WB1XWH-07-1-0716 and DARPA MTO N66001-11-1-4014. X.L. acknowledges the support of the National Natural Science Foundation of China (no. 21275087), the Natural Science Foundation of Shandong Provice of China (ZR2012BM008), and the Taishan Scholar Program of Shandong Province, China. The authors would like to thank Lacey Cirinelli for her assistance with the rat primary neuron culture and the RJ Lee Group for assistance with XPS analysis.
Footnotes
Electronic Supplementary Information (ESI) available: [supplementary figures for TEM characterization of GO and EIS of PEDOT/GO film].
Notes and references
- 1.Loh KP, Bao QL, Eda G, Chhowalla M. Nat Chem. 2010;2:1015–1024. doi: 10.1038/nchem.907. [DOI] [PubMed] [Google Scholar]
- 2.Zhu YW, Murali S, Cai WW, Li XS, Suk JW, Potts JR, Ruoff RS. Advanced Materials. 2010;22:3906–3924. doi: 10.1002/adma.201001068. [DOI] [PubMed] [Google Scholar]
- 3.Huang X, Yin Z, Wu S, Qi X, He Q, Zhang Q, Yan Q, Boey F, Zhang H. Small. 2011;7:1876–1902. doi: 10.1002/smll.201002009. [DOI] [PubMed] [Google Scholar]
- 4.Alwarappan S, Boyapalle S, Kumar A, Li CZ, Mohapatra S. The Journal of Physical Chemistry C. 2012;116:6556–6559. [Google Scholar]
- 5.Alwarappan S, Erdem A, Liu C, Li CZ. The Journal of Physical Chemistry C. 2009;113:8853–8857. [Google Scholar]
- 6.Alwarappan S, Joshi RK, Ram MK, Kumar A. Applied Physics Letters. 2010;96:263702. [Google Scholar]
- 7.Alwarappan S, Liu C, Kumar A, Li CZ. The Journal of Physical Chemistry C. 2010;114:12920–12924. doi: 10.1021/jp1025224. [DOI] [PubMed] [Google Scholar]
- 8.Kalbacova M, Broz A, Kalbac M. Journal of Biomedical Materials Research Part A. 2012;100A:3001–3007. doi: 10.1002/jbm.a.34231. [DOI] [PubMed] [Google Scholar]
- 9.Zhou K, Thouas GA, Bernard CC, Nisbet DR, Finkelstein DI, Li D, Forsythe JS. ACS Applied Materials & Interfaces. 2012;4:4524–4531. doi: 10.1021/am3007565. [DOI] [PubMed] [Google Scholar]
- 10.Depan D, Girase B, Shah JS, Misra RD. Acta Biomater. 2011;7:3432–3445. doi: 10.1016/j.actbio.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 11.Pan Y, Sahoo NG, Li L. Expert Opin Drug Deliv. 2012;9:1365–1376. doi: 10.1517/17425247.2012.729575. [DOI] [PubMed] [Google Scholar]
- 12.Ruiz ON, Fernando KAS, Wang B, Brown NA, Luo PG, McNamara ND, Vangsness M, Sun YP, Bunker CE. ACS Nano. 2011;5:8100–8107. doi: 10.1021/nn202699t. [DOI] [PubMed] [Google Scholar]
- 13.Zhou M, Zhai Y, Dong S. Analytical Chemistry. 2009;81:5603–5613. doi: 10.1021/ac900136z. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Li Z, Wang J, Li J, Lin Y. Trends Biotechnol. 2011;29:205–212. doi: 10.1016/j.tibtech.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Nayak TR, Hao H, Cai W. Nanoscale. 2012;4:3833–3842. doi: 10.1039/c2nr31040f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu W, Peng C, Lv M, Li X, Zhang Y, Chen N, Fan C, Huang Q. ACS Nano. 2011;5:3693–3700. doi: 10.1021/nn200021j. [DOI] [PubMed] [Google Scholar]
- 17.Wang K, Ruan J, Song H, Zhang J, Wo Y, Guo S, Cui D. Nanoscale Res Lett. 2011;6:1–8. doi: 10.1007/s11671-010-9751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee WC, Lim CHYX, Shi H, Tang LAL, Wang Y, Lim CT, Loh KP. ACS Nano. 2011;5:7334–7341. doi: 10.1021/nn202190c. [DOI] [PubMed] [Google Scholar]
- 19.Singh SK, Singh MK, Kulkarni PP, Sonkar VK, Grácio JJA, Dash D. ACS Nano. 2012;6:2731–2740. doi: 10.1021/nn300172t. [DOI] [PubMed] [Google Scholar]
