

Abstract
Mitogen-Activated Protein Kinase (MAPK) cascades play central roles in innate immune signaling networks in plants and animals1,2. In plants, however, the molecular mechanisms of how signal perception is transduced to MAPK activation remain elusive1. We report that pathogen-secreted proteases activate a previously unknown signaling pathway in Arabidopsis thaliana involving the Gα, Gβ and Gγ subunits of heterotrimeric G-protein complexes, which function upstream of a MAPK cascade. In this pathway, Receptor for Activated C Kinase 1 (RACK1) functions as a novel scaffold that binds to the Gβ subunit as well as to all three tiers of the MAPK cascade, thereby linking upstream G protein signaling to downstream activation of a MAPK cascade. The protease-G protein-RACK1-MAPK cascade modules identified in these studies are distinct from previously described plant immune signaling pathways such as the one elicited by bacterial flagellin, in which G proteins function downstream of or in parallel to a MAPK cascade without the involvement of the RACK1 scaffolding protein. The discovery of the novel protease-mediated immune signaling pathway described here was facilitated by the use of the broad host range, opportunistic bacterial pathogen Pseudomonas aeruginosa. The ability of P. aeruginosa to infect both plants and animals makes it an excellent model to identify novel types of immunoregulatory strategies that account for its niche adaptation to diverse host tissues and immune systems.
We found that culture filtrate of P. aeruginosa strain PA14 activates an Arabidopsis β-glucuronidase (GUS) reporter gene under the control of the pathogen-inducible CYP71A12 promoter (CYP71A12pro:GUS). Whereas the well-characterized immune elicitor flg22, a synthetic peptide that corresponds to the active epitope of bacterial flagellin, induces CYP71A12pro:GUS in the root elongation zone3, PA14 culture filtrate activates the reporter in the cotyledons and leaves of both wild-type Arabidopsis Col-0 and fls2 mutant seedlings in which the flagellin receptor is mutated (Fig. 1a).
Figure 1. Proteases trigger innate immune responses in Arabidopsis via proteolytic activity.
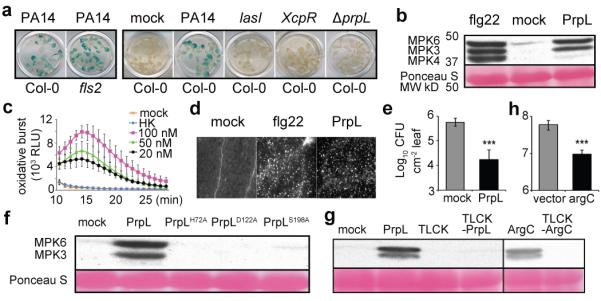
a, Activation of CYP71A12pro:GUS in wild-type Arabidopsis Col-0 or fls2 mutant cotyledons by culture filtrates from wild-type P. aeruginosa PA14, from PA14 mutants containing transposon insertion in lasI and xcpR, or from PA14/ΔprpL. b, Western blot depicting activation of MAPKs by PrpL or flg22. Numbers left of blot represent marker size in kDa.c, Chemiluminescence assay showing elicitation of an oxidative burst by PrpL. RLU: relative luminescence units. HK: 100 nM “heat-killed” PrpL.d, Callose formation in cotyledons elicited by PrpL or flg22 detected by aniline blue staining. e, Protection of 4-week old Arabidopsis leaves from P. syringae pv. tomato strain DC3000 infection by pre-infiltrated PrpL. f, Western blot depicting activation of MPK3 and MPK6 by PrpL and inactive variants of PrpL. The same molecular weight region of the blot is shown as in b. g, Western blot depicting activation of MPK3 and MPK6 by PrpL or ArgC or TLCK-treated PrpL or TLCK-treated ArgC. The same molecular weight region of the blot is shown as in b. h, Growth of X. campestris strains 8004/argC or 8004/vector in 3-week old Brassica oleracea (broccoli) leaves. The data represent the mean ± s.d., n = 16 individual seedlings (c), and n = 10 leaves from 5 plants (e, h); ***P < 0.001, Student’s t-test. The experiments in panels a and d were repeated three times with similar results and the representative images shown were selected from at least three images.
By screening a collection of 64 P. aeruginosa PA14 regulatory and secretion-related mutants, we found that the induction of the CYP71A12 promoter was dependent on the quorum-sensing gene lasI and on the Type II secretion apparatus-encoding genes XcpR, XcpT, XcpW, and XcpZ (Fig. 1a; Extended Data Table 1). Ion exchange chromatography fractionation (Extended Data Fig. 1a) followed by mass spectrometry (data not shown) identified the elicitor in the PA14 secretome as protease IV, a Type II-secreted, PvdS-regulated lysyl class serine protease encoded by the P. aeruginosa prpL gene (PA14_09900). Purified His-tagged PA14 protease IV (referred to as PrpL in the Figure Legends) activated CYP71A12pro:GUS (Extended Data Fig. 1b), whereas culture filtrate from an in-frame deletion mutant of prpL (PA14/ΔprpL) did not (Fig. 1a).
