

Abstract
Genetic predisposition and environmental factors influence the development of human autoimmune disease. Occupational exposure to crystalline silica (cSiO2) has been etiologically linked to increased incidence of autoimmunity, including systemic lupus erythematosus (SLE), but the underlying mechanisms are poorly understood. The purpose of this study was to test the hypothesis that early repeated short-term cSiO2 exposure will modulate both latency and severity of autoimmunity in the lupus-prone female NZBWF1 mouse. Weekly intranasal exposure to cSiO2 (0.25 and 1.0 mg) for 4 wk beginning at 9 wk of age both reduced latency and increased intensity of glomerulonephritis. cSiO2 elicited robust inflammatory responses in the lungs as evidenced by extensive perivascular and peribronchial lymphoplasmacytic infiltration consisting of IgG-producing plasma cells, and CD45R+ and CD3+ lymphocytes that were highly suggestive of ectopic lymphoid tissue (ELT). In addition, there were elevated concentrations of immunoglobulins and the cytokines MCP-1, TNF-α and IL-6 in bronchoalveolar lavage fluid. cSiO2-associated kidney and lung effects paralleled dose-dependent elevations of autoantibodies and proinflammatory cytokines in plasma. Taken together, cSiO2-induced pulmonary inflammation and ectopic lymphoid neogenesis in the NZBWF1 mouse corresponded closely to systemic inflammatory and autoimmune responses as well as the early initiation of pathological outcomes in the kidney. These findings suggest that following airway exposure to crystalline silica, in mice genetically prone to SLE, the lung serves as a platform for triggering systemic autoimmunity and glomerulonephritis.
Introduction
Development of autoimmunity is widely acknowledged to be impacted not only by genetic predisposition, but by environmental factors that potentially accelerate initiation of autoimmune responses and increase severity of organ-specific pathologies [1]. Systemic lupus erythematosus (SLE) is a prototypical autoimmune disease that affects 300,000 Americans, most of whom are women, and is often associated with glomerulonephritis and kidney failure [2]. In the United States, it is estimated that at least 1.7 million workers are employed in occupations (e.g. mining, construction, manufacturing and custodial industries) with potential exposure to crystalline silica (cSiO2) [3]. Human epidemiological studies have linked occupational exposure to crystalline silica to increased SLE incidence [4–6]. For example, a population-based, case-control study conducted with participants from the Carolina Lupus Cohort reported that development of SLE was more prevalent in individuals with occupations associated with cSiO2 exposure [5]. Similarly, in a cohort of 15,000 uranium miners heavily exposed to cSiO2, 28 men were diagnosed with SLE, whereas the predicted SLE prevalence for a group of this size was ≤ 1.5 individuals [7]. Finckh and coworkers [6] found that occupational exposure to cSiO2, but not organic solvents, contributed to increased SLE in urban women. Other studies report SLE-like symptoms in individuals with silicosis, including lymphocyte activation and elevated antinuclear antibodies, immunoglobulins, and immune complexes [8–11].
Both in vivo and in vitro studies have yielded several lines of evidence that cSiO2-induced cell death of alveolar macrophages is a critical event in cSiO2-induced exacerbation of autoimmunity [12–17]. Phagocytosis of cSiO2 particles by alveolar macrophages induces NF-κB activation [18,19]. This results in production of proinflammatory cytokines, including TNF-α [20], which are correlated with disease activity in individuals with SLE [21,22]. Aberrant apoptosis and/or necrosis of cSiO2-laden alveolar macrophages not only releases these particles, enabling continued pulmonary re-exposure to cSiO2 and sustained inflammation, but could also expose normally sequestered nuclear antigens (e.g. dsDNA). Both excessive apoptosis and defective clearance of apoptotic debris contribute to aberrant self-antigen presentation, driving consequent autoimmune sequealae [12,23].
Mouse strains that spontaneously develop SLE have been used to explore how toxicant exposure affects the pathogenesis of autoimmunity [24–27]. One such strain, NZM2410, bred from the widely used SLE model, NZBWF1 [24], exhibits rapid onset of lupus nephritis with most mice succumbing to glomerulonephritis by 25 wk of age [28]. When intranasally instilled with cSiO2, both male and female NZM2410 mice exhibit accelerated onset and increased severity of SLE-related responses including elevated autoreactive antibodies, proteinuria, and glomerulonephritis [29]. It was further determined that concentrations of TNF-α in bronchoalveolar lavage fluid of cSiO2-exposed NZM2410 mice were significantly higher than saline-treated control mice [30]. Autoantibodies isolated from sera of cSiO2-exposed NZM2410 mice were found to bind to apoptotic debris derived from alveolar macrophages [31]. Importantly, the effects of cSiO2 on autoimmunity could be attenuated in this strain in vivo by concurrent instillation with rottlerin, an inhibitor of apoptosis [32].
There are several issues that limit the use of the NZM2410 phenotype to uncover mechanisms by which cSiO2 triggers SLE. Not only does this strain develop rapid onset and progression of lupus nephritis compared to other lupus-prone mouse models [24], but the majority of NZM2410 mice succumb to glomerulonephritis within 2 wk of proteinuria appearance [33]. Furthermore, contrary to SLE prevalence in human population and other murine models [24], NZM2410 mice do not display female sex bias. Finally, while NZM2410 mice have been useful for delineating genetic contributions to SLE, most preclinical studies of putative therapeutic agents employ the NZBWF1 mouse, which displays slower development of autoimmunity [25]. Taken together, investigation of subtle underlying mechanisms and preventative measures to counter toxicant-triggered SLE might be enhanced by employing the less-penetrant NZBWF1 phenotype.
The purpose of this study was to test the hypothesis that short-term repeated intranasal cSiO2 exposure will both decrease latency to onset and increase severity of autoimmunity in female NZBWF1 mice. The results presented herein suggest that cSiO2 triggered robust autoimmune and inflammatory responses in the lung, including the formation of ectopic lymphoid tissue, that closely paralleled early onset of glomerulonephritis and exacerbated systemic autoimmune responses.
Materials and Methods
Mice
All experiments were approved by the Institutional Animal Care and Use Committee at Michigan State University in accordance with the National Institutes of Health guidelines for animal use. Female 7 wk old lupus-prone NZBWF1 and control C57Bl/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME), randomized into treatment groups, and allowed to acclimate for 2 wk prior to cSiO2 exposure. Upon arrival and throughout the study, mice were fed semi-purified American Institute of Nutrition (AIN)-93G diet containing high oleic safflower oil [34]. Mice were housed 4 per cage with free access to food and water and maintained at constant temperature and humidity (21°C–24°C and 40–55%, respectively) under a 12-h light/dark cycle.
cSiO2 exposure
The experimental design for cSiO2 exposure, summarized in Fig 1, was based on previous protocols [29–32,35]. cSiO2 (Min-U-Sil-5, average particle diameter 1.5–2.0 μm) was originally obtained from Pennsylvania Sand Glass Corporation (Pittsburgh, PA). Stock suspensions of each dose were prepared fresh in sterile phosphate buffered saline (PBS) just prior to intranasal exposure and sonnicated for 1 min before use. At 9 wk of age, mice were anesthetized with 4% isoflurane and instilled intranasally with 0, 0.25 and 1.0 mg of cSiO2 in 25 μl PBS. To mimic intense occupational exposure to cSiO2 over a short time period, mice were instilled once per wk for 4 wk. Urine was collected weekly and evaluated for proteinuria by 2P urine reagent strips (Cortez Diagnostics, Inc., Calabasas, CA). Mice were sacrificed and tissues collected 12 wk after the final exposure to cSiO2. At this time point, more than 50% of animals exposed to the high cSiO2 dose exhibited moderate proteinuria, defined as ≥ 300 mg/dl. In addition to NZBWF1, female C57Bl/6 mice were used as a control to discern histopathologic effects of high cSiO2 exposure in kidney and lung. C57Bl/6 mice were dosed with either 25 μl PBS or 1.0 mg cSiO2 in 25 μl PBS following the above protocol and terminated 12 wk after the final exposure.
Fig 1. Experimental design for intranasal cSiO2 exposure.

Beginning at 9 wk of age, NZBWF1 and C57Bl/6 mice were dosed intranasally with 25 μl PBS containing 0, 0.25 mg or 1.0 mg cSiO2 once per wk, for 4 wk. Proteinuria was monitored over the course of the experiment and all animals sacrificed at 24 wk of age.
Necropsy and tissue collection
Animals were euthanized by intraperitoneal injection with 56 mg/kg BW sodium pentobarbital and exsanguination via the abdominal aorta. Blood was collected using heparinized syringes and centrifuged at 3500 x g for 10 min at 4°C for separation of plasma. The collected plasma was stored at -80°C until future analysis. Following BALF collection as described previously [36], lungs were intratracheally fixed with 10% (v/v) neutral buffered formalin at constant pressure (30 cm H2O) for a minimum 1 h and stored in fixative until further processing for histology. The right kidney was excised and the cranial portion fixed for 24 h in 10% neutral buffered formalin.
