

Abstract
The treatment of impaired wounds requires the use of biomaterials that can provide mechanical and biological queues to the surrounding environment to promote angiogenesis, granulation tissue formation, and wound closure. Porous hydrogels have previously been shown to promote angiogenesis even in the absence of pro-angiogenic factors. We hypothesized that the added delivery of non-viral DNA encoding for pro-angiogenic growth factors could further enhance this effect. Here, 100 and 60 μm porous and non-porous (n-pore) hyaluronic acid-MMP hydrogels with encapsulated reporter (pGFPluc) or pro-angiogenic (pVEGF) plasmids were used to investigate scaffold-mediated gene delivery for local gene therapy in a diabetic wound healing mouse model. Porous hydrogels allowed for significantly faster wound closure compared to n-pore hydrogels, which did not degrade and essentially provided a mechanical barrier to closure. Interestingly, the delivery of pDNA/PEI polyplexes positively promoted granulation tissue formation even when the DNA did not encode for an angiogenic protein. And although transfected cells were present throughout the granulation tissue surrounding all hydrogels at 2 weeks, pVEGF delivery did not further enhance the angiogenic response. Despite this, the presence of transfected cells shows promise for the use of polyplex-loaded porous hydrogels for local gene delivery in the treatment of diabetic wounds.
Keywords: porous hydrogel, hyaluronic acid, non-viral gene delivery, poly(ethylene imine), impaired wound healing
1. Introduction
Treatment of hard-to-heal and chronic wounds as a result of diabetes mellitus, vessel disease and insufficiency, or gentic predisposition currently poses a significant medical challenge. In the United States alone approximately 70,000 non-traumatic lower-limb amputations are performed every year as necessitated by diabetes-induced non-healing foot ulcers [1]. Unlike normal human wounds which progress through phases of inflammation, new tissue formation, and remodeling, chronic wounds maintain a state of inflammation [2, 3]. During this time there is an imbalance of proteases and reactive oxygen species which leads to degradation of essential growth factors, impaired angiogenesis, and insufficient cellular recruitement to the wound site [4, 5]. This eventually results in skin ulcer formation and often leads to wound infection [5]. In these instances external therapies (e.g. tissue grafts, biomaterial-based scaffolds, protein delivery) can be used to facilitate tissue regeneration by providing pro-angiogenic cues to the injured tissue.
Hydrogels are suitable biomaterials to serve as scaffolds for cellular ingrowth and as signal delivery vehicles as they possess relatively similar mechanical properties to natural soft tissue (e.g. full-thickness skin). The type of natural or synthetic polymer used for hydrogel preparation is a critical factor in determining cell-material interactions, mechanical properties, fluid permeability and, subsequently, promotion of angiogenesis [6]. For example, inflammation has been directly associated with the host angiogenic response with evidence which indicates a slight pro-inflammatory response may be necessary for the stimulation of angiogenesis in the implant site [7]. Thus, glycosaminoglycans, which elicit relatively mild immune responses, are suitable materials for pro-angiogenic scaffolds, unlike natural collagen gels that have the potential to induce severe inflammation [8].
Hyaluronic acid (HA), an anionic, non-sulfated glycosaminoglycan and major component of the ECM, is a highly biocompatible biomaterial which, upon degradation, is also known to promote angiogenesis [9]. In our laboratory, soft (i.e. storage modulus <300 Pa) semi-synthetic hyaluronic acid (HA) hydrogels, which are degradable by hyaluronidases as well as matrix metalloproteinases (MMPs) via MMP-degradable peptide crosslinkers, have previously been developed for culturing mouse mesenchymal stem cells in vitro [10, 11], while stiffer hydrogels (i.e. storage modulus >300 Pa) have been made for long-term implantation in vivo [12]. The straightforward tunability of HA hydrogels, along with the availability and cost-effectiveness of HA production, makes them desirable for clinical use and, more specifically, wound repair [13].
In addition to the mechanical and biologcal cues introduced by the biomaterial, the effective delivery of tissue inductive signals is essential for angiogenesis and wound repair and regeneration. Signals, such as growth factors and genes, can be incorporated into scaffolds to achieve controlled localized protein delivery or expression. However, encapsulation of proteins within a biomaterial generally decreases their activity and long-tern bioactivity, while the direct delivery of proteins results in only short-term effects as they are quickly degraded by the protease-rich wound environment [3, 4]. For example, while the topical delivery of vascular endothelial growth factor (VEGF) has resulted in 100% wound closure in rodents, a daily dose of 20μg of VEGF was required to observe the effect [14]. As a result, large amounts of modified recombinant protein are required making this a very expensive therapy.
Alternatively plasmid DNA, polyplexes, lipoplexes, and viral vectors have been encapsulated in a variety of scaffolds including fibrin [15], enzymatically-degradable polyethylene glycol (PEG) [16], enzymatically-degradable hyaluronic acid (HA) [10, 17], gelatin [18], oligo(polyethylene glycol) fumarate (OPF) [19], alginate [20], and collagen [21] hydrogels to promote wound healing and angiogenesis. Physical encapsulation of nucleic acid-carrier complexes and viral vectors in a hydrogel can shield the vector from degradation from the harsh cellular microenvironment while also providing more sustained, localized transfection as compared to a bolus delivery of growth factors or genes. Specifically for wound healing when transient treatment is desirable, non-viral gene delivery from degradable hydrogels can be used to promote angiogenesis, granulation tissue formation and wound closure [22]. Additionally, micron-sized pores can be introduced into the hydrogel to allow for more rapid cellular infiltration [23] and gene transfer owing to the increased surface area available for cells to degrade the hydrogel and release encapsulated genetic payloads [12, 24].