Purified protease IV is a very strong elicitor of immune responses in Arabidopsis, comparable to flg22 in the activation of MPK3 and MPK6 (but not MPK4) (Fig. 1b), elicitation of an oxidative burst (Fig. 1c), deposition of callose in cotyledons (Fig. 1d), and protection of adult Arabidopsis leaves from P. syringae pv. tomato strain DC3000 infection (Fig. 1e). In contrast, trypsin, a well-characterized serine protease, failed to activate MAPK cascades or trigger an oxidative burst (Extended Data Fig. 2a, b). Global transcriptional profiling analysis (Extended Data Fig. 3a), confirmed by RT-qPCR analysis of selected defense-related genes (Extended Data Fig. 3b), showed a high degree of concordance between the genes activated or repressed by protease IV and genes previously shown to be regulated by flg22 or oligogalacturonides (OGs) in seedlings4 (Pearson correlation coefficients of 0.899 and 0.864 for protease IV-treated vs. flg22 and OGs, respectively).
Importantly, protease IV variants containing alanine substitutions at the proteolytic catalytic triad site (PrpLH72A, PrpLD122A, PrpLS198A), which exhibit no detectable proteolytic activity5, were impaired for MAPK activation (Fig. 1f), defense gene induction, and oxidative burst elicitation (Extended Data Fig. 4a, d). Treatment of protease IV with the protease inhibitor TLCK (Fig. 1g, Extended Data Fig. 4b, d) or with heat (Fig. 1c, Extended Data Fig. 4c) also resulted in a loss of elicitation ability.
The closest homolog of P. aeruginosa protease IV in sequenced bacterial genomes is encoded by the argC gene of Xanthomonas campestris, a bona fide plant pathogen (Extended Data Fig. 5a). Purified His-tagged ArgC protease exhibited protease activity in vitro and triggered the activation of MPK3 and MPK6 that is dependent on ArgC protease activity (Fig. 1g).
We noticed that there is a high rate of naturally-occurring null mutations in the Xanthomonas argC gene (8 out of 22 total alleles in sequenced Xanthomonas genomes; Extended Data Fig. 5b to d), suggesting that argC is likely under negative selection. Consistent with the sequence data, the culture filtrate of strain X. campestris pv. raphani strain 1946, from which the functional argC gene was cloned, activated the CYP71A12pro:GUS reporter, whereas culture filtrates from two X. campestris pv. campestris strains (8004 and BP109), which contain presumptive argC null frame shift mutations, failed to activate (Extended Data Fig. 5e). We complemented the null argC mutant in strain 8004 (Xcc 8004) with the functional argC gene from strain 1946 (8004/argC) (Extended Data Fig. 5e). Consistent with ArgC-mediated induction of a host immune response during an infection in a mature plant, the growth of 8004/argC in Brassica oleracea (broccoli), a natural host of X. campestris, was reduced about 6 fold compared to the 8004/vector control (Fig. 1h). The expression of HA-tagged ArgC was readily detected in broccoli leaves infected with 8004/argC (Extended Data Fig. 5e), indicating that ArgC is synthesized during infection.
Next, we sought to investigate the mechanism by which protease IV activates an immune response in Arabidopsis. Previous studies have shown that G proteins play a role in microbe-associated molecular pattern molecules (MAMPs)-mediated responses6. In the case of protease IV, we found reduced expression of defense-related genes in gα or gβ mutants (and in a gγ1gγ2 double mutant), reduced levels of the oxidative burst in a gα mutant and a gαβ double mutant, reduced MPK3 and MPK6 activation, and reduced protection against P. syringae infection in a gαβ double mutant (Fig. 2a to c; Extended Data Fig. 6a, b). The induction of CYP71A12 and activation of MPK3 and MPK6 by X. campestris ArgC was also diminished in the G protein mutants, similar to the pattern observed for protease IV (Fig. 2a and b). In contrast to protease IV and ArgC, in the case of flg22, defense gene expression was only reduced in gβ and gαβ double mutants, the oxidative burst was more severely affected in a gβ mutant than in a gα mutant, protection against P. syringae was only modestly affected in a gαβ double mutant, and the activation of MAPKs was not affected in any of the G protein mutants (Fig. 2a to c, Extended Data Fig 6b). These data show that G protein signaling is required to activate downstream MAPKs in response to protease IV and ArgC, but not flg22 (Fig. 2a), and that G proteins play different roles in canonical MAMP and protease-mediated signaling pathways.
Figure 2. Protease-mediated defense responses are coupled to G protein signaling.