Kidney histopathology
Formalin-fixed kidneys were paraffin-embedded, sectioned and stained with either hematoxylin and eosin (H&E) or Periodic acid-Schiff and hematoxylin (PASH). Stained sections were evaluated for lupus nephritis in a blinded fashion by a board-certified veterinary pathologist using a modified International Society of Nephrology/Renal Pathology (ISN/RPS) Lupus Nephritis Classification system [37]. Slide sections were graded as follows: (0) no tubular proteinosis and normal glomeruli; (1) mild tubular proteinosis with multifocal segmental proliferative glomerulonephritis and occasional early glomerular sclerosis and crescent formation; (2) moderate tubular proteinosis with diffuse segmental proliferative glomerulonephritis, early glomerular sclerosis and crescent formation; and (3) marked tubular proteinosis with diffuse global proliferative and sclerosing glomerulonephritis.
Lung histopathology
Randomly orientated, serial sections of the formalin-fixed left lung lobe were processed routinely and embedded in paraffin. Tissue sections (5 μm) were deparaffinized and stained with H&E for histopathology. Tissues were semi-quantitatively scored by a board-certified veterinary pathologist for the following lung lesions: (a) presence of lymphocytic cell infiltration within perivascular and peribronchial regions, (b) alveolitis defined as the presence of alveolar infiltration of vacuolated macrophages, neutrophils, and lymphocytes, granuloma formation in the alveolus, type II epithelial cell hyperplasia, and thickened alveolar wall, and (c) presence of alveolar proteinosis. Individual lungs were graded for these lesions using the following criteria (% of total pulmonary tissue examined): (0) no changes compared to control mice; (1) minimal (<10%); (2) slight (10–25%); moderate (26–50%); (4) severe (51–75%); or (5) very severe (>75%) of total area affected.
Immunohistochemistry for IgG was performed on paraffin-embedded lungs. Tissue sections (5 μm) were deparaffinized and subjected to heat-induced epitope retrieval with citrate buffer (pH 6.0) for 30 min at 100°C. Endogenous peroxidase was blocked with 3% (v/v) H2O2/methanol for 30 min at room temperature followed by blocking non-specific protein binding with normal horse serum (Vector Laboratories Inc., Burlingame, CA) for 30 min. Tissue endogenous avidin and biotin binding sites were blocked by incubation with Avidin D (Vector Laboratories) followed by d-Biotin (Sigma Aldrich, St. Louis, MO), each for 15 min. Goat anti-mouse IgG γ-chain specific (Alpha Diagnostic Inc., San Antonio, TX) was diluted 1:4000 in normal antibody diluent (ScyTek Laboratories Inc., Logan, UT) and incubated for 60 min followed by incubation with 11μg/ml biotinylated horse anti-goat IgG H+L (Vector Laboratories, Inc.) in normal antibody diluent for 30 min. Peroxidase enzyme was incubated on slides for 30 min with Ready-to-Use Elite Peroxidase Reagent (Vector Laboratories, Inc.). Finally, slides were incubated for 15 min with Nova Red substrate (Vector Laboratories, Inc.) and counterstained with hematoxylin.
B and T lymphocytes were identified using rat anti-mouse CD45R (BD Biosciences, San Jose, CA) (1:300) and rabbit anti-mouse CD3 (1:100), respectively on deparaffinized tissue sections (4 μm) that were subjected to heat-induced epitope retrieval with citrate buffer (pH 6.0) for 30 min at 100°C (CD45R) or 125°C for 15 min (CD3). Following pretreatments, formation of avidin—biotin complex and primary antibody staining were performed as described above. Bound CD45R and CD3 antibodies were detected by incubation with biotinylated rat and goat anti-rabbit in conjunction with Peroxidase Reagent and Nova Red substrate.
BALF processing and differential staining
Cytological slides from 150 μl BALF were prepared by centrifugation at 400 x g for 10 min using a Shandon Cytospin 3 (Shandon Scientific, Pittsburgh, PA), allowed to air dry, and stained with Diff-Quick (Fisher Scientific). A total of 200 cells were counted and cells identified as monocytes/macrophages, lymphocytes, and polymorphonuclear (PMN) leukocytes using morphological criteria. Remaining BALF was centrifuged at 2400 x g for 15 min and supernatant collected and stored at -80°C for IgG and cytokine analysis.
IgG and autoantibody measurement
Total IgG, IgA and IgM were quantitated in BALF using an ELISA method described previously [38]. Briefly, 96-well plates were coated with 50μL of 10 μg/ml goat anti-mouse IgA, IgM, or IgG, respectively (Alpha Diagnostics, Inc.). The standard curve was generated using mouse reference serum (Bethyl Laboratories, Inc., Montgomery, TX). Plates were read on an ELISA reader (Molecular Devices, Menlo Park, CA) at 650 nm and Ig concentrations calculated from the standard curves using Softmax software (Molecular Devices). Total Ig antibodies for nuclear antigens and double-stranded DNA (dsDNA) in plasma were measured according to manufacturer’s instructions using commercial ELISA kits (Alpha Diagnostics, Inc.).
Cytokine analysis
BALF and plasma were diluted two-fold and analyzed for the inflammatory cytokines IL-6, IL-10, MCP-1, IFN-γ, TNF, and IL-12p70 using a Mouse Inflammation Cytometric Bead Array (BD Biosciences). Data was acquired using a FACSCalibur flow cytometer (BD Biosciences) and cytokine concentrations were calculated from standard curves using FCAP Array Software (BD Biosciences).
Statistics
All treatment groups consisted of 8 mice and data presented as group mean ± SEM. Data were plotted and analyzed using SigmaPlot 11.0 for Windows (Jandel Scientific; San Rafael, CA). Grubb’s test was performed to identify and exclude outliers from statistical analysis. Differences between cSiO2- and vehicle-treated NZBWF1 mice were analyzed by one-way ANOVA on Ranks with Dunn’s method. Differences between cSiO2- and vehicle-treated C57Bl/6 mice, and NZBWF1 and C57Bl/6 histopathological lung grades were analyzed by Mann-Whitney Rank Sum Test. The Spearman rank-order correlation coefficient was used to correlate cSiO2 dose to experimental endpoint for NZBWF1. A P value of < 0.05 was considered statistically different for all study outcomes.
Results
cSiO2 elicits early onset of proteinuria and glomerulonephritis
The effects of weekly intranasal exposure to 0, 0.25 mg, or 1.0 mg cSiO2 from 9 to 12 wk of age on renal function were monitored by measuring urinary protein at weekly intervals. Proteinuria (≥ 300 mg/dl) was observed in 13% and 62% of NZBWF1 mice treated with 1.0 mg cSiO2 at 10 and 12 wk post-dosing, respectively (Fig 2). In contrast, proteinuria was not evident in NZBWF1 mice dosed with 0.25 mg cSiO2 or vehicle up to 12 wk post-treatment. Proteinuria was not observed in control C57Bl/6 mice treated with vehicle or 1.0 mg cSiO2 up to 12 wk post-dosing.
Fig 2. cSiO2 exposure accelerates development of proteinuria in NZBWF1 mice.

Proteinuria was monitored weekly until sacrifice 12 wk after the final cSiO2 exposure when most mice exposed to 1.0 mg cSiO2 were over threshold (≥ 300 mg/dl). Proteinuria was not detected in NZBWF1 mice dosed with 0.25 mg cSiO2 or vehicle. C57Bl/6 mice exposed to cSiO2 or vehicle did not develop detectable proteinuria over the course of the experiment.
To confirm nephritogenic effects of cSiO2, kidney sections from NZBWF1 mice at 12 wk post-treatment were assessed histopathologically following H&E and PASH staining (Fig 3A and 3B). Kidneys from NZBWF1 mice treated with 1.0 mg cSiO2 exhibited frequent tubular proteinosis and proliferative glomerulonephritis (Fig 3B). Lymphocytic infiltration at the renal pelvis as well as sclerosis and crescent formation were additional common findings in animals treated at this dose. These responses were largely absent in NZBWF1 mice treated with vehicle (Fig 3A) or 0.25 mg cSiO2 as well as control C57Bl/6 mice treated with vehicle or 1.0 mg cSiO2 (Fig 4A and 4B).
Fig 3. cSiO2 exposure increases severity of lupus nephritis in kidneys of NZBWF1 mice.

Representative light photomicrographs of PASH stained kidney sections in NZBWF1 mice at 24 wks of age exposed to vehicle (A) and 1.0mg cSiO2 (B). NZBWF1 mice exposed to 1.0mg cSiO2 developed extensive glomerulonephritis (black arrow) and tubular proteinosis (asterisk). NZBWF1 mice were individually graded for lupus nephritis following the modified ISN/RPS classification system as described in the Materials and Methods (C). Animals exposed to cSiO2 developed more severe lesions characteristic of lupus nephritis than vehicle-exposed mice. cSiO2 dose significantly correlated with lupus nephritis (Spearman rank-order coefficient = 0.64, p < 0.05).
Fig 4. cSiO2 exposure in C57Bl/6 mice induced kidney lesions resembling lupus nephritis.