Here we evaluated the effect of non-viral DNA delivery from porous HA-MMP hydrogels in full-thickness dermal wounds in diabetic mice. Homozygous db/db mice model Type II diabetes mellitus and have documented impairments in wound healing due to prolonged inflammatory responses, dysfunctional matrix deposition, and altered blood flow and angiogenesis [25]. Similar to previously published murine wound healing models, a splinted wound healing model in which the splint promotes wound closure by re-epithelialization and granulation tissue formation (i.e. the major mode of healing in humans [26]) instead of wound contraction (i.e. the major mode of healing in mice [27]) was used (Figure S1). The work described here aims to understand the role of pore size and hydrogel porosity on DNA delivery and how this correlates to wound closure and granulation tissue formation. Finally, the delivery of pro-angiogenic pVEGF is evaluated in this system.
2. Results and Discussion
2.1 Experimental design and assessment of matrix components
DNA delivery from porous and n-pore HA-MMP hydrogels was assessed in a relevant mouse wound healing model. When polyplexes (and, likewise, other DNA nanoparticles or growth factors) are encapsulated into hydrogel scaffolds, their release is dictated by diffusion out of the gel and gel degradation kinetics. For large polyplexes and DNA nanoparticles (>40 nm) hydrogel mesh size generally limits diffusion and requires that the gel degrade in order for release of encapsulated nanoparticles to occur. In the subcutaneous implant model described in Tokatlian, et. al. the degree of gel degradation and DNA release was too slow to produce an enhanced angiogenic response as a result of pVEGF/PEI gene transfer into infiltrating cells [12]. In diabetic wounds, however, protease and specifically MMP expression levels are elevated with respect to normal wounds [4, 5, 28]. Rapid cellular responses and elevated protease levels were anticipated to fully degrade the implanted hydrogels upon wound closure.
Initial studies were used to assess the role of matrix components on cellular infiltration and granulation tissue formation. Control 100 μm porous hydrogels that had no polyplexes and no agarose/sucrose or no polyplexes but with agarose/sucrose were implanted to ensure that the presence of polyplexes and/or agarose/sucrose were not influencing surrounding cells and inducing an undesirable immune response (Figure 1A). While no morphological differences of the surrounding tissue and infiltrating cells were observed between the various hydrogel conditions (Figure 1A, B), quantification of granulation tissue (i.e. cross-sectional area of granulation tissue at the center of the wound) revealed significantly reduced granulation tissue formation for 100 μm porous gels containing equal amounts of agarose/sucrose but without DNA polyplexes compared to those with pDNA/PEI polyplexes (Figure 1C). Granulation tissue is newly formed connective tissue composed of fibroblasts and capillaries, and its presence in wound beds over time is indicative of wound closure and healing. Granulation tissue was notably only present in areas where the hydrogel had degraded. These results suggest that either PEI or plasmid DNA at this specific dose had an effect on cellular infiltration and gel degradation by acting as a mild inflammatory signal, supporting cellular migration. The inflammatory nature of PEI has previously been reported [29] and even exploited for use as an adjuvant in vaccine formulations [30]. Interestingly, wounds treated with hydrogels containing no agarose/sucrose also resulted in increased granulation formation, most likely as a result of their significantly lower moduli [31]. Yet while softer hydrogels can allow for faster degradation, the incorporation of agarose/sucrose is essential for encapsulation of therapeutically relevant levels of non-aggregated pDNA/PEI polyplexes.
Figure 1.
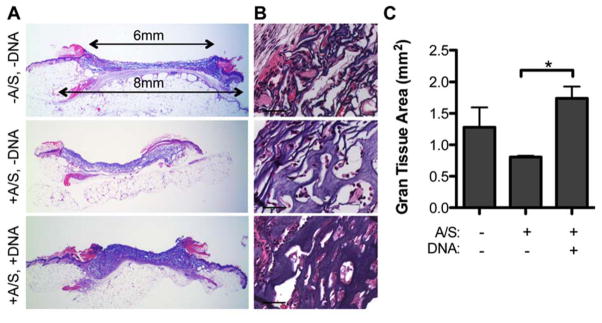
H&E stained full wound cross-sections (A, 1.6x magnification) with corresponding 40x magnification images (B, scale bar = 50 μm) of wounds with 100 μm porous hydrogels either with or without agarose/sucrose and/or pDNA/PEI polyplexes. (C) Quantification of granulation tissue (n = 3 – 4) reveals differences between granulation tissue formation surrounding porous hydrogels with various matrix components after 14 days of healing. A/S = agarose/sucrose.
2.2 Transfection from degraded hydrogels in newly formed granulation tissue
Next we aimed to study differences in granulation tissue formation and transfection as a function of pore size from hydrogels loaded with pGFPluc/PEI polyplexes. H&E staining of wound sections were used to observe morphological differences in the wound tissue. Full wound cross-sections demonstrated very clear differences between porous and n-pore hydrogels (Figure 2A). Despite the protease-rich wound environment, host cells were unable to effectively infiltrate the n-pore HA-MMP degradable hydrogels even after 14 days of continuous exposure. Many 100 and 60 μm porous hydrogels were highly infiltrated, partially or almost fully degraded, and surrounded by capillary-rich granulation tissue (Figure 2B). Quantification of granulation tissue revealed significantly less granulation tissue formation surrounding n-pore hydrogels compared to both 100 and 60 μm porous hydrogels (Figure 2D).
Figure 2.

H&E stained full wound cross-sections (A, 1.6x magnification) reveal significant differences in newly formed granulation tissue surrounding pGFPluc loaded n-pore and 60 and 100 μm porous hydrogels 2 weeks after continuous treatment. 40x magnification images show poor granulation tissue formation and cellular infiltration into n-pore hydrogels, unlike those of 60 and 100 μm porous hydrogels which show dense granulation tissue rich in small capillaries (B, scale bar = 50 μm). Immunofluorescence staining of GFP in pGFPluc loaded hydrogels confirm the presence of several transfected cells throughout the granulation tissue in each sample (C, scale bar = 50 μm; GFP positive cells = green, cell nuclei = blue, G and dotted line designate gel edge). Quantification of granulation tissue area (D) and GFP+ cells (E) reveals a correlation to hydrogel porosity (n = 4 – 5). Yet as a result of a gradient effect of transfection adjacent to the remaining hydrogel edges, n-pore hydrogel samples have an overall higher density of GFP+ cells compared to porous hydrogles after 14 days (F).