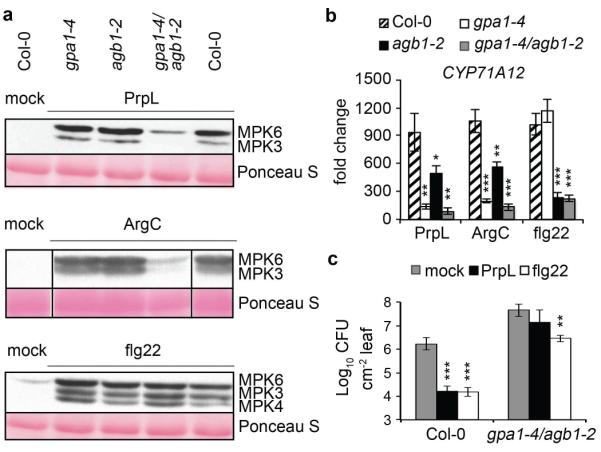
a, Western blot depicting activation of MAPKs by PrpL, ArgC or flg22 in wild-type Col-0 or G protein T-DNA mutants. The same molecular weight region of the blot is shown as in Fig. 1b. b, Induction of defense-related gene expression by PrpL, ArgC or flg22 in wild-type Col-0 or G protein T-DNA mutants measured by RT-qPCR. c, Protection of 4-week old wild-type Col-0 or gαβ double mutant leaves from P. syringae pv. tomato strain DC3000 infection by pre-infiltrated PrpL or flg22. gpa1-4 is a gα mutant; agb1-2 is a gβ mutant; and gpa1-4/agb1-2 is a gαβ double mutant. The data represent the mean ± s.d., n = 3 biological replicates with each experiment containing eight seedlings (b), and n = 10 leaves from 5 plants (c); *P < 0.05; **P < 0.01; ***P < 0.001, Student’s t-test vs. Col-0 (b) and vs. mock (c).
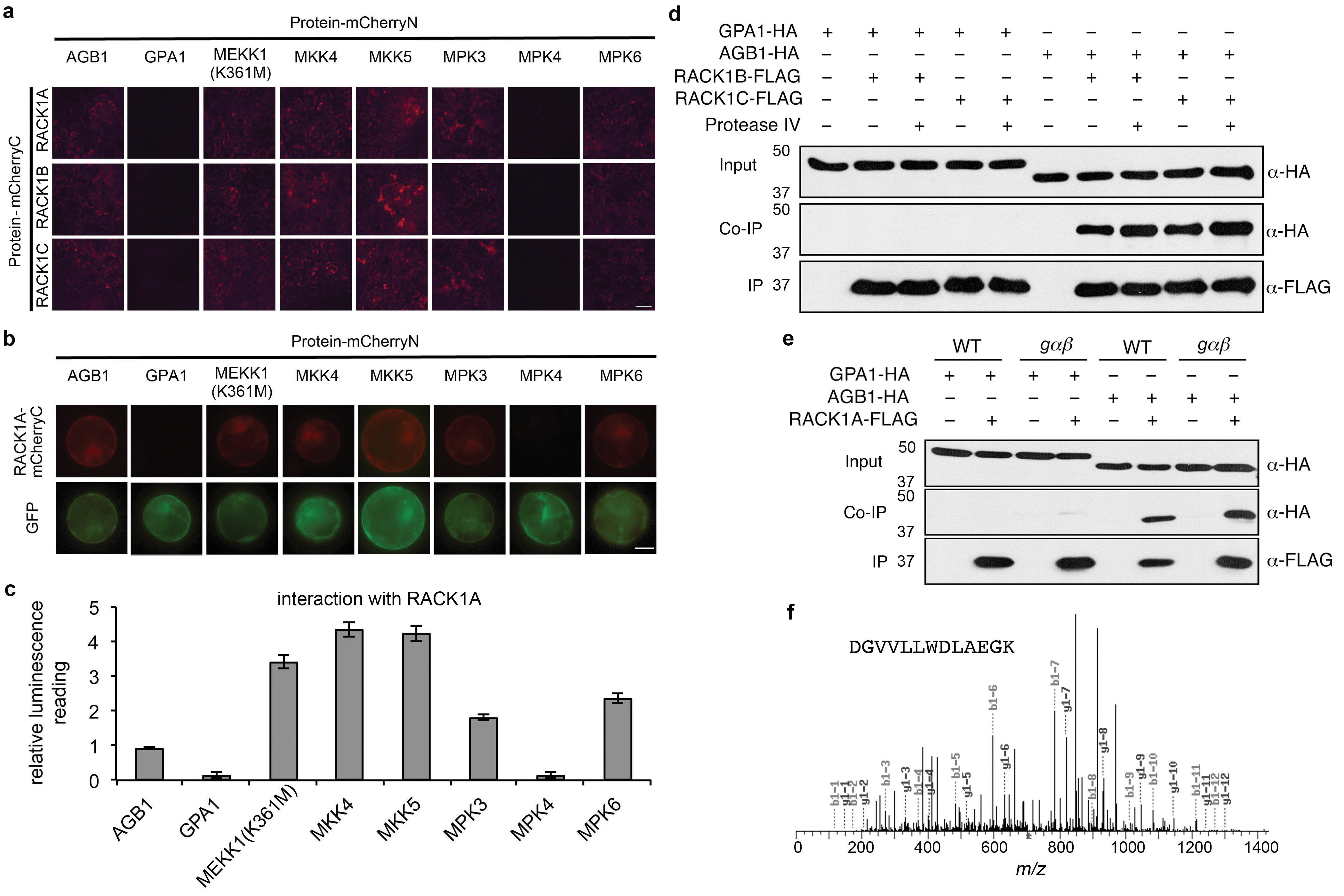
In a search of potential signaling components that could link the heterotrimeric G-protein complex to downstream MAPK cascades, we considered the conserved scaffold protein RACK17. The rationale was that RACK1 is homologous with Gβ7, interacts with Gβ in metazoans8, and functions in innate immune signaling in rice9. There are three RACK1 homologues in Arabidopsis: RACK1A, 1B, and 1C, which share 99% amino acid identity10.
