

Abstract
Highly enantioselective (up to 99.5% ee) arylation of cyclopropyl C–H bonds with aryl iodides has been developed using a chiral Pd(II) catalyst. Mono-protected amino acid ligands (MPAA) are demonstrated to be compatible with one of the most extensively studied C–H arylation reactions through a Pd(II)/Pd(IV) catalytic cycle.
Graphical Abstract

Pd-catalyzed enantioselective activation of prochiral C–H bonds has been demonstrated with both Pd(0)1,2 and Pd(II)3–9 catalysts. Achieving high levels of reactivity and enantioselectivity using different classes of substrates in these reactions remain a significant challenge. Previously, we demonstrated that Pd(II) complexes coordinated to a chiral mono-N-protected amino acid (MPAA) ligand catalyze both C(sp2)–H4–7 and C(sp3)–H4a,8,9,10 activation, leading to a range of enantioselective carbon–carbon, carbon–oxygen, and carbon–halogen bond forming reactions. Notably, although Pd(II)/Pd(IV) arylation is one of the most studied C–H activation reactions since the 1990s,11 an enantioselective version has not been demonstrated to date. In addition, our previous enantioselective C–H activation reactions had focused mainly on carboxylic acid-derived substrates, and only enantioselective C(sp2)–H iodination of benzylamine substrates has recently been reported. 7
Considering the widespread presence of cyclopropyl moieties in drug and agrochemical molecules (Figure 1),12 we launched efforts to develop enantioselective arylation of cyclopropyl C–H bonds in cyclopropylmethylamines.
Figure 1.
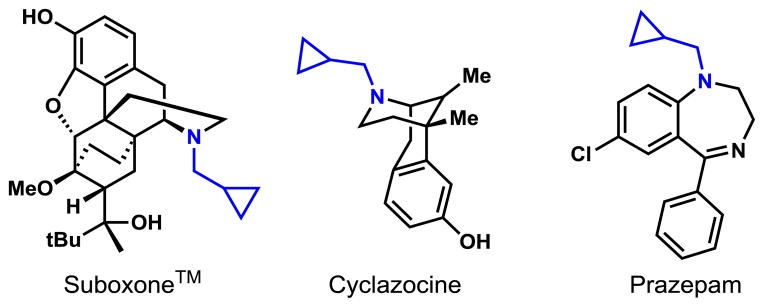
Drugs containing cyclopropylmethylamine moieties
Herein, we report a Pd(II)-catalyzed enantioselective arylation of cyclopropyl C–H bonds of triflyl-protected cyclopropylmethylamines using a chiral MPAA ligand, demonstrating the first example of C–H arylation via Pd(II)/Pd(IV) catalysis. Excellent yields of up to 99%, and enantiomeric excesses (ee) of up to 99.5% were obtained. The observed exclusive mono-selectivity is also a highly desirable feature in C–H arylation reactions. This reaction allows for the rapid generation of two chiral centers in a single step, yielding a single cis-functionalized enantiomer from a prochiral substrate, and provides a new route for the synthesis of enantiopure cyclopropylmethylamines. Remarkably, the chiral MPAA ligand can also override the innate diastereoselectivity of chiral substrates, thus providing a method to construct four different diastereomers, each with three chiral centers.
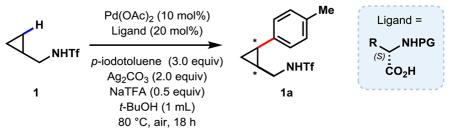
Following our recent success in using triflyl-protected amines as a weak coordinating directing group for C(sp3)–H activation,10 we first focused on reacting 1 with para-iodotoluene in the presence of acetyl-N-protected amino acids (Table 1, entries 1 to 5).