Immunofluorescence staining for GFP in pGFPluc loaded hydrogel samples was conducted to determine if released polyplexes from the degraded hydrogel portions were themselves not degraded and had transfected cells in the newly formed tissue. While there was some variability between animals, numerous transfected cells were present surrounding all 100 and 60 μm porous and n-pore hydrogels (Figure 2C, E). Quantification of total GFP+ cells per cross-sectional area indicates that pores have a positive effect on transfection, as more transfected cells were observed surrounding porous hydrogels when compared to non-porous counterparts. This trend may be a result of increased surface area contact for infiltrating cells to encounter released polyplexes. Transfection of cells within the non-degraded hydrogel pores and even in the n-pore hydrogels far from the hydrogel edges could not be detected indicating that there was minimal diffusion of incorporated polyplexes out of the hydrogel prior to degradation, mirroring the release trends observed in vitro [17]. Slow diffusion out of the hydrogel is attributed to electrostatic interactions between the highly negatively charged hyaluronic acid hydrogel and the positively charged DNA/L-PEI polyplexes. And although GFP-positive cells could be observed throughout the granulation tissue, a subtle gradient effect was present in all cross-sections, with a higher density of GFP-positive cells surrounding the degrading edges of each hydrogel. The authors hypothesize that since transfection with non-viral DNA polyplexes is transient, those cells in the granulation tissue far from the remaining hydrogel edge were most likely transfected early on and, after 14 days, only expressing the transgene at levels below detection by immunofluorescence. As a result the minimal granulation tissue surrounding n-pore hydrogels had the highest density of transfected cells compared to both 100 and 60 μm porous hydrogels (Figure 2F).
2.3 Wound closure is enhanced with porous hydrogels
Wound closure was monitored over the course of 14 days to determine if the enhanced granulation tissue formation surrounding porous hydrogels would result in more rapid wound closure. A short-term control experiment in normal balb/c mice comparing empty porous hydrogels (i.e. no DNA/PEI polyplexes and no agarose/sucrose) to no treatment further motivated the use of porous hydrogels as they modestly improved wound closure rates (Figure S2). Figure 3A shows representative live wound images of polyplex-loaded hydrogels over the course of the study. Upon visual inspection it was clear that 100 and 60 μm porous hydrogels allowed for rapid wound closure, while wounds treated with n-pore hydrogels remained almost unchanged even after 14 days. This timeframe has previously been shown to allow for 60–70 % wound closure for untreated wounds [27] and 100 % closure for wounds topically treated with 20μg of soluble VEGF delivered daily [14] in similar splinted full-thickness wound studies. Wound closure measurements showed all wounds remained relatively unchanged over the first six days (Figure 3B). However by days 8–10, porous hydrogel filled wounds started to exhibit evidence of closure and, by day 14, statistically significant differences between porous and n-pore hydrogels were apparent. Interestingly, 60 μm hydrogel wounds demonstrated a large degree of wound closure between day six and day eight. This resulted in significant differences in wound closure between the corresponding 100 μm porous hydrogel wounds (Figure 3B, C). This gap slowly started to close and by day 14 the difference in closure was not significant between the 100 and 60 μm conditions indicating that the eventual difference observed between porous and n-pore hydrogels may be a direct response to total porosity (i.e. porosity is independent of pore size; theoretical porosity for hexagonal close-packed bead template = 76 %, actual porosity = 45 – 65 %) and not specific pore size (Figure 3D). 60 μm porous hydrogels similarly allowed for faster cellular infiltration, corresponding to higher levels of early transfection and angiogenesis, in an analogous subcutaneous implant study [12]. However, by 6 weeks the differences between 60 and 100 μm porous hydrogels were once again indistinguishable and only significantly different to n-pore hydrogels.
Figure 3.

(A) Representative images of hydrogel treated wounds over the course of the study. Wound area was manually measured (n = 4 – 5) and reported as a percentage with respect to the wound area at t = 0 (B, triangles = n-pore, squares = 60 μm, circles = 100 μm porous hydrogels). Wound closure measurements of wounds with pDNA loaded hydrogels showed significant differences between porous and n-pore hydrogels. Direct comparison of wound closure at day 8 (C) and 14 (D). Times correspond to initial onset of closure and end of study, respectively.
Most wounds with n-pore hydrogels also showed signs of tissue maceration (i.e. whitening) around the wound edge. This was determined to be a result of excessive moisture retention, causing a buildup of leukocytes, albumin, macrophages, and cell debris around the inner edge of the wound. Tissue maceration can potentially prolong wound healing [32] and may, at least in part, explain the poor wound closure observed for n-pore hydrogels.
2.4 Angiogenesis within granulation tissue
Since transfected cells were mostly prevalent within the granulation tissue of all wound samples, qualitative and quantitative assessment of angiogenesis as a function of pore size and pVEGF/PEI delivery was conducted on the granulation tissue. VEGF is a known initiator of vessel branching and angiogenesis and the controlled delivery of VEGF both in vitro and in vivo has been shown to enhance angiogenic responses when compared to a single bolus delivery [33]. Vessels were present throughout the granulation tissue in all hydrogel samples containing either pVEGF or control pGFPluc polyplexes (Figure 4A, B). Numerous red blood cells could be seen filling capillaries, however, in a few areas they appeared as scattered cells most likely from bleeding and not aligned within defined, functional vessels. For both porous and n-pore hydrogels the number of vessels at the wound/hydrogel edges was higher than far below the hydrogel surface similar to the distribution of transfected cells at this time. And while transfection was observed for all hydrogel conditions (Figure 2C, E), quantitative differences in vasculature could not be attributed to the incorporation of pVEGF polyplexes (Figure 4B). Incorporation of pVEGF polyplexes also did not further enhance wound closure rates (data not shown). Both slow diffusion of polyplexes out of the hydrogels and insufficient gene expression could explain the lack of enhanced wound closure and angiogenesis. Independent of gene expression, vessel density was generally dictated by hydrogel pore size although variability among mice led to insignificant differences between most groups. Significant differences were only observed between pGFPluc loaded 60 μm porous and n-pore hydrogels. Interestingly, 60 μm porous exhibited the fastest wound closure (Figure 3B) suggesting that increased vessel density may correlate to faster wound closure.