We used three methods to determine whether Arabidopsis RACK1 proteins interact with G proteins and MAP kinases. In a bimolecular fluorescence complementation (BiFC) assay in Nicotiana benthamiana leaves, RACK1A, RACK1B, and RACK1C interacted with Gβ, MEKK1(K361M), MKK4, MKK5, MPK3, and MPK6, but not Gα or MPK4 (Extended Data Fig. 7a). A kinase-inactive version of MEKK1, MEKK1(K361M), was used in this experiment because the auto-activation of native MEKK1 destabilizes its interaction with RACK1 (data not shown). MEKK1, MKK4/5, and MPK3/6 are the Arabidopsis MAPKKK, MAPKKs, and MAPKs, respectively, that were proposed to constitute a MAP kinase-signaling cascade in the flg22/FLS2 signaling pathway11. Similar results to those obtained with the BiFC assay in N. benthamiana were obtained with BiFC and split firefly luciferase complementation (SFLC) assays for RACK1A interactors in Arabidopsis protoplasts (Extended Data Fig. 7b, c). The interaction between RACK1 proteins and MPK3/6, but not MPK4 is consistent with the data in Fig. 1b, showing that MPK6 and MPK3, but not MPK4, are strongly activated after protease IV treatment.
In co-immunoprecipitation (Co-IP) experiments in Arabidopsis mesophyll protoplasts using FLAG-tagged RACK1s as the bait and HA-tagged Gβ subunit as the prey, we observed binding between all three Arabidopsis RACK1 proteins and Gβ (Fig. 3a and Extended Data Fig. 7d). In contrast to Gβ, HA-tagged Gα was not pulled down by FLAG-tagged RACK1 proteins (Fig. 3b and Extended Data Fig. 7d). In the co-IP experiments, the interaction of Gβ with RACK1A was not dependent on Gα, because the interaction was still present in the gαβ double mutant (Extended Data Fig. 7e). Finally, consistent with the BiFC and SFLC assays, HA-tagged MEKK1(K361M), MKK5, MPK3, and MPK6 all co-immunoprecipitated with FLAG-tagged RACK1A, whereas MPK4 did not under the same condition (Fig. 3c to f). The amounts of the MAPKK and MAPKs that were pulled down by RACK1A in the co-IP experiments clearly decreased in the presence of protease IV (Fig. 3d to f), suggesting that protease IV releases the activated MAPKs from the RACK1-MAPK cascade complex to execute their downstream cellular functions. In the case of the MAPKKK MEKK1, we also identified endogenous RACK1 proteins by mass spectrometric analysis as binding partners of MEKK1(K361M) (Extended Data Fig. 7f) in a transgenic line in which FLAG-tagged MEKK1(K361M) is expressed under the control of the 3.9-kb MEKK1 native promoter in a mekk1 null mutant background.
Figure 3. RACK1A interacts with Gβ and MAPKs.