Representative light photomicrographs of PASH stained kidney sections in C57Bl/6 mice at 24 wks of age exposed to vehicle (A) and 1.0mg cSiO2 (B). Some renal histopathological lesions were observed in cSiO2-exposed C57Bl/6 mice (B) with notable renal tubular proteinosis (asterisk) and several glomeruli with global mesangial hypercellularity (white arrows). C57Bl/6 mice were individually graded for lupus nephritis as described in the Materials and Methods (C). Animals exposed to cSiO2 developed more severe lesions characteristic of lupus nephritis than vehicle exposed mice.
When mice were graded individually for glomerulonephritis severity, only 13% (1/8) of NZBWF1 mice treated with vehicle were ranked 1 due to the occurrence of minimal tubular proteinosis and rare glomeruli with segmental mesangial hypercellularity, whereas the remaining vehicle-treated mice were ranked 0 (Fig 3C). In contrast, cSiO2 elicited renal lesions characteristic of lupus nephritis, with 38% (3/8) and 63% (5/8) of NZBWF1 mice treated with 0.25 mg and 1.0 mg of the agent, respectively, being ranked 1. Additionally, 25% (2/8) of NZBWF1 mice treated with 1.0 mg cSiO2 had renal lesions ranked 2 or 3, whereas no mice treated with vehicle or 0.25 mg cSiO2 developed such severe lesions. Interestingly, while all C57Bl/6 mice exposed to vehicle were ranked 0, indicating no renal lesions consistent with glomerulonephritis, 38% of mice dosed with 1.0 mg cSiO2 developed renal lesions ranked 1 or 2 (Fig 4C). Thus, cSiO2 appeared to induce modest glomerulonephritis in some of the C57Bl/6 control mice without onset of evident proteinuria.
cSiO2 induces marked lymphoplasmacytic infiltration in lung parenchyma
Histologically, conspicuous lymphoplasmacytic infiltrates were present in interstitial tissues surrounding bronchioles and associated blood vessels throughout the lung in NZBWF1 mice exposed to 1.0 mg cSiO2 (Fig 5C). This response was less severe in NZBWF1 mice treated with 0.25 mg cSiO2 and minimal in NZBWF1 mice treated with vehicle (Fig 5A and 5B; Table 1). C57Bl/6 mice exposed to 1.0 mg cSiO2 had minimal perivascular and peribronchial lymphoplasmacytic infiltrates as compared to vehicle controls (Fig 5D and 5E) and NZBWF1 mice treated with cSiO2 (Table 1).
Fig 5. cSiO2 elicits perivascular and peribronchiolar lymphocytic infiltration in lungs of NZBWF1 and C57Bl/6 mice.

Representative light photomicrographs of H&E stained lung sections from NZBWF1 mice exposed to vehicle (A), 0.25 mg cSiO2 (B), or 1.0 mg cSiO2 (C) and C57Bl/6 mice given vehicle (D), or 1.0mg cSiO2 (E). Br = bronchiole, bv = bronchial vasculature, ap = alveolar parenchyma. Black arrows indicate lymphocytic infiltration in perivascular and peribronchial regions. Lymphocytic infiltration was semi-quantitatively graded as described in the Materials and Methods (Table 1).
Table 1. cSiO2 exposure increases severity of lymphocytic cell infiltration in lungs of NZBWF1 mice relative to C57Bl/6 mice.
| Histopathological Lesion | cSiO2 dose | ||||
|---|---|---|---|---|---|
| NZBWF1 | C57Bl/6 | ||||
| 0.0 mg | 0.25 mg | 1.0 mg | 0.0 mg | 1.0 mg | |
| Lymphocytic cell infiltration | 0.8 ± 0.2 | 2.5 ± 0.3* | 3.4 ± 0.2*, b | 0.0 ± 0.0 | 2.3 ± 0.2*, a |
| Alveolitis | 0.0 ± 0.0 | 1.5 ± 0.2* | 2.5 ± 0.3*, a | 0.0 ± 0.0 | 2.3 ± 0.3*, a |
| Alveolar proteinosis | 0.0 ± 0.0 | 1.4 ± 0.2* | 2.6 ± 0.2*, a | 0.0 ± 0.0 | 2.6 ± 0.2*, a |
Mice were graded individually for severity of lung inflammation (% of total pulmonary tissue examined) as follows: 0, no changes; 1, minimal (<10%); 2, slight (10–25%); 3, moderate (26–50%); 4, severe (51–75%) 5; very severe (>75%) of total area affected. Data are mean ± SEM (n = 8/gp). NZBWF1 mice were analyzed by One-way ANOVA on Ranks followed by Spearman rank-order correlation. C57Bl/6 mice were analyzed by Mann-Whitney Rank Sum Test. Comparisons between strains at 1.0mg cSiO2 were analyzed for statistical differences by Mann-Whitney Rank Sum Test. Asterisks indicate statistical difference between silica treatment and vehicle control (p < 0.05). Different letters indicate statistical difference in histopathological lesions between strains (p < 0.05). cSiO2 dose in NZBWF1 significantly correlated (p < 0.05) with lymphocytic cell infiltration (Spearman rank-order correlation coefficient = 0.87), alveolitis (Spearman rank-order correlation coefficient = 0.90), and alveolar proteinosis (Spearman rank-order correlation coefficient = 0.94)
In addition to the pronounced inflammatory cell infiltration observed in the interstitial tissue of the lung, diffuse alveolar lesions were histologically evident after cSiO2 exposure in NZBWF1 and C57Bl/6 mice. In contrast to the aforementioned lymphocytic response in the lung tissues, the severity of alveolar lesions in NZBWF1 mice was comparable to C57Bl/6 mice given the same amount of cSiO2 (Table 1). Dosing of NZBWF1 mice with 0.25 mg cSiO2 also elicited the responses observed with 1.0 mg cSiO2 in this strain, albeit to a more modest extent.
cSiO2 elicits IgG producing plasma cells in lung
Based on the prominent lymphoplasmacytic response in the lungs of cSiO2-treated NZBWF1 mice, the effects of intranasal cSiO2 on IgG production in these tissues was assessed immunohistochemically. Numerous IgG-laden lymphocytes were found present in the perivascular and peribronchiolar lymphoplasmacytic aggregates in cSiO2-treated but not vehicle-treated NZBWF1 mice (Fig 6B and 6D). In addition, H&E staining of lung sections from NZBWF1 mice treated with cSiO2 revealed occasional plasma cells containing Russell bodies (conspicuous cytoplasmic accumulation of hyaline material) that were immunohistochemically identified as containing IgG globulin. Taken together, these results suggest that both production and secretion of IgG by infiltrating plasma cells is induced in both lungs and airways after intranasal cSiO2 exposure in NZBWF1 mice.
Fig 6. Marked accumulation of IgG producing plasma cells occurs in lungs of cSiO2-exposed NZBWF1 mice.

Representative immunohistochemical photomicrographs of IgG in lungs of NZBWF1 mice. Photomicrographs taken at low magnification are shown in A and B whereas images at high magnification are shown in C and D. Vehicle-exposed mice did not indicate positive IgG staining (A,C). cSiO2 induced marked infiltration of IgG-laden lymphocytes peripheral to both blood vessels (bv) and bronchiole airways (BA) (black arrows in B, white arrows in D). IgG was also detected extracellularly within alveolar parenchyma (ap) (stippled arrows in B, black arrows in D).
CD45R+ and CD3+ infiltrating lymphocytes in lung organize into ectopic lymphoid tissue after cSiO2 exposure in NZBWF1 mice
Infiltrating lymphocytes induced by cSiO2 in lungs of NZBWF1 mice were further characterized by immunohistochemistry for B and T lymphocytes. Distinct aggregates of CD45R+ cells (B lymphocytes) (Fig 7B, 7F and 7J) interspersed with CD3+ cells (T lymphocytes) (Fig 7C, 7G and 7K) were observed peripheral to both bronchiolar airways and blood vessels in lung interstitial tissue of NZBWF1 mice exposed to 1.0 mg cSiO2. The distinct localization of CD45R+ aggregated cells in conjunction with infiltrating CD3+ lymphocytes in the lungs following cSiO2 exposure in NZBWF1 mice structurally resembled the spatial architecture of ectopic lymphoid tissue (ELT). The identification of ELT in parallel with the observation of IgG-producing plasma cells suggest that these structures functionally contribute to initiation of antigen-specific adaptive immune responses in lungs of NZBWF1 mice following intranasal cSiO2 exposure.
Fig 7. Intranasal cSiO2 exposure induces infiltration of CD45R+ and CD3+ lymphocytes in lungs that resemble ectopic lymphoid tissue.