Figure 4.

Staining for endothelial marker CD31 confirms the presence of small capillaries within the granulation tissue of 60 and 100 μm porous hydrogel implants loaded with pVEGF (A, scale bar = 50 μm; PECAM positive staining (CD31+ endothelial cells) = red, cell nuclei = blue). 100 μm porous hydrogel implants loaded with pGFPluc are shown for comparison. Quantification of vessel density (n = 4 – 5) reveals a slight correlation to hydrogel porosity (B, black bars = pVEGF, grey bars = pGFPluc loaded hydrogels). Vessel diameters were also measured (C, solid bars = pVEGF, striped bars = pGFPluc loaded hydrogels). Approximately 40–50 % of vessels in all samples were less than 6 μm in diameter.
Vessel size was also measured to determine if VEGF expression could have influenced this factor. To characterize vessel size distribution, the diameters of all vessels in images spanning across the total granulation tissue area in two separate sections were manually measured. In all samples about 40–50 % of all vessels were <6 μm in diameter, or the width of a single red blood cell (Figure 4C). Approximately 30–40 % of all vessels were characterized to be 6–12 μm in diameter. In general, the pVEGF loaded hydrogels had on average ~10 % less small capillaries (<6 μm) and subsequently an increased number of larger vessels (>12 μm) than their corresponding pGFPluc loaded hydrogels. Due to degree of variability in all samples, however, observed differences were not statistically significant and no clear conclusions could be made regarding the vessel size enhancement provided by VEGF expression. Combined these results suggest that either increased VEGF expression, a combination of burst and sustained VEGF expression, or expression of multiple growth factors (i.e. VEGF and PDGF) will be necessary to further enhance the angiogenic response already facilitated by the presence of a porous hydrogel structure.
3. Conclusion
Using a human relevant splinted mouse wound healing model, porous and n-pore HA hydrogels loaded with reporter (pGFPluc) or pro-angiogenic (pVEGF) polyplexes were tested for their ability to promote wound closure and induce an enhanced angiogenic response by transfecting infiltrating cells. 100 and 60 μm porous hydrogels allowed for significantly faster wound closure than n-pore hydrogels, which did not degrade and provided a mechanical barrier to closure. Further, pore size seemed to be the dominant factor in determining wound closure rates, with 60 μm porous hydrogels initially allowing for much faster wound closure compared to 100 μm porous hydrogels. GFP-expressing transfected cells were present throughout the granulation tissue surrounding all hydrogel samples after 14 days of continuous treatment. Transfection levels of pVEGF, however, did not seem to be high enough to enhance angiogenesis by statistically increasing vessel density or size. Yet, these results show promise for the use of polyplex loaded porous hydrogels to transfect infiltrating cells in vivo. Future wound healing studies will need to incorporate hydrogels with the ability to deliver either more DNA or deliver the polyplexes more rapidly for increased transfection and angiogenesis. Likewise, it will be important to determine the optimal hydrogel degradation rate to allow for overall faster wound closure while still allowing for sustained polyplex release and mechanical support to the wound bed.
4. Experimental Section
4.1 Materials
Peptides Ac-GCRDGPQGIWGQDRCG-NH2 (HS-MMP-SH) and Ac-GCGYGRGDSPG-NH2 (RGD) were purchased from Genscript (Piscataway, NJ). Sodium hyaluronan (HA) was a gift from Genzyme Corporation (60 kDa, Cambridge, MA). Linear poly(ethylene imine) (PEI, 25 kDa) was purchased from Polysciences (Warrington, PA). Vectors expressing mammalian GFP-firefly luciferase (pGFPluc) and human VEGF-165 (pVEGF) were obtained from New England Biolabs (Ipswich, MA) and expanded using a Giga Prep kit from Qiagen following the manufacturer’s protocol. All other chemicals were purchased from Fisher Scientific (Pittsburgh, PA) unless otherwise noted.
4.2 Hyaluronic acid modification
Sodium hyaluronan was modified to contain acrylate functionalities. Briefly, hyaluronic acid (2.00 g, 60kDa, 5.28 mmol carboxylic acids) was reacted with 36.77 g (211.07 mmol) adipic acid dihydrazide (ADH) at pH 4.75 in the presence of 4.00 g (20.84 mmol) 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) overnight and purified through dialysis (8000 MWCO) in a 100 mM to 0 mM salt gradient for 1 day followed by dialysis in DI water for 4–5 days. The purified intermediate (HA-ADH) was lyophilized and stored at −20 °C until used. Approximately 54 % of the carboxyl groups were modified with ADH, which was determined using 1H NMR (D2O) by taking the ratio of peaks at δ = 1.6 and 2.3 corresponding to the 8 hydrogens of the methylene groups on the ADH to the singlet peak of the acetyl methyl protons in HA (δ = 1.88). All of the modified HA-ADH was reacted with N-Acryloxysuccinimide (NHS-Ac) (4.46 g, 26.38 mmol) in HEPES buffer (10 mM HEPES, 150 mM NaCl, 10 mM EDTA, pH 7.2) overnight and purified through dialysis in a 100 mM to 0 mM salt gradient for 1 day followed by dialysis in DI water for 3–4 days before lyophilization. The degree of acrylation was determined to be ~12 % using 1H-NMR (D2O) by taking the ratio of the multiplet peak at δ = 6.2 corresponding to the cis and trans acrylate hydrogens to the singlet peak of the acetyl methyl protons in HA (δ = 1.88).