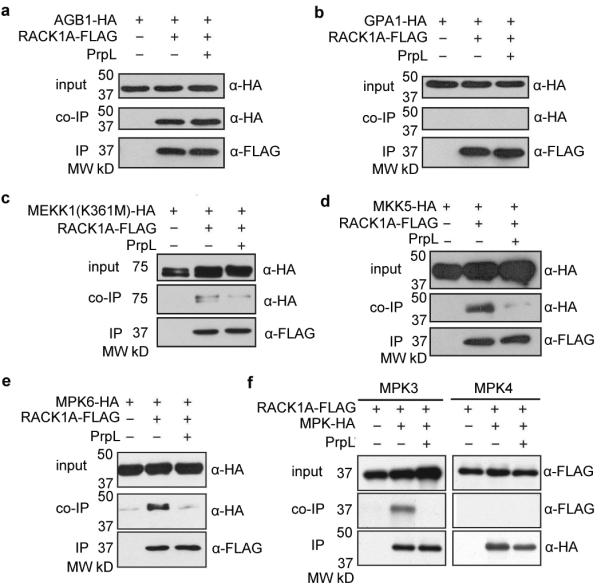
a to f, Co-immunoprecipitation assays in Arabidopsis protoplasts. Protoplasts were treated with 100 nM purified PrpL for 15 minutes. Target proteins were detected in Western blots using anti-HA or anti-FLAG antibodies. Numbers left of blots represent marker size in kDa.
To confirm the physiological relevance of the observed interactions between RACK1 and MAPK cascade components (Fig. 3 and Extended Data Fig. 7), we tested a variety of loss-of-function MAPK mutants and knockdowns. We found that the activation of the defense-related genes WRKY30 and WRKY33 by protease IV was almost completely blocked in two independent mpk3,6-es transgenic lines in which mpk3 is silenced with an estradiol inducible MPK3-RNAi construct in a null mpk6 mutant background (Extended Data Fig. 8a). We also found that both protease IV-triggered MPK3/6 activation and WRKY30 and WRKY33 gene induction were disrupted in mkk4,5-es transgenic lines (Extended Data Fig. 8a, b), which utilize a single estradiol inducible RNAi construct to target both MKK4 and MKK5 mRNAs. Finally, we observed a significant decrease in protease IV-triggered induction of WRKY30 and WRKY33 mRNA accumulation in two mekk1 mutants, an mekk1 null mutant and the mekk1 null mutant complemented with an MEKK1(K361M) construct [mekk1/pMEKK1::MEKK1(K361M)] (Extended Data Fig. 8c, d). As previously reported, MEKK1(K361M), which is deficient in kinase activity, rescues the severe growth defect of an mekk1 null mutant12. In contrast to the mkk4,5 knockdown lines, we did not consistently observe a decreased level of protease IV-triggered MPK3/6 phosphorylation in either of the mekk1 mutants (Extended Data Fig. 8e). One explanation for the partial decrease in WRKY gene induction but not in MPK3/6 phosphorylation in the mekk1 mutants is that multiple MAPKKKs13 function additively to activate MPK3/6 but that the phosphorylation assay is not sensitive enough to detect a partial loss of MAPKKK activity.
Obtaining genetic evidence that RACK1 is required for protease-mediated signaling is challenging because of the functional redundancy of the three RACK1 proteins in Arabidopsis. T-DNA mutants corresponding to insertions in individual rack1 genes did not show any decrease in protease IV- or flg22-activated MAPK levels (Extended Data Fig. 9a), and only moderate decreases in protease IV- but not flg22-triggered defense gene induction (Extended Data Fig. 9b). Because rack1a rack1b rack1c triple null mutants have a dwarf phenotype and do not set seeds14, we generated two independent transgenic lines, amiR-rack1-es1 and amiR-rack1-es2, which express a previously described artificial microRNA (amiR-RACK1-4)15 under the control of an estradiol-inducible promoter. These transgenic lines showed dramatically decreased transcript levels of all three rack1 genes following estradiol treatment (Extended Data Fig. 9c). Following protease IV or ArgC treatment, amiR-rack1-es1 and amiR-rack1-es2 seedlings that had been induced with estradiol exhibited markedly decreased levels of activated MPK3 and MPK6 (Fig. 4a). Protoplasts transfected with constitutively expressed amiR-RACK1-4 also showed reduced levels of protease IV-mediated MPK3 and MPK6 activation (Extended Data Fig. 9d, e). Similarly, knockdown of the rack1 genes blocked protease IV or ArgC-mediated defense gene induction (Fig. 4b) and protease IV-mediated protection against P. syringae infection (Fig. 4c). In contrast to protease IV and ArgC, flg22-mediated activation of MAPKs or defense gene expression or protection against P. syringae, were not affected by knock down of the rack1 genes (Fig. 4a to c; Extended Data Fig. 9e). These data are consistent with the conclusion that RACK1 proteins function in the protease IV and ArgC signaling pathway but not the flg22 pathway.
The RACK1 proteins studied here are the first MAPK cascade scaffolding proteins discovered for the large family of plant genes encoding MAPK cascade components. In yeast, the scaffolding protein Ste5 links a MAPK cascade to G-protein signaling in the mating pathway that is mediated by G protein coupled receptor (GPCR) stimulation by yeast pheromone16. In mammals, the scaffolding protein β-arrestin 2 brings MAPK cascade activity under the control of upstream GPCRs16. However, since plants do not have canonical GPCRs or orthologs of Ste5 and β-arrestin6,16, our data suggest that the linkage of G-proteins to MAPKs via RACK1 is mechanistically distinct from G protein signaling in metazoans and yeast.