Table 1.
| ||||
|---|---|---|---|---|
| Entry | Ligand
|
Yield (%)a | ee (%) | |
| R | PG | |||
| 1 | Me | Ac | 84 | 76.3 |
| 2 | n-Pr | Ac | 89 | 93.6 |
| 3 | i-Bu | Ac | 87 | 96.0 |
| 4 |
|
Ac | 94 | 96.0 |
| 5 | i-Pr | Ac | 94 | 87.8 |
| 6 | i-Pr | Formyl | 40 | 55.8 |
| 7 | i-Pr |
|
64 | 89.9 |
| 8 | i-Pr |

|
37 | 56.3 |
| 9 | i-Pr |
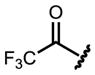
|
70 | 94.2 |
| 10 | i-Pr |

|
70 | 98.1 |
| 11 |
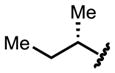
|
Boc | 94 | 98.5 |
| 12 | i-Pr | Boc | 91 | 98.8 |
| 13 | i-Pr | Fmoc | 93 | 98.2 |
| 14 | i-Pr | Cbz | 85 | 98.5 |
| 15 | i-Pr | Troc | 76 | 95.3 |
Experiments were performed with 1 (0.2 mmol), p-iodotoluene (0.6 mmol), Pd(OAc)2 (0.02 mmol), Ag2CO3 (0.4 mmol), sodium trifluoroacetate (0.1 mmol), ligand (0.04 mmol) in t-BuOH (1 mL) for 18 hours at 80 ºC under air.
Yields were determined by 1H NMR spectroscopy using CH2Br2 as an internal standard.
We found that bulky substituents on the amino acid side chain improved the ees without affecting the yields (entries 1 to 5). Linear substituents (n-alkyl) did not perform as well as branched chains. We then explored the protecting groups on the amino group. Formyl protection performed poorly (entry 6). Increasing bulk did not improve either reactivity or selectivity (entries 7 and 8). The electron-withdrawing trifluoroacetyl group substantially improved the ee to 94.2% (entry 9). Carbamates are most effective for this reaction, affording both high yields, and excellent ees (entries 11 to 15), with Boc-protected valine reaching the highest ee of 98.8% (entry 12). Notably, the high enantioselectivities are obtained in spite of the relatively high reaction temperature (80 ºC).
With the optimized reaction conditions in hand, we explored the enantioselective arylation reaction of various substrates with para-iodotoluene in the presence of Boc-L-Val-OH (Table 2). The unsubstituted cyclopropylmethylamine 1 gave a single mono-arylated product 1a in good yield, and excellent ee of 98.8%. The observed exclusive mono-selectivity is rare with C–H arylation reactions using aryl iodides. Excellent mass balance was retained with unreacted starting material fully recovered. In the presence of competing terminal methyl C(sp3)–H bonds, benzylic C(sp3)–H bonds, and aryl C(sp2)–H bonds (substrates 2 to 4), the regio-selectivity for the cyclopropyl C(sp3)–H bonds remained exclusive. Although the yield was significantly lower for 4a, mass balance remained excellent and unreacted starting material was fully recovered. To explore this anomaly further, we subjected substrate 5, an electron-deficient analog of substrate 4, to the same reaction conditions. To our delight, nearly quantitative yield of the desired product in 98.9% ee was obtained. We hypothesized that the electron-rich π-ring in 4 or 4a could interfere with the active catalyst, but the electronegative F atoms in the rings in 5 or 5a reduced the electron density in the rings so that the reaction could proceed. The TBS-protected 1,3-amino alcohol derivative 6 reacted in excellent yield and correspondingly high ee. The β-amino acid 7 was compatible with the reaction conditions, and in spite of lower yields, the excellent ee of 98.7% was obtained. The lowered yield of 7a could be due to possible competing coordination modes of the ester moiety, which may prevent the Pd center from approaching the C–H bond.
Table 2.