- 20.Wojtoniszak M, Chen X, Kalenczuk RJ, Wajda A, Lapczuk J, Kurzewski M, Drozdzik M, Chu PK, Borowiak-Palen E. Colloids and Surfaces B: Biointerfaces. 2012;89:79–85. doi: 10.1016/j.colsurfb.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 21.Bendrea AD, Cianga L, Cianga I. J Biomater Appl. 2011;26:3–84. doi: 10.1177/0885328211402704. [DOI] [PubMed] [Google Scholar]
- 22.Abidian MR, Ludwig KA, Marzullo TC, Martin DC, Kipke DR. Advanced Materials. 2009;21:3764–3770. doi: 10.1002/adma.200900887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Istamboulie G, Sikora T, Jubete E, Ochoteco E, Marty JL, Noguer T. Talanta. 2010;82:957–961. doi: 10.1016/j.talanta.2010.05.070. [DOI] [PubMed] [Google Scholar]
- 24.Ludwig KA, Langhals NB, Joseph MD, Richardson-Burns SM, Hendricks JL, Kipke DR. Journal of Neural Engineering. 2011;8:014001. doi: 10.1088/1741-2560/8/1/014001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rozlosnik N. Analytical and Bioanalytical Chemistry. 2009;395:637–645. doi: 10.1007/s00216-009-2981-8. [DOI] [PubMed] [Google Scholar]
- 26.Akoudad S, Roncali J. Electrochemistry Communications. 2000;2:72–76. [Google Scholar]
- 27.Ali EM, Kantchev EAB, Yu HH, Ying JY. Macromolecules. 2007;40:6025–6027. [Google Scholar]
- 28.Arias-Pardilla J, Otero TF, Blanco R, Segura JL. Electrochim Acta. 2010;55:1535–1542. [Google Scholar]
- 29.Luo SC, Ali EM, Tansil NC, Yu HH, Gao S, Kantchev EAB, Ying JY. Langmuir. 2008;24:8071–8077. doi: 10.1021/la800333g. [DOI] [PubMed] [Google Scholar]
- 30.Segura JL, Gomez R, Blanco R, Reinold E, Bauerle P. Chem Mater. 2006;18:2834–2847. [Google Scholar]
- 31.Xie H, Luo SC, Yu HH. Small. 2009;5:2611–2617. doi: 10.1002/smll.200900312. [DOI] [PubMed] [Google Scholar]
- 32.Povlich LK, Cho JC, Leach MK, Corey JM, Kim J, Martin DC. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbagen.2012.10.017. http://dx.doi.org/10.1016/j.bbagen.2012.1010.1017. [DOI] [PubMed]
- 33.Roncali J. J Mater Chem. 1999;9:1875–1893. [Google Scholar]
- 34.Brisset H, Navarro AE, Moustrou C, Perepichka IF, Roncali J. Electrochemistry Communications. 2004;6:249–253. [Google Scholar]
- 35.Doherty WJ, Wysocki RJ, Armstrong NR, Saavedra SS. Macromolecules. 2006;39:4418–4424. [Google Scholar]
- 36.Yidiz E, Camurlu P, Tanyeli C, Akhmedov I, Toppare L. J Electroanal Chem. 2008;612:247–256. [Google Scholar]
- 37.Abidian MR, Martin DC. Adv Funct Mater. 2009;19:573–585. [Google Scholar]
- 38.Asplund M, von Holst H, Inganas O. Biointerphases. 2008;3:83–93. doi: 10.1116/1.2998407. [DOI] [PubMed] [Google Scholar]
- 39.Kim DH, Richardson-Burns SM, Hendricks JL, Sequera C, Martin DC. Adv Funct Mater. 2007;17:79–86. [Google Scholar]
- 40.Jeong HK, Lee YP, Lahaye RJWE, Park MH, An KH, Kim IJ, Yang CW, Park CY, Ruoff RS, Lee YH. Journal of the American Chemical Society. 2008;130:1362–1366. doi: 10.1021/ja076473o. [DOI] [PubMed] [Google Scholar]
- 41.Shen JF, Hu YZ, Shi M, Lu X, Qin C, Li C, Ye MX. Chem Mater. 2009;21:3514–3520. [Google Scholar]
- 42.Dreyer DR, Park S, Bielawski CW, Ruoff RS. Chemical Society Reviews. 2010;39:228–240. doi: 10.1039/b917103g. [DOI] [PubMed] [Google Scholar]
- 43.Agarwal S, Zhou X, Ye F, He Q, Chen GCK, Soo J, Boey F, Zhang H, Chen P. Langmuir. 2010;26:2244–2247. doi: 10.1021/la9048743. [DOI] [PubMed] [Google Scholar]
- 44.Li N, Zhang X, Song Q, Su R, Zhang Q, Kong T, Liu L, Jin G, Tang M, Cheng G. Biomaterials. 2011;32:9374–9382. doi: 10.1016/j.biomaterials.2011.08.065. [DOI] [PubMed] [Google Scholar]