The protease-activated signaling pathway is summarized in the model shown in Fig. 4d. It remains to be determined whether the cleavage of protein targets by protease IV directly or indirectly activates downstream responses. In the latter scenario, pathogen-secreted proteases could release host polypeptides that function as damage-associated molecular patterns (DAMPs) that are subsequently recognized by corresponding DAMP immune receptors. In either case, an evolutionary and physiological interpretation of our findings is that plants evolved a novel surveillance system to recognize and respond to pathogen-encoded proteases that disrupt host homeostasis via their proteolytic activity.
Methods
Bacterial strains
P. aeruginosa strains used in this work were wild type and mutants of UCBPP-PA1417,18 and PAO ADD197619. The latter strain carries the chromosomally incorporated gene for T7 RNA polymerase under the control of the lac repressor and was used for production of His-tagged PrpL and His-tagged ArgC. Xanthomonas campestris strains were described previously20.
A PA14/ΔprpL in-frame deletion mutant was constructed using a method described previously21 that employed a sequence that contained regions immediately flanking the coding sequence of the prpL gene. This fragment was generated by a standard 3-step PCR protocol using Phusion DNA polymerase (New England Biolabs) and then cloned into the BamHI and HindIII sites of pEX18Ap22, resulting in plasmid pEX18-ΔprpL. pEX18-ΔprpL was used to introduce the deleted region into the wild-type PA14 genome by homologous recombination. Escherichia coli strain SM10 λpir was used for triparental mating23.
For the purification of His-tagged protease IV or ArgC, the P. aeruginosa PA14 prpL gene or the X. campestris strain 1946 argC gene were cloned into the EcoRI and XhoI sites of pETP3024, creating plasmids pETP-prpL or pETP-argC, which encode 6xHis-tagged PrpL and 6xHis-tagged ArgC, respectively. The resulting plasmids were transformed into P. aeruginosa PAO ADD1976 by electroporation25 to generate the strains ADD/pETP-prpL or ADD/pETP-argC for purification of His-tagged protease IV or His-tagged ArgC, respectively.
For argC complementation in Xanthomonas, the X. campestris strain 1946 argC gene was cloned into the BamHI site of pVSP6126, creating plasmid pVSP61-argC. An HA-tag was incorporated at the C-terminal of the argC gene for the detection of the complemented protein. The resulting plasmid and empty pVSP61 vector were transformed into X. campestris strain 8004 by triparental conjugation23.
Antibiotics were supplemented as needed: ampicillin or carbenicillin, 50 μg/ml for E. coli or 300 μg/ml for P. aeruginosa; kanamycin 50 μg/ml for E. coli and Xanthomonas campestris or 200 μg/ml for P. aeruginosa; and rifampicin 100 μg/ml.
Construction of Arabidopsis transgenic lines
Construction of amiR-rack1-es transgenic lines and the mekk1/pMEKK1::MEKK1(K361M) transgenic line was carried out as follows: the BamHI/PstI fragment of pre-amiR-RACK1-415 was inserted between the estradiol-inducible promoter27 and the NOS terminator in a modified pUC119-RCS vector28. The pre-amiR-RACK1-4 expression cassette was then cut out by AscI digestion and inserted into AscI-digested binary vector pFGC19-XVE-RCS28, which expresses the XVE transcriptional activator29 under the 35S promoter, to obtain pFGC-EST-RACK1. This latter plasmid was introduced into Agrobacterium tumefaciens GV3101 cells by electroporation, and GV3101/ pFGC-EST-RACK1 was used to generate transgenic Arabidopsis plants with inducible amiR-RACK1 expression using the floral dip technique30. To generate mekk1/pMEKK1::MEKK1(K361M) transgenic Arabidopsis, a ~9.4 kb MEKK1 genomic fragment was used to complement a mekk1 null mutant (Salk_052557). This genomic fragment contains a ~3.9 kb promoter sequence upstream of the start codon, an “AAGG” to “ATGG” mutation in exon 2 (corresponding to K361M mutation in MEKK1) to disrupt MEKK1 kinase activity, and a double FLAG-tag coding sequence upstream of the stop codon.