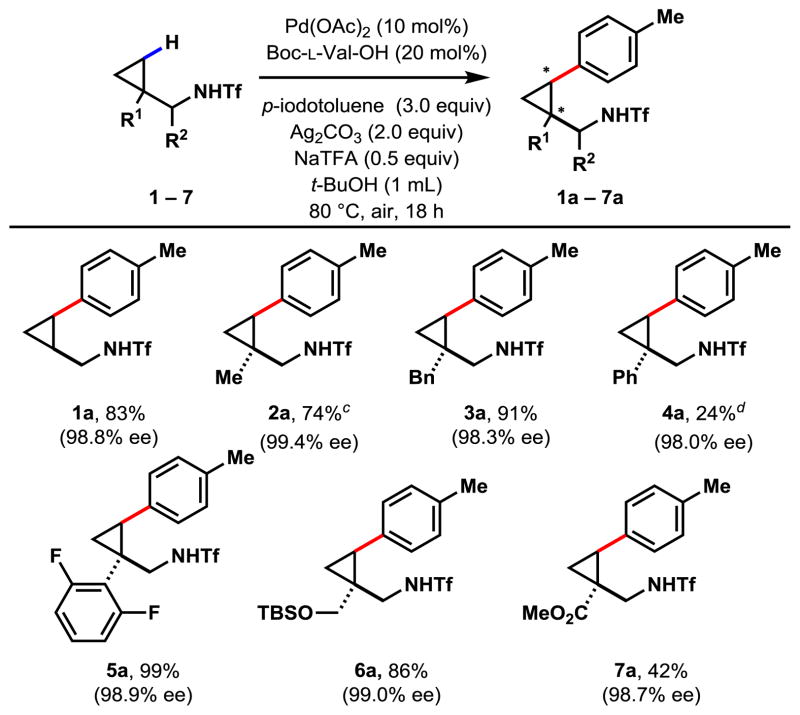
|
Experiments were performed with substrate (0.2 mmol), p-iodotoluene (0.6 mmol), Pd(OAc)2 (0.02 mmol), Ag2CO3 (0.4 mmol), sodium trifluoroacetate (0.1 mmol), Boc-L-Val-OH (0.04 mmol) in t-BuOH (1 mL) for 18 hours at 80 ºC under air.
Isolated yields.
Substrate 2 was run at 0.179 mmol scale.
Substrate 4 was run at 0.25 mmol scale.
In order to derive diversified chiral cycloproanes from simple cyclopropylmethylamine 1, it is crucial that a wide range of aryl iodides can be used as coupling partners. We were delighted to find that a wide range of electron-donating and electron-withdrawing substituents in para-, meta-, and ortho-positions on the aryl iodides are compatible affording good to excellent yields (Table 3). In all but one example, the enantioselectivity is higher than 98% ee. Electron-donating substituents at the para-position of the aryl iodide appeared to lower the yield slightly (products 1d–1e), but did not compromise the ee of the reaction. The highest ee of >99.5% was obtained in the reaction with 1-iodo-3,4-methylenedioxybenzene affording 1e. Electron-withdrawing substituents such as ester, acetyl, cyano, nitro, and trifluoromethyl groups at the para-position gave good yields and excellent ees (1f–j). Aryl iodides containing halogens at the para-positions performed well (1k–n), with yields ranging from 81% to quantitative yields. Both electron-donating (1o), and electron-withdrawing substituents such as ester, acetyl, nitro, and 3,5-di(trifluoromethyl) (1p–s) at the meta-position afforded good to excellent yields and ees. Although the ortho-fluoro substituent afforded the lowest yield of 38% and lowest ee of 95.3%, we were particularly delighted to find that aryl iodide containing an ortho-ester substituent is an excellent coupling partner (95% yield, 98.8% ee). At this stage of development, heterocyclic iodides are incompatible with the reaction (see Supporting Information).
Table 3.

|
Experiments were performed with 1 (0.2 mmol), aryl iodide (0.6 mmol), Pd(OAc)2 (0.02 mmol), Ag2CO3 (0.4 mmol), sodium trifluoroacetate (0.1 mmol), Boc-L-Val-OH (0.04 mmol) in t-BuOH (1 mL) for 18 hours at 80 ºC under air.
Isolated yields.
The excellent enantiocontrol of this ligand prompted us to test whether we can override substrate-controlled diastereoselectivity in substrates containing a chiral center, thereby providing access to all four possible diastereomers. When we subjected the cyclopropylglycine (S)-8 to the optimized conditions with Boc-L-Val-OH (Table 4 entry 1), a >20:1 diastereomeric ratio (dr) favoring product 8ai was obtained. To discern whether the observed stereoselectivity was controlled by the substrate or ligand, we performed a series of control experiments. The use of achiral Boc-Gly-OH (entry 2) resulted in a drastic decrease of stereoselectivity, suggesting that substrate control is not responsible for the observed high selectivity. The use of Boc-D-Val-OH afforded a >20:1 dr favoring the other diastereomer 8aii (entry 3), suggesting that the external chiral ligand is overriding substrate control of stereoselectivity. These results indicate that the careful choice of ligands allow access to all four diastereomers in high dr >20:1. It is therefore possible to enantioselectively arylate racemic 8 to give two diastereomers which can be separated by chromatography or recrystallization.