Representative light photomicrographs of lung tissue sections from mice treated with 0.0 mg cSiO2 (vehicle controls; A, D, E, H, I and L) or 1.0 mg cSiO2 (B, C, F, G, J and K). Some lung sections were immunohistochemically stained to identify B lymphocytes (CD45R+) (A, B, E, F, I, and J), while others (C, D, G, H, K, and L) were immunohistochemically stained to identify T lymphocytes (CD3+). Photomicrographs taken at high magnifications of blood vessels (v) and bronchioles (b) are illustrated in E, F, G, H and I, J, K, L, respectively. In cSiO2-treated mice, both bronchiolar airways and blood vessels were fully or partially circumscribed by thick interstitial infiltrates of mononuclear cells (arrows in B and C). These infiltrates were primarily comprised of B lymphocytes (solid arrows in F and J) and T lymphocytes (stippled arrows in G and K). B cells tended to form distinct focal aggregates (solid arrows in F and J) and T cells (stippled arrows in G and K) were more diffusely distributed throughout the peribronchiolar and perivascular lymphoid infiltrates. Control mice had only a few widely scattered B (solid arrow in E, I) and T (stippled arrow in H, L) cells present in the alveolar parenchyma (a), but no distinct lymphoid cell cuffing around the bronchioles or blood vessels. All tissues were counterstained with hematoxylin. e, airway epithelium.
cSiO2 induces leukocyte infiltration and elevates IgG, IgA, and IgM in BALF
Effects of enhanced lymphocytic infiltration following intranasal cSiO2 exposure were also evident in the airways. In the recovered BALF, treatment with 0.25 mg and 1.0 mg cSiO2 in NZBWF1 mice elicited increased recruitment of lymphocytes (33% and 43%, respectively) relative to vehicle control where the majority of cells were monocytes/macrophages in both NZBWF1 and C57Bl/6 mice (Fig 8). Relative to NZBWF1 mice, 1.0 mg cSiO2 in C57Bl/6 mice elicited a modest increase of lymphocytes in BALF (23%). Similar PMN leukocyte responses were observed after 1.0mg cSiO2 exposure in both NZBWF1 (19%) and C57Bl/6 mice (17%).
Fig 8. Intranasal cSiO2 exposure induces infiltration of lymphocytes and PMN leukocytes in BALF of NZBWF1 and C57Bl/6 mice.

Cytometric slides prepared from BALF were stained with Diff-Quick and 200 cells per slide identified as monocytes/macrophages (Mo), lymphocytes (Lymph) or polymorphonuclear (PMN) cells. In NZBWF1 mice, group mean ± SEM at 0.0 mg cSiO2 were 97.2 ± 0.4%, 2.4 ± 0.3%, and 0.5 ± 0.1% for Mo, Lymph and PMN, respectively. Group mean ± SEM for 0.25 mg cSiO2 group were 50.8 ± 2.9%, 32.8 ± 3.5%, and 14.0 ± 1.8%, respectively and for the 1.0 mg cSiO2 group were 39.3 ± 1.7%, 43.0 ± 2.5%, and 18.7 ± 1.8%, respectively. In C57Bl/6 mice, group mean ± SEM at 0.0 mg cSiO2 were 95.9 ± 0.9%, 3.1 ± 0.7% and 1.0 ± 0.2% for Mo, Lymph, and PMN, respectively. Group mean ± SEM for 1.0mg cSiO2 were 59.9 ± 3.7%, 22.6 ± 3.7, and 17.3 ± 2.1% for Mo, Lymph, and PMN, respectively.
Based on the observed increase in lymphocytes following intranasal cSiO2 exposure, we performed ELISA to quantitate IgG, IgA and IgM in BALF of NZBWF1 and C57Bl/6 mice. IgG in BALF of NZBWF1 was substantially elevated in a dose-dependent manner following cSiO2 treatment (Fig 9A). cSiO2 also increased IgA and IgM in BALF of both NZBWF1 and C57Bl/6 mice, albeit to a lesser extent than IgG (Fig 9B and 9C).
Fig 9. cSiO2 exposure elevates IgG (A), IgA (B), and IgM (C) concentrations in BALF of NZBWF1 and C57Bl/6 mice.
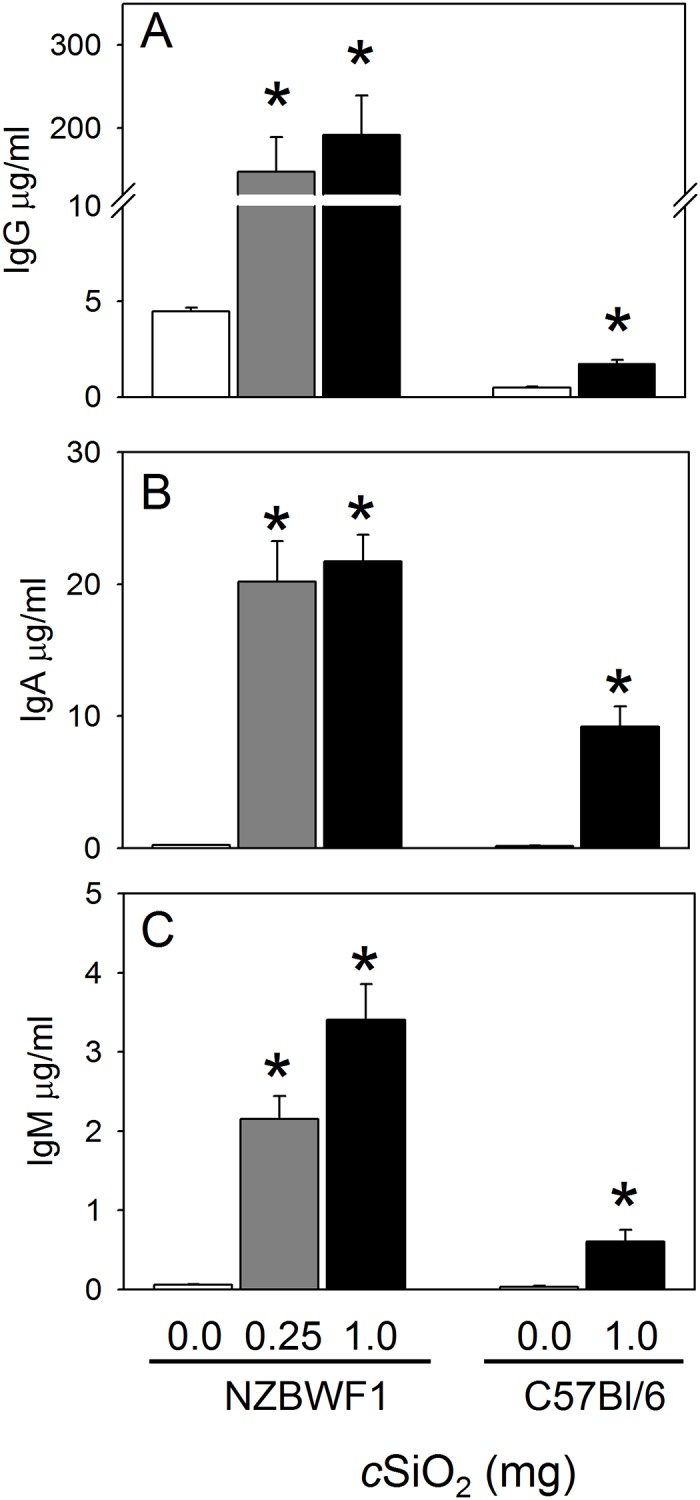
Total immunoglobulins in BALF were measured by ELISA. Data are group mean ± SEM (n = 7–8/gp) and were analyzed by one-way ANOVA on Ranks with Dunn’s method (NZBWF1) or Mann-Whitney Rank Sum Test (C57Bl/6). Asterisk indicates a statistically significant difference between cSiO2 treatment and vehicle control (p < 0.05). In BALF of NZBWF1 mice, cSiO2 dose correlated significantly (p < 0.05) with IgG (Spearman rank-order correlation coefficient = 0.80), IgA (Spearman rank-order correlation coefficient = 0.72), and IgM (Spearman rank-order correlation coefficient = 0.85).
cSiO2 induces elevation of MCP-1, TNF-α, and IL-6 in BALF
BALF of NZBWF1 mice was further analyzed for the cytokines IL-6, IL-10, MCP-1, IFN-γ, TNF-α, and IL-12p70. While effects on IFN-γ, IL-10 or IL-12p70 were not detectable, cSiO2 treatment dose-dependently induced elevation of the proinflammatory cytokines MCP-1, TNF-α, and IL-6 (Fig 10A–10C).
Fig 10. cSiO2 exposure increases proinflammatory cytokines MCP-1 (A, D), TNF-α (B, E), and IL-6 I (C, F) in BALF and plasma of NZBWF1 mice.

Proinflammatory cytokine concentrations in BALF and plasma were determined by cytometric bead array. Data are group mean ± SEM (n = 5–8/gp) and were analyzed by one-way ANOVA on Ranks with Dunn’s method. Asterisk indicates a statistically significant difference in analyte between cSiO2 treatment and vehicle control (p < 0.05). cSiO2 dose correlated significantly (p < 0.05) with BALF MCP-1 (Spearman rank-order correlation coefficient = 0.90), BALF TNF-α (Spearman rank-order correlation coefficient = 0.89), and BALF IL-6 (Spearman rank-order correlation coefficient = 0.82). cSiO2 dose also correlated significantly (p < 0.05) with plasma TNF-α (Spearman rank-order correlation coefficient = 0.60), and IL-6 (Spearman rank-order correlation coefficient = 0.49). n.d. indicates not detected.
cSiO2 triggers systemic autoantibody and proinflammatory cytokine responses
Marked elevations of total Ig anti-dsDNA antibodies (Fig 11A) and anti-nuclear antibodies (Fig 11B) were detected in plasma of cSiO2-dosed NZBWF1 mice compared to vehicle-treated mice. No changes in plasma anti-dsDNA antibodies were detected in C57Bl/6 mice exposed to cSiO2 or vehicle (S1 Fig). cSiO2 dose-dependently induced elevation of the proinflammatory cytokines MCP-1, TNF-α, and IL-6 (Fig 11D–11F) in NZBWF1 mice whereas effects on plasma cytokines IFN-γ, IL-10 or IL-12p70 were undetectable.