Fractionation of the PA14 secretome
One liter of PA14 cells grown in M9 minimal medium (6.8 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 1 g/L NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, 10 μM FeCl3, 0.4 % glucose, 10 mg/L thiamine) was centrifuged at 20,000 × g at 4°C for 30 minutes and the pellet was discarded. The supernatant was filtered through a 0.22 μm low protein-binding filter (Corning). Secreted PA14 proteins in the filtrate were precipitated with ammonium sulfate (85% saturation) at 4°C overnight, followed by centrifugation at 20,000 × g at 4°C for 1 hour. The pellet was resuspended in 30 mL buffer A (20 mM Tris, pH 8.8), concentrated to 150 μL using Centrion Plus-70 filter (Millipore) to remove the excess ammonium sulfate, and diluted again into 10 mL buffer A. The protein sample was loaded onto a 1-mL DEAE anion exchange chromatography column (GE Healthcare) that was washed with buffer B (20 mM Tris, pH 8.8, 1 M NaCl) and equilibrated with buffer A. Proteins were separated into 1-mL fractions with a linear gradient of buffer B (0-60% within 20-column volumes). The fractionation was carried out at 4°C with a flow rate of 1 mL/minute.
Purification of P. aeruginosa protease IV and X. campestris ArgC
Secreted proteins from ADD1976/pETP-prpL were precipitated as described above and resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0). The sample was loaded onto a 5-mL HisTrap Affinity Column (GE Healthcare) and the 6×His tagged PA14 protease IV was purified according to the manufacturer’s instructions. The eluted protease IV was concentrated to 150 μL and immediately subjected to a Superdex 200 gel filtration column (GE Healthcare). Purified protease IV was exchanged into M9 minimal medium and filter-sterilized using a 0.22 μm low protein-binding filter (Millipore). The concentration of the purified protease IV was adjusted to 20 μM, aliquoted, and stored at −80°C before being used for plant treatments. X. campestris protease ArgC was purified using the same protocol.
Protease assay
The protease activity assay of protease IV and its homolog ArgC was determined as previously described31. Protease IV and ArgC were inactivated by TLCK as previously described31.
Plant growth
Seeds were sterilized in 20% bleach for 2 min and washed three times with sterile water. Seedlings were grown in liquid MS medium (Murashige and Skoog basal medium with vitamins from Phytotechnology Laboratories supplemented with 0.5 g/L MES hydrate and 0.5% sucrose at pH 5.7) in either 24-well assay plates (BD Falcon) (8 seeds and 0.5 mL medium/well) for MAP kinase assays, microarray and RT-qPCR analysis, callose induction and GUS expression, or 96-well plates (Greiner Bio-One) (1 seed and 0.2 mL medium/well) for oxidative burst measurements. Plates were sealed with Micropore tape and placed on grid-like shelves over water trays on a Floralight cart in a plant growth chamber for 10 days at 21°C with 75% relative humidity under 16 hours of daylight (65-70 μE·m−2·s−1). The media in 24-well plates was exchanged for fresh media on day 8, whereas the media in 96-well plates was exchanged for sterile water on day 9.
Elicitor treatments
The synthetic peptide flg22 was synthesized by Genscript, New Jersey. Experimentally determined optimal concentrations of protease IV were: 20 nM for oxidative burst measurements, microarray and RT-qPCR analyses; 40 nM for MAP kinase activation; 100 nM for GUS expression; 500 nM for callose elicitation and the infection protection assay. For direct comparison, the same concentrations of flg22 and protease IV or ArgC were used in the same assays. Ten-day old seedlings were treated with different elicitors for the following times unless otherwise specified: 6 hours for GUS assays in reporter line CYP71A12pro:GUS; 10 minutes for MAP kinase activation assays; 1 hour or 6 hours for RT-qPCR analysis of selected genes; and 18 hours for callose induction.
Transient silencing of MAPK or MAPKK genes in transgenic plants
In two independent mpk3,6-es transgenic lines, MPK3 was silenced with an estradiol inducible MPK3-RNAi construct in a null mpk6 mutant (Salk_062471) background. In two mkk4,5-es transgenic lines, a single estradiol inducible RNAi construct was utilized to target both MKK4 and MKK5 mRNAs. Details concerning the construction of the mpk3,6-es and mkk4,5-es transgenic lines will be described elsewhere. The transgenic and control plants were grown in MS medium in a 24-well plate as described above for 4 days. Then the medium was changed to MS medium containing 10 μM estradiol (Sigma, 100 mM stock in DMSO). After exposure to estradiol for 3 days, the seedlings were treated with water and 40 nM purified protease IV for 10 minutes (for MAPK assays) or 20 nM purified protease IV for 1 hour (for RT-qPCR assays).
Transient silencing of rack1 genes in protoplasts and transgenic plants
Mesophyll protoplasts isolated from leaves of 4-week old Arabidopsis plants (4×104 cells in 200 μL) were transfected with 40 μg (20 μL) of amiR-RACK1-4 construct or empty amiRNA expression vector15 as a control. After 24 hours of expression, 100 nM flg22 or 100 nM purified protease IV was added to the protoplasts followed by incubation for 10 minutes before the cells were harvested for MAPK assays and rack1 gene silencing confirmation by RT-qPCR.