4.3 Polyplex lyophilization
Similar to previously described methods for caged nanoparticle encapsulation (CnE) [12, 31], plasmid DNA (250 μg) and L-PEI (228.3 μg, N/P = 7) were mixed in 3.5 mL water with 35 mg (0.10 mmol) of sucrose (Ultra pure, MP Biomedicals, Santa Ana, CA) and incubated at room temperature for 15 min. Low-melting point agarose (1.0 mg, UltraPureTM Agarose, Tm = 34.5–37.5 °C, Invitrogen, Grand Islands, NY) in 1.5 mL water was added before lyophilization. Each aliquot was intended for a 100 μL hydrogel. For smaller hydrogel volumes, both sucrose and agarose were scaled down proportionally.
4.4 Design template using PMMA microspheres
Microsphere templates for porous hydrogels were prepared using dry PMMA microspheres (53–63 and 90–106 μm, Cospheric, Santa Barbara, CA). Approximately 20 mg of microspheres (1.19 mg/μL) were mixed with DI water for a final concentration of 20 mg per 100 μL. Then 100 μL of the microsphere solution was pipetted into each well in a glass-bottom silicon well mold (wells = 6 mm × 2 mm, D x H). The microspheres were then allowed to dry and pack (by naturally settling) over 3–4 h at 37 °C. The glass-bottom silicon wells were then placed into an oven and the microspheres were sintered for 22 h at 150 °C.
4.5 Porous (and n-porous) HA hydrogel formation
Hydrogels were formed by Michael-type addition of acrylate-functionalized HA (HA-Ac) with bis-cysteine containing MMP peptide crosslinkers at pH 8.0–8.2. Prior to reaction, a hydrogel precursor solution was made by mixing a fraction of the total HA-Ac with a lyophilized aliquot of cell adhesion peptide, RGD, in .3 M TEOA pH 8.2 for 30 min at 37 °C. After incubation, HA-RGD was mixed with the remaining HA-Ac and .3 M TEOA pH 8.2 for a final gel concentration of 3.0 w/v% HA and 100 μM RGD. Finally lyophilized aliquots of the crosslinker (HS-MMP-SH) were diluted in .3 M TEOA pH 8.2 immediately before addition to a mixture of lyophilized (CnE) DNA/PEI polyplexes and the hydrogel precursor solution for a final r ratio (SH/Ac) = 0.4. For porous hydrogels, 20 μL of gel solution was then added directly on top of a PMMA microsphere template, covered with a glass slide, and perfused into the template by centrifugation at 1500 rpm for 6 min at 4 °C. The slide was then incubated at 37 °C for 30–45 min to induce polymerization. Once complete, the gels were removed from the silicon wells and placed directly into 100 % acetone for 48 h to dissolve the PMMA microsphere template. The acetone solution was replaced 2–3x during this incubation. The gels were then serially hydrated into sterile PBS and left in PBS until ready for use. For n-pore hydrogels, the gel solution was sandwiched between two Sigmacoted slides using 1 mm thick plastic spacers and incubated at 37 °C for 30–45 min to induce polymerization. Once complete, the gels were placed directly into sterile PBS and left in PBS until ready for use. Prior to surgery all gels were placed in sterile PBS with 1 % P/S overnight.
4.6 Splinted wound healing model
All in vivo studies were conducted in compliance with the NIH Guide for Care and Use of Laboratory Animals and UCLA ARC standards. 10 to 12-week old female db/db mice each 30–40 grams were used to study cellular infiltration and blood vessel formation in HA hydrogels since this strain and size has been previously used for wound healing and angiogenesis assays [14, 27, 34]. Only for control experiments were 8 to 10-week old female balb/c mice each 20–30 grams used. Porous or n-pore hydrogels were made exactly as described above and cut to 6 mm in diameter using a biopsy punch, for final overall dimensions of 6 mm × 1 mm, D x H. In fabricating the hydrogels, the starting reagents were sterilized through filtering with a 0.22 μm filter. After scaffold fabrication, the hydrogels were washed with sterile PBS and kept in PBS with 1 % P/S. Immediately prior to surgery, mice were anesthetized with 4–5 % isoflurane through a nose cone inhaler. After anesthesia induction, the isoflurane concentration was lowered to 1.5–2.5 % for the remainder of the surgery. The back of the mouse was subsequently shaved, all remaining hair was removed with Nair (1 min), and finally sterilized with Betadine and 70 % ethanol. Two full-thickness wounds were then be generated using a 6 mm biopsy punch (4 mm for wounds on smaller balb/c mice) and the hydrogels were placed directly into the wounds. Sterilized 8 mm (D) silicon rings (0.5 mm thick) sandwiched between two sterile pieces of Tegaderm (i.e. splints with non-stick, clear windows) were fixed to the outside of the wound using a tissue adhesive, Mastisol. The splints were then lightly pressed down to come into contact with the hydrogel. Eight (5–0) interrupted sutures were also utilized to hold the splint in place. Finally, additional adhesive was placed around the outer edges of the splints and used to secure strips of Tegaderm to cover the splint edges and sutures, preventing the mice from removing the splints during the study (see Figure S1 for representative images of animals immediately following surgery). All animals were observed daily for signs of inflammation and pain and also administered Buprenorphine injections every 12 h for the first 48 h post survival surgery. At the end of each study (14 days), animals were sacrificed with isoflurane overdose. Two 8 mm diameter pieces of tissue were collected from each mouse containing the implant and the surrounding tissue and skin using a biopsy punch, fixed in 2 % PFA overnight at 4 °C, dehydrated in 70 % ethanol, and finally paraffin embedded.