Fig 11. cSiO2 exposure increases anti-dsDNA antibodies (A) and anti-nuclear antibodies (B) in plasma of NZBWF1 mice.

Autoreactive antibodies in plasma at sacrifice were measured by ELISA. Data are group mean ± SEM (n = 7–8/gp) and were analyzed by One-Way ANOVA on Ranks with Dunn’s method. Asterisk indicates statistically significant difference in antibody concentration between cSiO2 treatment and vehicle control (p < 0.05). cSiO2 dose significantly correlated (p < 0.05) with plasma anti-dsDNA Ab’s (Spearman rank-order correlation coefficient = 0.62) and anti-nuclear Ab’s (Spearman rank-order correlation coefficient = 0.58).
Discussion
Human epidemiological findings support the contention that airway exposure to cSiO2 is etiologically linked to development of autoimmunity [4–6,39–43]. However, such studies shed little light on the mechanisms that underlie this putative association. Towards that end, we investigated the effects of short-term repeated cSiO2 exposure on autoimmune disease development in the NZBWF1 female mouse, a widely used experimental animal model for SLE. Several novel observations regarding this lupus-prone mouse strain were made 12 wk after the final cSiO2 exposure. First, cSiO2 triggered the onset of and exacerbated glomerulonephritis. Second, concurrent with kidney effects, cSiO2 induced marked lymphocyte infiltration in the lungs as well as dramatic increases in total immunoglobulins (IgG, IgA and IgM), and proinflammatory cytokine concentrations. Third, a striking observation in the lungs of NZBWF1 mice was the identification of extensive ELT that likely contributes to early triggering of SLE after cSiO2 exposure. Finally, in plasma, we found that cSiO2 exposure elicited marked elevations in autoreactive antibodies and proinflammatory cytokine concentrations. Collectively, these findings suggest intensive cSiO2 exposure via the airways over a discrete period evoked long-lasting injurious inflammatory and autoimmune responses that were evident at the site of exposure (lung), systemically, and at a distal tissue site (kidney).
Elevated plasma titers of autoreactive antibodies are an immunological hallmark for human SLE. Systemic autoantibodies form immune complexes with circulating self-antigens (e.g. dsDNA and nucleosome fractions). When deposited in the kidney [29,44–46], these immune complexes promote proinflammatory cytokine and chemokine production that mediate infiltration by mononuclear cells and consequent tissue injury [47]. Congruent with these mechanisms, heightened severity of glomerulonephritis was observed here in cSiO2-treated NZBWF1 mice concurrently with early onset of proteinuria. These findings are consistent with previous observations in cSiO2-exposed NZM2410 mice [29].
Several epidemiological studies have identified an association between occupational crystalline silica exposure and development of end-stage renal disease [39,48,49], particularly glomerulonephritis [50,51]. In a previous study by our laboratory, NZBWF1 mice fed diet identical to that employed here (safflower oil-based AIN-93G) spontaneously developed proteinuria at 30 wk of age [34]. Herein, NZBWF1 dosed with 1.0 mg cSiO2 exhibited proteinuria beginning at 22 wk of age. Thus, intranasal exposure to cSiO2 likely decreased the time to onset of lupus nephritis by 8 wk. Interestingly, kidney lesions were also increased in cSiO2-treated C57Bl/6 mice, albeit to a more modest extent. The observation of renal pathology consistent with lupus nephritis in C57Bl/6 mice supports the notion of an association between cSiO2 exposure and kidney disease. Consistent with these findings, autoimmune-like responses in kidneys including IgG deposition have been previously described after intratracheal administration of fibrogenic particles in C57Bl/6 mice [52,53]. While overt proteinuria (≥ 300 mg/dl) did not develop over the 12 wk duration of our study, it might be speculated that given sufficient time, short-term repeated airway exposure to cSiO2 might induce autoimmune nephritis in C57Bl/6 and other mice without evident genetic predisposition to SLE.
Although a prior study of cSiO2-induced pulmonary inflammation in NZM2410 autoimmune mice qualitatively described inflammatory cell infiltration in the lung [29], this report is the first to semi-quantitatively grade this response. A critical finding in this study was that NZBWF1 mice exposed to cSiO2 exhibited extensive lymphoplasmacytic infiltration in peribronchial and perivasculature regions of the lungs. Similar but more modest effects were observed in C57Bl/6 mice. This observation is consistent with the findings of Singh et al. [54] who suggested that the magnitude of perivasculature and peribronchial inflammation induced by pulmonary toxicant exposure is dependent on genetic background.
A critical question relates to the relevance of the doses employed in this study to actual human exposure. Using the NIOSH limit for respirable cSiO2 established at 0.05mg/m3/d and the assumption that human ventilation rate is 6.0L/min, we calculate that humans could be exposed to 1433mg silica in 40 years of work (8h/d for 5 d/wk) with respirable cSiO2. Based on the assumption that mice ventilation rate is 0.03L/min, we estimate that an equivalent lifetime exposure to crystalline silica would be 8.28 mg in mouse. Therefore, the two cumulative doses of cSiO2 used in this study, 1 and 4 mg, represent approximately one eighth and one half, respectively, of a human lifetime exposure to cSiO2 at the recommended NIOSH exposure limit.
We opted here to employ intranasal instillation of cSiO2 based on its successful use in previous studies with NZM2410 mice [29,30,32,35]. Other routes of cSiO2 administration in murine species have been described including inhalation, intratracheal and trans-oral instillation (oropharyngeal aspiration) [55–57]. Development of silicosis in four in-bred mouse strains, including lupus-prone MRL/MpJ mice, was assessed 16 wks after inhalation exposure to 70 mg m-3 cSiO2 for 5 h for 12 consecutive days [58]. MRL/MpJ mice developed extensive pulmonary lymphocytic infiltrates, similar to those observed herein with lupus-prone NZBWF1 mice, whereas Balb/c mice given the same exposure exhibited only mild pulmonary histopathological lesions.
Although inhalation exposure of rodent species is the gold standard for modeling human airway exposure to respirable toxicants such as crystalline silica, the need for specialized inhalation exposure chambers, rigorous technical considerations and high operational costs limit these experiments to only a few laboratory facilities. A comparative study of intratracheal, trans-oral, and intranasal instillation in Balb/c mice receiving a single 1.0 mg dose of cSiO2 revealed that each method was capable of eliciting significant pulmonary inflammation 4 h after exposure [59]. Intranasal instillation resulted in comparatively less inflammation relative to both trans-oral and intratracheal instillation. This could be attributed to trapping of cSiO2 particles in the nasal cavity, resulting in delayed passage to the lower airways [59]. However, this effect is not likely to be an issue in long-term studies of intranasal exposure to cSiO2, as mice treated in this manner eventually develop pulmonary fibrosis, a hallmark of cSiO2 exposure [35,60,61]. These latter studies support the contention that the high-dose, short-term repeated intranasal exposure employed here is a suitable model for investigations of environmentally triggered onset of autoimmunity in lupus-prone mice. Nevertheless, in the future it will be of interest to compare the effects of other routes of cSiO2 exposure, especially inhalation, on development of autoimmunity in NZBWF1 mice.
An unexpected observation in this study was extensive ectopic lymphoid neogenesis in the lung after cSiO2 exposure in NZBWF1 mice. ELT morphologically and functionally resemble secondary lymphoid tissues (e.g. lymph nodes) by supporting maturation and differentiation of B cells into plasma cells that produce class-switched antibodies [62]. ELT have been previously identified in target tissues of several autoimmune diseases, including the kidneys of aged NZWBF1 mice [63], synovial joints in rheumatoid arthritis [64,65], and salivary glands in Sjögren’s syndrome [66–68], where they contribute to local manifestations of autoimmunity. Although events that drive formation of ELT remain poorly defined, neogenesis of ectopic lymphoid structures is indicative of tissue-specific chronic inflammation [69] and importantly, may influence systemic autoimmunity [70]. Taken together, cSiO2-triggered formation of pulmonary ELT likely contributed to the exacerbated systemic autoimmunity observed in NZBWF1 mice following cSiO2 exposure.