Table 4.

| ||||
|---|---|---|---|---|
| Entry | Ligand | Major Diastereomer | Yield | dr (major:minor) |
| 1 | Boc-L-Val-OH | 8ai | 35% | >20:1 |
| 2 | Boc-Gly-OH | 8ai | 11% | 2:1 |
| 3 | Boc-D-Val-OH | 8aii | 28% | >20:1 |
Experiments were performed with 8 (0.2 mmol), p-iodotoluene (0.6 mmol), Pd(OAc)2 (0.02 mmol), Ag2CO3 (0.4 mmol), sodium trifluoroacetate (0.1 mmol), ligand (0.04 mmol) in t-BuOH (1 mL) for 18 hours at 80 ºC under air.
Isolated yields reported are combined yields of diastereomers. Relative stereoconfiguration is reported.
To test the scalability of the reaction, we proceeded to run a gram-scale reaction of the simple cyclopropylmethylamine 1 with methyl-2-iodobenzoate (Scheme 1a). Scaled by 25 times to 5 mmol, the reaction proceeded as expected, affording 89% isolated yield and excellent ee (98.6% ee). The triflamide could also be removed in a two-step reaction to yield the free amine in excellent yields (Scheme 1b). For simplicity in product isolation, we decided to trap the free amine with benzoyl chloride, and the reaction yielded 91% over three-steps with unchanged ee.
Scheme 1.
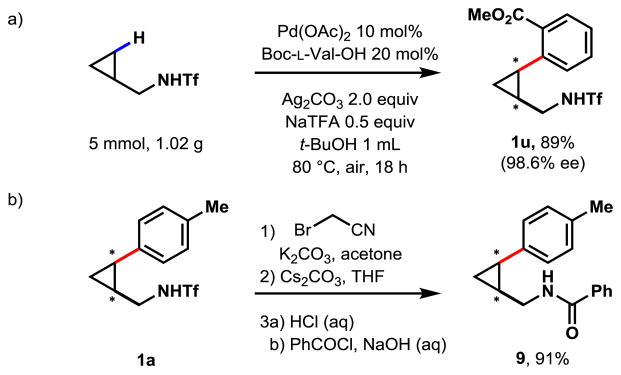
Scale up and deprotection of triflamide.
In summary, Pd(II)-catalyzed highly enantioselective arylation of cyclopropylmethylamines with aryl iodides has been achieved using Pd(II)-MPAA catalysts in excellent yields and ees. This reaction represents the first example of enantioselective C–H arylation through a Pd(II)/Pd(IV) catalytic manifold. The extension of this method to effect other C(sp3)–H bonds is currently underway in our laboratory.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute and the National Institutes of Health (NIGMS, 2R01GM084019) for financial support. K.S.L.C. thanks the Agency for Science, Technology and Research (A*STAR) Singapore for a predoctoral fellowship. This is The Scripps Research Institute (TSRI) manuscript number xxxxx.
Footnotes
Notes
The authors declare no competing financial interest.
Detailed experimental procedure, characterization for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For intramolecular examples of enantioselective Pd(0)-catalyzed C(sp3)–H activation, see: Nakanishi M, Katayev D, Besnard C, Kündig EP. Angew Chem, Int Ed. 2011;50:7438. doi: 10.1002/anie.201102639.Anas S, Cordi A, Kagan HB. Chem Commun. 2011;47:11483. doi: 10.1039/c1cc14292e.Martin N, Pierre C, Davi M, Jazzar R, Baudoin O. Chem— Eur J. 2012;18:4480. doi: 10.1002/chem.201200018.Saget T, Lemouzy SJ, Cramer N. Angew Chem, Int Ed. 2012;51:2238. doi: 10.1002/anie.201108511.Nakanishi M, Katayev D, Besnard C, Kündig EP. Chimia. 2012;66:241. doi: 10.2533/chimia.2012.241.