4.7 Immunofluorescence and immunohistochemistry
Paraffin embedded sections (5 μm) were deparaffinized by incubation in multiple xylene washes followed by serial hydration from 100 % ethanol into 100 % water. For CD31 staining, antigen-retrieval was conducted with 15 min incubation at 37 °C in 0.1 mg/mL proteinase K solution. Sections were then washed with PBS and incubated in blocking buffer (1 % goat serum (Invitrogen, Grand Islands, NY) + 0.05 % Tween-20 in PBS) for 1 h at RT before being incubated in primary antibody solution (1:100 dilution in blocking buffer of rat anti-mouse CD31 (BD Pharmingen, San Diego, CA)) overnight at 4 °C. Sections were again washed with PBS and incubated in blocking buffer for 10 min at RT before being incubated for 2 h at RT in secondary antibody solution (1:100 dilution in blocking buffer of goat anti-rat Alexa 568 (Invitrogen, Grand Islands, NY) which also contained DAPI nuclear stain (1:400 dilution, Invitrogen). Sections were then washed twice in PBS, mounted and imaged using an inverted Zeiss fluorescence microscope. For GFP staining, neither protease nor heat-mediated antigen-retrieval was conducted as these were found to eliminate any GFP signal. All other procedures were conducted exactly as described above with a 1:50 dilution of the primary GFP antibody (Invitrogen, Grand Islands, PA) and 1:100 and 1:400 dilutions of the goat anti-rabbit Alexa 488 secondary antibody (Invitrogen, Grand Islands, PA) and DAPI, respectively. All hematoxylin and eosin staining of sections was conducted by the Translational Pathology Core Laboratory (TPCL) at UCLA. Granulation tissue area was manually measured from two separate full-thickness cross-sections at least 50 – 100 μm apart from each mouse (n = 4 – 5) using ImageJ software.
4.8 GFP characterization and statistical analysis
Two separate sections at least 50 – 100 μm apart were analyzed for each sample. Twenty randomly chosen areas were imaged from each section. GFP positive cells were counted manually in each section, totaled from all sections, and finally normalized to the total imaged area for each mouse (n = 4 – 5).
All statistical analysis was performed using Prism (GraphPad, San Diego, CA). Experiments were statistically analyzed using a one-way ANOVA followed by a Tukey test to compare all pairs of columns using a 95 % confidence interval. All error bars represent the standard error of the mean (SEM).
4.9 Live imaging and wound closure analysis
Every two days, each mouse (n = 4 – 5) was anesthetized (as described above) and both wounds were imaged using a Zeiss inverted stereomicroscope. Wound area was manually measured using ImageJ software. The fixed splint area was used to calibrate measurements in each image.
4.10 Granulation vessel quantification, characterization, and statistical analysis
Two separate sections at least 50 – 100 μm apart were analyzed for each sample. Five to ten randomly chosen areas within the newly formed granulation tissue were imaged in each section. Vessels were counted manually in each section, totaled from all sections, and finally normalized to the total imaged area for each mouse (n = 4 – 5). For all samples the diameter of each vessel was manually measured using Zen imaging and analysis software (Zeiss). Percentages were determined using the entire vessel set.
All statistical analysis was performed using Prism (GraphPad, San Diego, CA). Experiments were statistically analyzed using a one-way ANOVA followed by a Tukey test to compare all pairs of columns using a 95 % confidence interval. All errors bars represent the standard error of the mean (SEM).
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Joanne Zahorsky-Reeves and Dr. Joanne Sohn for their continuous help with the animal models, as well as Dr. Gregory Lawson and Dr. Lloyd Miller for technical support with histology and Dr. David Gjertson for assistance with statistical analysis. T. Tokatlian would especially like to thank Suwei Zhu, Shayne Siegman, Shiva Gojgini, Jonathan Lam and Dr. Lina Nih for their assistance at one point or another with animal experiments and Dr. Antoni Torres Collado for his advice and helpful discussions. The authors acknowledge NIH R01 HL110592 (TS), T32 GM067555NSF (TT) and NSF (CAREER 0747539).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Center for Disease Control and Prevention. Atlanta, GA: US Department of Health and Human Services; 2014. [Google Scholar]
- 2.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Nature. 2008;453:314. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 3.McCarty SM, Percival SL. Advances in Wound Care. 2013;2:438. doi: 10.1089/wound.2012.0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung AW, Hsiang YN, Matzke LA, McManus BM, van Breemen C, Okon EB. Circulation Research. 2006;99:140. doi: 10.1161/01.RES.0000232352.90786.fa. [DOI] [PubMed] [Google Scholar]
- 5.Blakytny R, Jude E. Diabetic Medicine. 2006;23:594. doi: 10.1111/j.1464-5491.2006.01773.x. [DOI] [PubMed] [Google Scholar]