Elevated BALF concentrations of TNF-α, MCP-1, and IL-6 observed here in cSiO2-exposed NZBWF1 mice are key observations because these cytokines play critical roles in both initiating innate and amplifying adaptive immune responses. The sustained elevation of these cytokines in BALF up to 12 wk after cSiO2 exposure suggests that the initial innate immune response was insufficient to contain the inflammation, supporting a previously described model for continuous alveolar macrophage apoptosis after cSiO2 exposure with ensuing self-antigen presentation [12]. Robust elevation of TNF-α in BALF has been similarly reported in cSiO2-exposed NZM2410 mice [30]. TNF-α induces apoptosis in alveolar macrophages and therefore could contribute to increased apoptotic debris containing autoantigens [12,31]. This cytokine can also stimulate production of additional proinflammatory cytokines, which could further amplify the immune response to cSiO2 [71–74]. Released predominately by monocytes/macrophages, MCP-1 recruits monocytes from systemic circulation to local sites of inflammation after tissue injury in attempt to contain or resolve further inflammation. The heightened elevation of this chemokine in BALF 12 wks following cSiO2 exposure further supports the notion that cSiO2 induces a continuous cycle of alveolar macrophage apoptosis, as sustained recruitment of circulating monocytes to the lungs by MCP-1 is likely an innate attempt to repopulate lost alveolar macrophages and resolve inflammation. We observed robust increases in the percent of lymphocytes in BALF recovered from cSiO2-treated NZBWF1 mice. Previous studies in cSiO2 exposed NZM2410 mice have indicated that both B and T cells contribute to increased lymphocytes in BALF [30]. IL-6 has a well-established role in both CD4+ T cell survival and B-cell differentiation to Ig-secreting plasma cells [75]. Taken together, the increase of lymphocytes in the presence of IL-6 in BALF likely promotes antigen-specific effector responses in the lungs. Indeed, in the lung, cSiO2-induced elevation of IL-6 corresponded to significant increases of IgG in BALF of NZBWF1 mice.
Mechanistic studies have indicated that alveolar macrophages are key to not only cSiO2-induced pulmonary pathogenesis [12], but are also essential for development of IgG immune complex-mediated inflammation in the lungs [76,77]. Following exposure to chrysotile asbestos, in vitro production of superoxide anion by alveolar macrophages was enhanced by stimulation with IgG [78]. Proinflammatory cytokines in lungs were markedly reduced in C57Bl/6 mice deficient in FcγRIII, which mediates activation of mononuclear phagocytes by binding the Fc region of the IgG molecule [79]. The notion that IgG both initiates and enhances lung inflammatory responses through the alveolar macrophage is therefore of possible significance to cSiO2 triggering of SLE in autoimmune-prone mice. Extensive deposition/production of IgG in lungs of cSiO2-exposed NZBWF1 mice could act synergistically with defective clearance of apoptotic macrophages, further contributing to acceleration of autoimmunity.
Airway exposure to cSiO2 stimulates alveolar macrophages, epithelial cells, and fibroblasts that mediate recruitment of circulating monocytes, neutrophils, and lymphocytes by releasing an array of inflammatory mediators including cytokines [57,80–82]. The close proximity of these inflammatory cells to both the airways and vasculature in this study suggest that these cells are not only capable of mediating production of proinflammatory mediators that impact the lung, but that may also be secreted into systemic circulation, thereby exacerbating development of systemic autoimmunity. It is notable that cSiO2-induced plasma increases of TNF-α and IL-6 mirrored elevations of these cytokines in BALF. Importantly, systemic concentrations of TNF-α and IL-6 correlate with SLE disease activity in humans [83] and treatment with exogenous IL-6 exacerbates glomerulonephritis in NZBWF1 mice [84]. There was also a trend towards elevated MCP-1 in plasma of NZBWF1 mice exposed to cSiO2. Urinary MCP-1 concentration has been identified as a biomarker of disease activity in lupus nephritis [85,86], and one study indicated that renal expression of MCP-1 correlates with NF-ΚB activation in kidney [87]. Overall, these results suggest that elevated plasma proinflammatory cytokines induced after cSiO2 exposure might further contribute to production of plasma autoantibodies as well as exacerbated renal pathology. Interestingly, cytokine array analysis of cSiO2-exposed NZM2410 mice failed to reveal any significant difference in plasma cytokines IL-4, IFN-γ, IL-10, IL-12, and TNF-α [30] suggesting some inherent differences in the response to intranasal cSiO2 might exist between that strain and the NZBWF1 employed here.
To summarize, the results presented here suggest that following airway exposure to cSiO2, the lung serves as a platform for the early triggering and exacerbation of systemic autoimmunity and glomerulonephritis in the NZBWF1 mouse. This model can serve as a starting point for further studies to gain insight into toxicant-triggered autoimmunity. First, it will be essential to characterize antigen-presenting cell and lymphocyte subpopulations recruited to and migrating out of the lung after cSiO2 exposure. These cells have the potential to drive subsequent tissue-specific homing of effector cell populations that mediate pathological outcomes in the lung and kidney. Second, while it is apparent that cSiO2 induces plasma elevation of proinflammatory cytokines, further studies are warranted to ascertain if these originate from lung ELT, inflamed kidneys, and/or immune tissues such as spleen. Third, this model of cSiO2-accelerated lupus can be used to study potential approaches for prevention and intervention in occupationally exposed human populations. A particularly attractive approach is the consumption of n-3 polyunsaturated fatty acids in fish oil which have been shown to delay onset and severity of autoimmune nephritis in NZBWF1 and other models [34,88]. Finally, the strategy described herein could be used to investigate whether other airborne toxicants that cause inflammatory responses in the lung might similarly exacerbate autoimmunity.
Supporting Information
Antibodies in plasma at sacrifice were measured by ELISA. Data are mean ± SEM (n = 7–8/gp) and were analyzed by Mann-Whitney Rank Sum Test.
(TIFF)
Acknowledgments
We would like to thank Dr. Hui-Ren Zhou and Lori Bramble for their excellent technical advice and support, and Amy Porter and Kathy Joseph from the Michigan State University Histopathology Laboratory.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by National Institute of Environmental Health Sciences (ES021265) to JP. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.National Institute of Allergy and Infectious Diseases (NIAID). Autoimmune Diseases Coordinating Committee. Progress in autoimmune diseases research. NIAID Report to Congress. 2005. Available: https://www.niaid.nih.gov/topics/autoimmune/Documents/adccfinal.pdf.
- 2. Sacks JJ, Luo Y-H, Helmick CG. Prevalence of specific types of arthritis and other rheumatic conditions in the ambulatory health care system in the United States, 2001–2005. Arthritis Care Res. 2005;62: 460–464. [DOI] [PubMed] [Google Scholar]
- 3.National Institude for Occupational Safety and Health. Health effects of occupational exposure to respirable crystalline silica. 2002. Available: http://www.cdc.gov/niosh/docs/2002-129/pdfs/2002-129.pdf
- 4. Cooper GS, Parks CG. Occupational and environmental exposures as risk factors for systemic lupus erythematosus. Curr Rheumatol Rep. 2004;6: 367–374. [DOI] [PubMed] [Google Scholar]
- 5. Parks CG, Cooper GS, Nylander-French LA, Sanderson WT, Dement JM, Cohen PL, et al. Occupational exposure to crystalline silica and risk of systemic lupus erythematosus—A population-based, case-control study in the southeastern United States. Arthritis Rheum. 2002;46: 1840–1850. [DOI] [PubMed] [Google Scholar]
- 6. Finckh A, Cooper GS, Chibnik LB, Costenbader KH, Watts J, Pankey H, et al. Occupational silica and solvent exposures and risk of systemic lupus erythematosus in urban women. Arthritis Rheum. 2006;54: 3648–3654. [DOI] [PubMed] [Google Scholar]
- 7. Conrad K, Mehlhorn J, Luthke K, Dorner T, Frank K-H. Systemic lupus erythematosus after heavy exposure to quartz dust in uranium mines: clinical and serological characteristics. Lupus. 1996;5: 62–69. [DOI] [PubMed] [Google Scholar]
- 8. Rocha-Parise M, Santos LM, Damoiseaux JG, Bagatin E, Lido AV, Torello CO, et al. Lymphocyte activation in silica-exposed workers. Int J Hyg Environ Health. 2014;217: 586–591. 10.1016/j.ijheh.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 9. Zaghi G, Koga F, Nisihara RM, Skare TL, Handar A, Rosa Utiyama SR, et al. Autoantibodies in silicosis patients and in silica-exposed individuals. Rheumatol Int. 2010;30: 1071–1075. 10.1007/s00296-009-1116-z [DOI] [PubMed] [Google Scholar]