- 2.For an intermolecular example of enantioselective Pd(0)-catalyzed C(sp3)–H activation, see: Renaudat A, Jean-Gérard L, Jazzar R, Kefalidis CE, Clot E, Baudoin O. Angew Chem, Int Ed. 2010;49:7261. doi: 10.1002/anie.201003544.
- 3.For an example of Pd-catalyzed atropselective arylation, see: Yamaguchi K, Yamaguchi J, Studer A, Itami K. Chem Sci. 2012;3:2165.
- 4.For intermolecular examples of enantioselective Pd(II)/MPAA-catalyzed C(sp2)–H activation and subsequent coupling with organoboron reagents, see: Shi BF, Maugel N, Zhang YH, Yu JQ. Angew Chem, Int Ed. 2008;47:4882. doi: 10.1002/anie.200801030.Gao DW, Shi YC, Gu Q, Zhao ZL, You SL. J Am Chem Soc. 2013;135:86. doi: 10.1021/ja311082u.
- 5.For intermolecular examples of enantioselective Pd(II)/MPAA-catalyzed C(sp2)–H activation and subsequent olefination, see: Shi BF, Zhang YH, Lam JK, Wang DH, Yu JQ. J Am Chem Soc. 2009;132:460. doi: 10.1021/ja909571z.Pi C, Li Y, Cui X, Zhang H, Han Y, Wu Y. Chem Sci. 2013;4:2675.
- 6.For an intermolecular example of enantioselective Pd(II)/MPAA-catalyzed C(sp2)–H activation and subsequent oxidation, see: Cheng XF, Li Y, Su YM, Yin F, Wang JY, Sheng J, Vora HU, Wang XS, Yu JQ. J Am Chem Soc. 2013;135:1236. doi: 10.1021/ja311259x.
- 7.For an intermolecular example of enantioselective Pd(II)/MPAA-catalyzed C(sp2)–H activation and subsequent iodination, see: Chu L, Wang XC, Moore CE, Rheingold AL, Yu JQ. J Am Chem Soc. 2013;135:16344. doi: 10.1021/ja408864c.
- 8.For an intermolecular example of enantioselective Pd(II)/MPAA-catalyzed cyclopropyl C–H activation and subsequent coupling with organoboron reagents, see: Wasa M, Engle KM, Lin DW, Yoo EJ, Yu JQ. J Am Chem Soc. 2011;133:19598. doi: 10.1021/ja207607s.
- 9.For an intermolecular example of enantioselective Pd(II)-catalyzed cyclobutyl C(sp3)–H activation and subsequent coupling with organoboron reagents, using a MPAA analog (mono-protected amino-O-methylhydroxamic acid) ligand, see: Xiao KJ, Lin DW, Miura M, Zhu RY, Gong W, Wasa M, Yu JQ. J Am Chem Soc. 2014;136:8138. doi: 10.1021/ja504196j.
- 10.For an example of Pd(II)/MPAA-catalyzed C(sp3)–H activation, see: Chan KSL, Wasa M, Chu L, Laforteza BN, Miura M, Yu JQ. Nat Chem. 2014;6:146. doi: 10.1038/nchem.1836.
- 11.For examples of Pd(II)/Pd(IV) C–H arylation, see: Dyker G. Angew Chem, Int Ed. 1994;33:103.Zaitsev VG, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154. doi: 10.1021/ja054549f.Rodríguez N, Romero-Revilla JA, Fernández-Ibáñez MÁ, Carretero JC. Chem Sci. 2013;4:175.
- 12.For reviews of transition metal-mediated transformations for the synthesis of enantiopure cyclopropanes, see: Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. Wiley; New York: 1998. Lebel H, Marcoux JF, Molinaro C, Charette AB. Chem Rev. 2003;103:977. doi: 10.1021/cr010007e.Davies HML, Antoulinakis EG. Org React. 2001;57:1.Denmark SE, Beutner G. In: Cycloaddition Reactions in Organic Synthesis. Kobayashi S, Jørgensen KA, editors. Wiley-VCH; Weinheim, Germany: 2002. p. 85.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.