- 6.a) Andreadis ST, Geer DJ. Trends in Biotechnology. 2006;24:331. doi: 10.1016/j.tibtech.2006.05.001. [DOI] [PubMed] [Google Scholar]; b) Bramfeldt H, Sabra G, Centis V, Vermette P. Current Medicinal Chemistry. 2010;17:3944. doi: 10.2174/092986710793205327. [DOI] [PubMed] [Google Scholar]; c) Fischbach C, Mooney DJ. Biomaterials. 2007;28:2069. doi: 10.1016/j.biomaterials.2006.12.029. [DOI] [PubMed] [Google Scholar]; d) Iyer P, Walker KJ, Madihally SV. Biotechnology and Bioengineering. 2011;109:1314. doi: 10.1002/bit.24396. [DOI] [PubMed] [Google Scholar]
- 7.Laschke MW, Menger MD. European Surgical Research. 2012;48:85. doi: 10.1159/000336876. [DOI] [PubMed] [Google Scholar]
- 8.a) Friedlaender GE, Perry CR, Cole JD, Cook SD, Cierny G, Muschler GF, Zych GA, Calhoun JH, LaForte AJ, Yin S. The Journal of Bone & Joint Surgery. 2001;83:151. [PMC free article] [PubMed] [Google Scholar]; b) Rucker M, Laschke MW, Junker D, Carvalho C, Schramm A, Mulhaupt R, Gellrich NC, Menger MD. Biomaterials. 2006;27:5027. doi: 10.1016/j.biomaterials.2006.05.033. [DOI] [PubMed] [Google Scholar]; c) Gilmartin DJ, Alexaline MM, Thrasivoulou C, Phillips AR, Jayasinghe SN, Becker DL. Advanced Healthcare Materials. 2013;2:1151. doi: 10.1002/adhm.201200357. [DOI] [PubMed] [Google Scholar]
- 9.a) Fraser JR, Laurent TC, Laurent UB. Journal of Internal Medicine. 1997;242:27. doi: 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]; b) Gaffney J, Matou-Nasri S, Grau-Olivares M, Slevin M. Molecular BioSystems. 2010;6:437. doi: 10.1039/b910552m. [DOI] [PubMed] [Google Scholar]; c) Eldridge L, Moldobaeva A, Wagner EM. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2011;301:L782. doi: 10.1152/ajplung.00079.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gao F, Liu Y, He Y, Yang C, Wang Y, Shi X, Wei G. Matrix Biology. 2010;29:107. doi: 10.1016/j.matbio.2009.11.002. [DOI] [PubMed] [Google Scholar]; e) Voelcker V, Gebhardt C, Averbeck M, Saalbach A, Wolf V, Weih F, Sleeman J, Anderegg U, Simon J. Experimental Dermatology. 2008;17:100. doi: 10.1111/j.1600-0625.2007.00638.x. [DOI] [PubMed] [Google Scholar]; f) Price RD, Myers S, Leigh IM, Navsaria HA. American Journal of Clinical Dermatology. 2005;6:393. doi: 10.2165/00128071-200506060-00006. [DOI] [PubMed] [Google Scholar]
- 10.Lei Y, Gojgini S, Lam J, Segura T. Biomaterials. 2011;32:39. doi: 10.1016/j.biomaterials.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gojgini S, Tokatlian T, Segura T. Molecular Pharmaceutics. 2011;8:1582. doi: 10.1021/mp200171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tokatlian T, Cam C, Segura T. Biomaterials. 2014;35:825. doi: 10.1016/j.biomaterials.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prestwich GD. Advances in Wound Care. 2010;1:394. [Google Scholar]
- 14.Galiano RD, Tepper OM, Pelo CR, Bhatt KA, Callaghan M, Bastidas N, Bunting S, Steinmetz HG, Gurtner GC. The American Journal of Pathology. 2004;164:1935. doi: 10.1016/S0002-9440(10)63754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) des Rieux A, Shikanov A, Shea LD. Journal of Controlled Release. 2009;136:148. doi: 10.1016/j.jconrel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kidd ME, Shin S, Shea LD. Journal of Controlled Release. 2012;157:80. doi: 10.1016/j.jconrel.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lei P, Padmashali RM, Andreadis ST. Biomaterials. 2009;30:3790. doi: 10.1016/j.biomaterials.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Lei Y, Ng QKT, Segura T. Microscopy Research and Technique. 2010;73:910. doi: 10.1002/jemt.20840. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lei Y, Segura T. Biomaterials. 2009;30:254. doi: 10.1016/j.biomaterials.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shepard JA, Huang A, Shikanov A, Shea LD. Journal of Controlled Release. 2010;146:128. doi: 10.1016/j.jconrel.2010.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tokatlian T, Cam C, Siegman SN, Lei Y, Segura T. Acta Biomaterialia. 2012;8:3921. doi: 10.1016/j.actbio.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Kasahara H, Tanaka E, Fukuyama N, Sato E, Sakamoto H, Tabata Y, Ando K, Iseki H, Shinozaki Y, Kimura K. Journal of the American College of Cardiology. 2003;41:1056. doi: 10.1016/s0735-1097(02)03007-3. [DOI] [PubMed] [Google Scholar]; b) Kushibiki T, Tomoshige R, Fukunaka Y, Kakemi M, Tabata Y. Journal of Controlled Release. 2003;90:207. doi: 10.1016/s0168-3659(03)00197-4. [DOI] [PubMed] [Google Scholar]
- 19.a) Dadsetan M, Szatkowski J, Shogren K, Yaszemski M, Maran A. Journal of Biomedical Materials Research Part A. 2009;91:1170. doi: 10.1002/jbm.a.32291. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kasper FK, Jerkins E, Tanahashi K, Barry MA, Tabata Y, Mikos AG. Journal of Biomedical Materials Research Part A. 2006;78A:823. doi: 10.1002/jbm.a.30736. [DOI] [PubMed] [Google Scholar]