- 10. Albuquerque De Castro H, Gimenes Da Silva C, Lemle A. Immunoglobulins, complements and autoantibodies in 58 workers exposed to silica. Jornal Brasileiro de Pneumologia. 2003;30: 201–206. [Google Scholar]
- 11. Doll NJ, Stankus RP, Hughes J, Weill H, Gupta RC, Rodriguez M et al. Immune complexes and autoantibodies in silicosis. J Allergy Clin Immunol. 1981;68: 281–285. [DOI] [PubMed] [Google Scholar]
- 12. Hamilton RF Jr., Thakur SA, Holian A. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med. 2008;44: 1246–1258. 10.1016/j.freeradbiomed.2007.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamilton RF Jr., Thakur SA, Mayfair JK, Holian A. MARCO mediates silica uptake and toxicity in alveolar macrophages from C57BL/6 mice. J Biol Chem. 2006;281: 34218–34226. [DOI] [PubMed] [Google Scholar]
- 14. Holian A, Uthman MO, Goltsova T, Brown SD, Hamilton RF Jr. Asbestos and silica-induced changes in human alveolar macrophage phenotype. Environ Health Perspect. 1997;105 Suppl 5: 1139–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iyer R, Hamilton RF, Li L, Holian A. Silica-induced apoptosis mediated via scavenger receptor in human alveolar macrophages. Toxicol Appl Pharmacol. 1996;141: 84–92. [DOI] [PubMed] [Google Scholar]
- 16. Migliaccio CT, Hamilton RF Jr., Holian A. Increase in a distinct pulmonary macrophage subset possessing an antigen-presenting cell phenotype and in vitro APC activity following silica exposure. Toxicol Appl Pharmacol. 2005;205: 168–176. [DOI] [PubMed] [Google Scholar]
- 17. Thakur SA, Beamer CA, Migliaccio CT, Holian A. Critical role of MARCO in crystalline silica-induced pulmonary inflammation. Toxicol Sci. 2009;108: 462–471. 10.1093/toxsci/kfp011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen F, Sun SC, Kuh DC, Gaydos LJ, Demers LM. Essential role of NF-kappa B activation in silica-induced inflammatory mediator production in macrophages. Biochem Biophys Res Commun. 1995;214: 985–992. [DOI] [PubMed] [Google Scholar]
- 19. Di Giuseppe M, Gambelli F, Hoyle GW, Lungarella G, Studer SM, Richards T, et al. Systemic inhibition of NF-KB activation protects from silicosis. PLoS ONE. 2009;4: e5689 10.1371/journal.pone.0005689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piguet PF, Collart MA, Grau GE, Sappino AP, Vassalli P. Requirement of tumour necrosis factor for development of silica-induced pulmonary fibrosis. Nature. 1990;344: 245–247. [DOI] [PubMed] [Google Scholar]
- 21. Gabay C, Cakir N, Moral F, Roux-Lombard P, Meyer O, Dayer JM, et al. Circulating levels of tumor necrosis factor soluble receptors in systemic lupus erythematosus are significantly higher than in other rheumatic diseases and correlate with disease activity. J Rheumatol. 1997;24: 303–308. [PubMed] [Google Scholar]
- 22. Davas EM, Tsirogianni A, Kappou I, Karamitsos D, Economidou I, Dantis PC, et al. Serum IL-6, TNFalpha, p55 srTNFalpha, p75srTNFalpha, srIL-2alpha levels and disease activity in systemic lupus erythematosus. Clin Rheumatol. 1999;18: 17–22. [DOI] [PubMed] [Google Scholar]
- 23. Brown JM, Pfau JC, Pershouse MA, Holian A. Silica, apoptosis, and autoimmunity. J Immunotoxicol. 2005;1: 177–187. 10.1080/15476910490911922 [DOI] [PubMed] [Google Scholar]
- 24. Rudofsky UH, Lawrence DA. New Zealand mixed mice: a genetic systemic lupus erythematosus model for assessing environmental effects. Environ Health Perspect. 1999;107 Suppl 5: 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perry D, Sang A, Yin Y, Zheng YY, Morel L. Murine models of systemic lupus erythematosus. J Biomed Biotechnol. 2011;2011: 271694 10.1155/2011/271694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sang A, Yin Y, Zheng YY, Morel L. Animal models of molecular pathology systemic lupus erythematosus. Prog Mol Biol Transl Sci. 2012;105: 321–370. 10.1016/B978-0-12-394596-9.00010-X [DOI] [PubMed] [Google Scholar]
- 27. Mohan C, Yu Y, Morel L, Yang P, Wakeland EK. Genetic dissection of Sle pathogenesis: Sle3 on murine chromosome 7 impacts T cell activation, differentiation, and cell death. J Immunol. 1999;162: 6492–6502. [PubMed] [Google Scholar]
- 28. Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Lab Invest. 1993;68: 419–426. [PubMed] [Google Scholar]
- 29. Brown JM, Archer AI, Pfau IC, Holian A. Silica accelerated systemic autoimmune disease in lupus-prone New Zealand mixed mice. Clin Exp Immunol. 2003;131: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brown JM, Pfau JC, Holian A. Immunoglobulin and lymphocyte responses following silica exposure in New Zealand mixed mice. Inhal Toxicol. 2004;16: 133–139. [DOI] [PubMed] [Google Scholar]
- 31. Pfau JC, Brown JM, Holian A. Silica-exposed mice generate autoantibodies to apoptotic cells. Toxicology. 2004;195: 167–176. [DOI] [PubMed] [Google Scholar]
- 32. Brown JM, Schwanke CM, Pershouse MA, Pfau JC, Holian A. Effects of rottlerin on silica-exacerbated systemic autoimmune disease in New Zealand mixed mice. Am J Physiol Lung Cell Mol Physiol. 2005;289: L990–998. [DOI] [PubMed] [Google Scholar]
- 33. Berthier CC, Bethunaickan R, Gonzalez-Rivera T, Nair V, Ramanujam M, Zhang W, et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol. 2002;189: 988–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pestka JJ, Vines LL, Bates MA, He K, Langohr I. Comparative effects of n-3, n-6 and n-9 unsaturated fatty acid-rich diet consumption on lupus nephritis, autoantibody production and CD4+ T cell-related gene responses in the autoimmune NZBWF1 Mouse. PLoS ONE. 2014;9: e100255 10.1371/journal.pone.0100255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beamer CA, Migliaccio CT, Jessop F, Trapkus M, Yuan D, Holian A. Innate immune processes are sufficient for driving silicosis in mice. J Leukoc Biol. 2010;88: 547–557. 10.1189/jlb.0210108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brandenberger C, Rowley NL, Jackson-Humbles DN, Zhang Q, Bramble LA, Lewandowski RP, et al. Engineered silica nanoparticles act as adjuvants to enhance allergic airway disease in mice. Part Fibre Toxicol. 2013;10: 26 10.1186/1743-8977-10-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weening JJ, D'Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65: 521–530. [DOI] [PubMed] [Google Scholar]
- 38. Yan D, Zhou HR, Brooks KH, Pestka JJ. Role of macrophages in elevated IgA and IL-6 production by Peyer's patch cultures following acute oral vomitoxin exposure. Toxicol Appl Pharmacol. 1998;148: 261–273. [DOI] [PubMed] [Google Scholar]
- 39. Steenland K, Sanderson W, Calvert GM. Kidney disease and arthritis in a cohort study of workers exposed to silica. Epidemiology. 2001;12: 405–412. [DOI] [PubMed] [Google Scholar]
- 40. Makol A, Reilly MJ, Rosenman KD. Prevalence of connective tissue disease in silicosis (1985–2006)—a report from the state of michigan surveillance system for silicosis. Am J Ind Med. 2011;54: 255–262. 10.1002/ajim.20917 [DOI] [PubMed] [Google Scholar]
- 41. Speck-Hernandez CA, Montoya-Ortiz G. Silicon, a possible link between environmental exposure and autoimmune diseases: the case of rheumatoid arthritis. Arthritis. 2012;2012: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steenland NK, Thun MJ, Ferguson CW, Port FK. Occupational and other exposures associated with male end-stage renal disease: a case/control study. Am J Public Health. 1990;80: 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rosenman KD, Moore-Fuller M, Reilly MJ. Connective tissue disease and silicosis. Am J Ind Med. 1999;35: 375–381. [DOI] [PubMed] [Google Scholar]
- 44. Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279: 1052–1054. [DOI] [PubMed] [Google Scholar]
- 45. Gonzalez ML, Waxman FJ. Glomerular deposition of immune complexes made with IgG2a monoclonal antibodies. J Immunol. 2000;164: 1071–1077. [DOI] [PubMed] [Google Scholar]
- 46. Su W, Madaio MP. Recent advances in the pathogenesis of lupus nephritis: autoantibodies and B Cells. Semin Nephrol. 2003;23: 564–568. [DOI] [PubMed] [Google Scholar]
- 47. Nowling TK, Gilkeson GS. Mechanisms of tissue injury in lupus nephritis. Arthritis Res Ther. 2001;13: 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vupputuri S, Parks CG, Nylander-French LA, Owen-Smith A, Hogan SL, Sandler DP et al. Occupational silica exposure and chronic kidney disease. Ren Fail. 2012;34: 40–46. 10.3109/0886022X.2011.623496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Steenland K. One agent, many diseases: exposure-response data and comparative risks of different outcomes following silica exposure. Am J Ind Med. 2005;48: 16–23. [DOI] [PubMed] [Google Scholar]