- 45.Lv M, Zhang Y, Liang L, Wei M, Hu W, Li X, Huang Q. Nanoscale. 2012;4:3861–3866. doi: 10.1039/c2nr30407d. [DOI] [PubMed] [Google Scholar]
- 46.Ameen S, Akhtar MS, Shin HS. Sensors and Actuators B: Chemical. 2012;173:177–183. [Google Scholar]
- 47.Zhu C, Zhai J, Wen D, Dong S. J Mater Chem. 2012;22:6300–6306. [Google Scholar]
- 48.Matsuzawa M, Liesi P, Knoll W. J Neurosci Methods. 1996;69:189–196. doi: 10.1016/S0165-0270(96)00052-0. [DOI] [PubMed] [Google Scholar]
- 49.Liesi P, Narvanen A, Soos J, Sariola H, Snounou G. Febs Letters. 1989;244:141–148. doi: 10.1016/0014-5793(89)81180-9. [DOI] [PubMed] [Google Scholar]
- 50.Stauffer WR, Cui XT. Biomaterials. 2006;27:2405–2413. doi: 10.1016/j.biomaterials.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 51.Zhang L, Stauffer WR, Jane EP, Sammak PJ, Cui XYT. Macromol Biosci. 2010;10:1456–1464. doi: 10.1002/mabi.201000176. [DOI] [PubMed] [Google Scholar]
- 52.Hummers WS, Offeman RE. J Am Chem Soc. 1958;80:1339–1339. [Google Scholar]
- 53.Mohanty N, Berry V. Nano Lett. 2008;8:4469–4476. doi: 10.1021/nl802412n. [DOI] [PubMed] [Google Scholar]
- 54.Chen D, Li L, Guo L. Nanotechnology. 22:325601. doi: 10.1088/0957-4484/22/32/325601. [DOI] [PubMed] [Google Scholar]
- 55.Cui X, Wiler J, Dzaman M, Altschuler RA, Martin DC. Biomaterials. 2003;24:777–787. doi: 10.1016/s0142-9612(02)00415-5. [DOI] [PubMed] [Google Scholar]
- 56.Asplund M, Thaning E, Lundberg J, Sandberg-Nordqvist AC, Kostyszyn B, Inganas O, von Holst H. Biomed Mater. 2009;4:045009. doi: 10.1088/1748-6041/4/4/045009. [DOI] [PubMed] [Google Scholar]
- 57.Richardson-Burns SM, Hendricks JL, Foster B, Povlich LK, Kim DH, Martin DC. Biomaterials. 2007;28:1539–1552. doi: 10.1016/j.biomaterials.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cui XY, Martin DC. Sensor Actuat B-Chem. 2003;89:92–102. [Google Scholar]
- 59.Powell SK, Kleinman HK. Int J Biochem Cell Biol. 1997;29:401–414. doi: 10.1016/s1357-2725(96)00110-0. [DOI] [PubMed] [Google Scholar]
- 60.Huber M, Heiduschka P, Kienle S, Pavlidis C, Mack J, Walk T, Jung G, Thanos S. Journal of Biomedical Materials Research. 1998;41:278–288. doi: 10.1002/(sici)1097-4636(199808)41:2<278::aid-jbm13>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 61.Walsh JF, Manwaring ME, Tresco PA. Tissue Engineering. 2005;11:1085–1094. doi: 10.1089/ten.2005.11.1085. [DOI] [PubMed] [Google Scholar]
- 62.Khan SP, Auner GG, Newaz GM. Nanomedicine: Nanotechnology, Biology and Medicine. 2005;1:125–129. doi: 10.1016/j.nano.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 63.Hsiao MC, Liao SH, Yen MY, Liu PI, Pu NW, Wang CA, Ma CCM. ACS Applied Materials & Interfaces. 2010;2:3092–3099. doi: 10.1021/am100597d. [DOI] [PubMed] [Google Scholar]
- 64.Jeong HK, Noh HJ, Kim JY, Jin MH, Park CY, Lee YH. EPL (Europhysics Letters) 2008;82:67004. [Google Scholar]
- 65.Xiao SJ, Textor M, Spencer ND, Wieland M, Keller B, Sigrist H. Journal of materials science: Materials in Medicine. 1997;8:867–872. doi: 10.1023/a:1018501804943. [DOI] [PubMed] [Google Scholar]
- 66.Cui XY, Hetke JF, Wiler JA, Anderson DJ, Martin DC. Sensor Actuat a-Phys. 2001;93:8–18. [Google Scholar]
- 67.Pancrazio JJ. Nanomedicine (Lond) 2008;3:823–830. doi: 10.2217/17435889.3.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng C, Jin J, Chen GZ. Electrochim Acta. 2007;53:525–537. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.