- 20.a) Kong HJ, Kim ES, Huang YC, Mooney DJ. Pharmaceutical Research. 2008;25:1230. doi: 10.1007/s11095-007-9526-7. [DOI] [PubMed] [Google Scholar]; b) Neumann AJ, Schroeder J, Alini M, Archer CW, Stoddart MJ. Molecular Biotechnology. 2013;53:207. doi: 10.1007/s12033-012-9522-y. [DOI] [PubMed] [Google Scholar]; c) Wegman F, Bijenhof A, Schuijff L, Oner F, Dhert W, Alblas J. European Cells and Materials Journal. 2011;21:230. doi: 10.22203/ecm.v021a18. [DOI] [PubMed] [Google Scholar]
- 21.a) Bonadio J, Smiley E, Patil P, Goldstein S. Nature Medicine. 1999;5:753. doi: 10.1038/10473. [DOI] [PubMed] [Google Scholar]; b) Capito R, Spector M. Gene Therapy. 2007;14:721. doi: 10.1038/sj.gt.3302918. [DOI] [PubMed] [Google Scholar]; c) Scherer F, Schillinger U, Putz U, Stemberger A, Plank C. The Journal of Gene Medicine. 2002;4:634. doi: 10.1002/jgm.298. [DOI] [PubMed] [Google Scholar]
- 22.a) Cam C, Segura T. Current Opinion in Biotechnology. 2013;24:855. doi: 10.1016/j.copbio.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rimann M, Hall H. Gene Therapy and Molecular Biology. 2009:13. [Google Scholar]; c) Layliev J, Wilson S, Warren SM, Saadeh PB. Advances in Wound Care. 2012;1:218. doi: 10.1089/wound.2011.0322. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nelson CE, Gupta MK, Adolph EJ, Guelcher SA, Duvall CL. Advances in Wound Care. 2013;2:93. doi: 10.1089/wound.2011.0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiu YC, Cheng MH, Engel H, Kao SW, Larson JC, Gupta S, Brey EM. Biomaterials. 2011;32:6045. doi: 10.1016/j.biomaterials.2011.04.066. [DOI] [PubMed] [Google Scholar]
- 24.a) Jang JH, Rives CB, Shea LD. Molecular Therapy. 2005;12:475. doi: 10.1016/j.ymthe.2005.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang YC, Riddle K, Rice KG, Mooney DJ. Human Gene Therapy. 2005;16:609. doi: 10.1089/hum.2005.16.609. [DOI] [PubMed] [Google Scholar]; c) Saul JM, Linnes MP, Ratner BD, Giachelli CM, Pun SH. Biomaterials. 2007;28:4705. doi: 10.1016/j.biomaterials.2007.07.026. [DOI] [PubMed] [Google Scholar]; d) Shepard JA, Virani FR, Goodman AG, Gossett TD, Shin S, Shea LD. Biomaterials. 2012;33:7412. doi: 10.1016/j.biomaterials.2012.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultz GS, Wysocki A. Wound Repair and Regeneration. 2009;17:153. doi: 10.1111/j.1524-475X.2009.00466.x. [DOI] [PubMed] [Google Scholar]
- 26.Brown NJ, Smyth EAE, Cross SS, Reed MWR. Wound Repair and Regeneration. 2002;10:245. doi: 10.1046/j.1524-475x.2002.10408.x. [DOI] [PubMed] [Google Scholar]
- 27.Galiano RD, Michaels J, Dobryansky M, Levine JP, Gurtner GC. Wound Repair and Regeneration. 2004;12:8. doi: 10.1111/j.1067-1927.2004.12404.x. [DOI] [PubMed] [Google Scholar]
- 28.a) Galkowska H, Olszewsk WL, Wojewodzka U, Mijal J, Filipiuk E. Surgery. 2003;134:213. doi: 10.1067/msy.2003.223. [DOI] [PubMed] [Google Scholar]; b) Lobmann R, Ambrosch A, Schultz G, Waldmann K, Schiweck S, Lehnert H. Diabetologia. 2002;45:1011. doi: 10.1007/s00125-002-0868-8. [DOI] [PubMed] [Google Scholar]; c) Silva JAF, Lorencini M, Peroni LA, Hoz CLRDL, Carvalho HF, Stach-Machado DR. Journal of Periodontal Research. 2008;43:48. doi: 10.1111/j.1600-0765.2007.00993.x. [DOI] [PubMed] [Google Scholar]
- 29.a) Godbey WT, Wu KK, Mikos AG. Journal of Controlled Release. 1999;60:149. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]; b) Beyerle A, Braun A, Banerjee A, Ercal N, Eickelberg O, Kissel TH, Stoeger T. Biomaterials. 2011;32:8694. doi: 10.1016/j.biomaterials.2011.07.082. [DOI] [PubMed] [Google Scholar]
- 30.Wegmann F, Gartlan KH, Harandi AM, Brinckmann SA, Coccia M, Hillson WR, Kok WL, Cole S, Ho LP, Lambe T, Puthia M, Svanborg C, Scherer EM, Krashias G, Williams A, Blattman JN, Greenberg PD, Flavell RA, Moghaddam AE, Sheppard NC, Sattentau QJ. Nature Biotechnology. 2012;30:883. doi: 10.1038/nbt.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lei Y, Huang S, Sharif-Kashani P, Chen Y, Kavehpour P, Segura T. Biomaterials. 2010;31:9106. doi: 10.1016/j.biomaterials.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hilton JR, Williams DT, Beuker B, Miller DR, Harding KG. Clinical Infectious Diseases. 2004;39:S100. doi: 10.1086/383270. [DOI] [PubMed] [Google Scholar]
- 33.a) Carmeliet P, Jain RK. Nature. 2011;473:298. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ehrbar M, Zeisberger SM, Raeber GP, Hubbell JA, Schnell C, Zisch AH. Biomaterials. 2008;29:1720. doi: 10.1016/j.biomaterials.2007.12.002. [DOI] [PubMed] [Google Scholar]; c) Moon JJ, Saik JE, Poché RA, Leslie-Barbick JE, Lee SH, Smith AA, Dickinson ME, West JL. Biomaterials. 2010;31:3840. doi: 10.1016/j.biomaterials.2010.01.104. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Silva EA, Mooney DJ. Journal of Thrombosis and Haemostasis. 2007;5:590. doi: 10.1111/j.1538-7836.2007.02386.x. [DOI] [PubMed] [Google Scholar]; e) Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmokel H, Bezuidenhout D, Djonov V, Zilla P, Hubbell JA. Faseb Journal. 2003;17:2260. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- 34.Martino MlM, Tortelli F, Mochizuki M, Traub S, Ben-David D, Kuhn GA, Muller R, Livne E, Eming SA, Hubbell JA. Science Translational Medicine. 2011;3:100ra89. doi: 10.1126/scitranslmed.3002614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.