- 50. Calvert GM, Steenland K, Palu S. End-stage renal disease among silica-exposed gold miners. A new method for assessing incidence among epidemiologic cohorts. J Am Med Assoc. 1997;277: 1219–1223. [PubMed] [Google Scholar]
- 51. Steenland K, Rosenman K, Socie E, Valiante D. Silicosis and end-stage renal disease. Scand J Work Environ Health. 2002;28: 439–442. [DOI] [PubMed] [Google Scholar]
- 52. Pfau JC, Sentissi JJ, Li S, Calderon-Garciduenas L, Brown JM, Blake DJ, et al. Asbestos-induced autoimmunity in C57BL/6 mice. J Immunotoxicol. 2008;5: 129–137. 10.1080/15476910802085756 [DOI] [PubMed] [Google Scholar]
- 53. Zebedeo CN, Davis C, Pena C, Ng KW, Pfau JC. Erionite induces production of autoantibodies and IL-17 in C57BL/6 mice. Toxicol Appl Pharmacol. 2014;275: 257–264. 10.1016/j.taap.2014.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Singh B, Shinagawa K, Taube C, Gelfand EW, Pabst R. Strain-specific differences in perivascular inflammation in lungs in two murine models of allergic airway inflammation. Clin Exp Immunol. 2005;141: 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gossart S, Cambon C, Orfila C, Seguelas MH, Lepert JC, Rami J, et al. Reactive oxygen intermediates as regulators of TNF-alpha production in rat lung inflammation induced by silica. J Immunol. 1996;156: 1540–1548. [PubMed] [Google Scholar]
- 56. Lakatos HF, Burgess HA, Thatcher TH, Redonnet MR, Hernady E, Williams JP, et al. Oropharyngeal aspiration of a silica suspension produces a superior model of silicosis in the mouse when compared to intratracheal instillation. Exp Lung Res. 2006;32: 181–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu F, Liu J, Weng D, Chen Y, Song L, He Q, et al. CD4+CD25+Foxp3+ regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLoS One. 2010;5: e15404 10.1371/journal.pone.0015404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davis GS, Leslie KO, Hemenway DR. Silicosis in mice: effects of dose, time, and genetic strain. J Environ Pathol Toxicol Oncol. 1998;17: 81–97. [PubMed] [Google Scholar]
- 59. Lacher SE, Johnson C, Jessop F, Holian A, Migliaccio CT. Murine pulmonary inflammation model: a comparative study of anesthesia and instillation methods. Inhal Toxicol. 2010;22: 77–83. 10.3109/08958370902929969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Beamer CA, Holian A. Scavenger receptor class A type I/II (CD204) null mice fail to develop fibrosis following silica exposure. Am J Physiol Lung Cell Mol Physiol. 2005;289: L186–195. [DOI] [PubMed] [Google Scholar]
- 61. Ferreira TP, de Arantes AC, do Nascimento CV, Olsen PC, Trentin PG, Rocco PR, et al. IL-13 immunotoxin accelerates resolution of lung pathological changes triggered by silica particles in mice. J Immunol. 2013;191: 5220–5229. 10.4049/jimmunol.1203551 [DOI] [PubMed] [Google Scholar]
- 62. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014;35: 571–580. 10.1016/j.it.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 63. Schiffer L, Kumpers P, Davalos-Misslitz AM, Haubitz M, Haller H, Anders HJ, et al. B-cell-attracting chemokine CXCL13 as a marker of disease activity and renal involvement in systemic lupus erythematosus (SLE). Nephrol Dial Transplant. 2009;24: 3708–3712. 10.1093/ndt/gfp343 [DOI] [PubMed] [Google Scholar]
- 64. Takemura S, Klimiuk PA, Braun A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. 2001;167: 4710–4718. [DOI] [PubMed] [Google Scholar]
- 65. Wengner AM, Hopken UE, Petrow PK, Hartmann S, Schurigt U, Brauer R et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56: 3271–3283. [DOI] [PubMed] [Google Scholar]
- 66. Hansen A, Lipsky PE, Dorner T. B cells in Sjogren's syndrome: indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis Res Ther. 2007;9: 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bombardieri M, Barone F, Lucchesi D, Nayar S, van den Berg WB, Proctor G, et al. Inducible tertiary lymphoid structures, autoimmunity, and exocrine dysfunction in a novel model of salivary gland inflammation in C57BL/6 mice. J Immunol. 2012;189: 3767–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stott DI, Hiepe F, Hummel M, Steinhauser G, Berek C. Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease. The salivary glands of patients with Sjogren's syndrome. J Clin Invest. 1998;102: 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grogan JL, Ouyang W. A role for Th17 cells in the regulation of tertiary lymphoid follicles. Eur J Immunol. 2012;42: 2255–2262. 10.1002/eji.201242656 [DOI] [PubMed] [Google Scholar]
- 70. Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic Lymphoid Structures Support Ongoing Production of Class-Switched Autoantibodies in Rheumatoid Synovium. Plos Medicine. 2009;6: 59–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Satriano J, Schlondorff D. Activation and attenuation of transcription factor NF-kB in mouse glomerular mesangial cells in response to Tumor Necrosis Factor-alpha, Immunoglobulin G, and adenosine 3':5'-cyclic monophosphate. J Clin Invest. 1994;94: 1629–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sanz AB, Sanchez-Nino MD, Ramos AM, Moreno JA, Santamaria B, Ruiz-Ortega M, et al. NF-kappaB in renal inflammation. J Am Soc Nephrol. 2010;21: 1254–1262. 10.1681/ASN.2010020218 [DOI] [PubMed] [Google Scholar]
- 73. Zheng L, Sinniah R, Hsu SI. Pathogenic role of NF-kappaB activation in tubulointerstitial inflammatory lesions in human lupus nephritis. J Histochem Cytochem. 2008;56: 517–529. 10.1369/jhc.7A7368.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320: 674–677. 10.1126/science.1156995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Maeda K, Mehta H, Drevets DA, Coggeshall KM. IL-6 increases B-cell IgG production in a feed-forward proinflammatory mechanism to skew hematopoiesis and elevate myeloid production. Blood. 2010;115: 4699–4706. 10.1182/blood-2009-07-230631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lentsch AB, Czermak BJ, Bless NM, Ward PA. NF-kappaB activation during IgG immune complex-induced lung injury: requirements for TNF-alpha and IL-1beta but not complement. Am J Pathol. 1998;152: 1327–1336. [PMC free article] [PubMed] [Google Scholar]
- 77. Johnson KJ, Ward PA. Acute immunologic pulmonary alveolitis. J Clin Invest. 1974;54: 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Scheule RK, Holian A. IgG specifically enhances chrysotile asbestos-stimulated superoxide anion production by the alveolar macrophage. Am J Respir Cell Mol Biol. 1989;1: 313–318. [DOI] [PubMed] [Google Scholar]
- 79. Chouchakova N, Skokowa J, Baumann U, Tschernig T, Philippens KM, Nieswandt B, et al. Fc gamma RIII-mediated production of TNF-alpha induces immune complex alveolitis independently of CXC chemokine generation. J Immunol. 2001;166: 5193–5200. [DOI] [PubMed] [Google Scholar]
- 80. Davis GS, Leslie KO, Schwarz JE, Pfeiffer LM, Hill-Eubanks L, Hemenway DR, et al. Altered patterns of lung lymphocyte accumulation in silicosis in cytokine-sufficient (C3H/HeN) and cytokine-deficient (C3H/HeJ-LPSd) mice. Chest. 1993;103: 120s–121s. [PubMed] [Google Scholar]
- 81. Davis GS, Holmes CE, Pfeiffer LM, Hemenway DR. Lymphocytes, lymphokines, and silicosis. J Environ Pathol Toxicol Oncol. 2001;20 Suppl 1: 53–65. [PubMed] [Google Scholar]
- 82. Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. 1998;157: 1666–1680. [DOI] [PubMed] [Google Scholar]
- 83. Sabry A, Sheashaa H, El-Husseini A, Mahmoud K, Eldahshan KF, George SK, et al. Proinflammatory cytokines (TNF-alpha and IL-6) in Egyptian patients with SLE: its correlation with disease activity. Cytokine. 2006;35: 148–153. [DOI] [PubMed] [Google Scholar]
- 84. Ryffel B, Car BD, Gunn H, Roman D, Hiestand P, Mihatsch MJ, et al. Interleukin 6 exacerbates glomerulonephritis in (NZBXNZW)F-1 mice. Am J Pathol. 1994;144: 927–937. [PMC free article] [PubMed] [Google Scholar]
- 85. Abujam B, Cheekatla S, Aggarwal A. Urinary CXCL-10/IP-10 and MCP-1 as markers to assess activity of lupus nephritis. Lupus. 2013;22: 614–623. 10.1177/0961203313484977 [DOI] [PubMed] [Google Scholar]
- 86. Barbado J, Martin D, Vega L, Almansa R, Goncalves L, Nocito M, et al. MCP-1 in urine as biomarker of disease activity in Systemic Lupus Erythematosus. Cytokine. 2012;60: 583–586. 10.1016/j.cyto.2012.07.009 [DOI] [PubMed] [Google Scholar]
- 87. Rovin BH, Dickerson JA, Tan LC, Hebert CA. Activation of nuclear factor-KB correlates with MCP-1 expression by human mesangial cells. Kidney Int. 1995;48: 1263–1271. [DOI] [PubMed] [Google Scholar]
- 88. Pestka JJ. n-3 Polyunsaturated fatty acids and autoimmune-mediated glomerulonephritis. Prost Leukotriene Essent Fatty Acids. 2010;82: 251–258. 10.1016/j.plefa.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Antibodies in plasma at sacrifice were measured by ELISA. Data are mean ± SEM (n = 7–8/gp) and were analyzed by Mann-Whitney Rank Sum Test.
(TIFF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.