For estradiol-induced rack1 silencing in transgenic amiR-rack1-es lines, the wild-type Col-0 and transgenic plants were grown in MS medium in a 24-well plate as described above for 3 days. Then the medium was changed to MS medium containing 10 μM estradiol (Sigma, 100 mM stock in DMSO). After exposure to estradiol for 2 days, the seedlings were treated with water and 40 nM flg22 or 40 nM purified protease IV for 10 minutes (MAPK assay) or 20 nM flg22 or 20 nM purified protease IV for 6 hours (RT-qPCR measuring transcript levels of CYP71A12, GST6 and the three rack1 genes). For the protease IV-mediated protection assay against P. syringae DC3000, 20 μM estradiol was infiltrated into 4-week old control Col-0 and transgenic amiR-rack1-es1 and amiR-rack1-es2 leaves 24 hour prior to the mock treatment or treatment with 500 nM purified protease IV.
Mutant seed stocks
T-DNA insertion lines gpa1-4 (CS6534), agb1-2 (CS6536), agg1-1c (CS16550), agg2-1 (SALK_022447), gpa1-4/agb1-2 (CS6535), agg1-1c/agg2-1 (CS16551), mekk1 (SALK_052557), rack1a-3 (CS862351), rack1b-2 (SALK_145920), rack1b-3 (CS863092), rack1c-2 (SALK_017913), and rack1c-3 (SALK_001973) were obtained from the Arabidopsis Biological Resource Center.
GUS histochemical assay
After treatment with 100 nM flg22 or 100 nM purified protease IV for 6 hours, plants were washed with 50 mM sodium phosphate (pH 7) and 0.5 mL of GUS substrate solution (50 mM sodium phosphate, pH 7, 10 mM EDTA, 0.5 mM K4[Fe(CN)6], 0.5 mM K3[Fe(CN)6], 0.5 mM X-Gluc, and 0.1% v/v Triton X-100) was added to each well. The plants were vacuum-infiltrated for 5 minutes and then incubated at 37°C for 4 hours. Tissues were fixed with a 3:1 ethanol:acetic acid solution at 4°C overnight and placed in 95% ethanol. Tissues were cleared in lactic acid and then examined using a Discovery V12 microscope (Zeiss). For the screen of PA14 secretome fractions, 100 μL of buffer A (20 mM Tris, pH 8.8) or different DEAE fractions were added to each well.
MAP kinase activity
Total proteins in seedling or protoplast lysates were resolved on a 10% SDS PAGE gel and transferred to a PVDF membrane. Western blot analysis was conducted by using anti-phospho ERK antibodies (Cell Signaling) as the primary antibody at 1:10,000 dilution in 5% BSA and HRP-conjugated anti-rabbit antibodies as the secondary antibody at 1:10,000 dilution in 5% non-fat milk. The immunoblot signal was visualized by using the SuperSignal West Femto kit (Thermo Scientific).
Oxidative burst measurement
H2O2 was detected using a luminol-HRP (horseradish peroxidase) based chemiluminescence assay. A 10 mg/mL 500× HRP (Sigma-Aldrich) stock solution was prepared by dissolving 10 mg HRP in water. A 20 mg/mL 500× luminol (Sigma-Aldrich) stock solution was prepared by dissolving 20 mg luminol in 100 mM KOH. For each elicitor, a master reaction mixture was prepared by diluting individual elicitor, HRP and luminol stocks with water. The plates were kept in the dark for 1 hour before elicitation. The following procedures were carried out in the dark. Liquid was removed at the end of the 1-hour pretreatment and 200 μL of master reaction mixture was added into each well. Plates were placed into a 96-well scintillation reader immediately and light emission was monitored using a 96-well scintillation counter (1450 Microbeta Wallac TriLux Scintillation/Luminescence counter). Every plate was read for about 30 cycles. Kinetics of H2O2 production was determined by plotting the average chemiluminescence counts from all the seedlings under the same condition over the reading period. Every time point is the mean value of 16 seedlings.
RNA isolation and microarray and RT-qPCR analysis
Total RNA was isolated according to the manufacturer’s instructions using the RNeasy Plant Mini Kit (Qiagen). DNA was removed using the DNA-free kit (Ambion), and reverse transcription reactions were carried out using the iScript cDNA synthesis kit (Bio-Rad). cDNA concentrations were measured using a Nano-drop instrument (Thermo Scientific). RT-qPCR was carried out using a CFX96 real-time PCR machine (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad). The following PCR reaction program was used: 95°C for 3 minutes followed by 50 cycles of 95°C for 30 seconds and 55°C for 30 seconds. Fold-change was calculated relative to plants treated with M9 buffer. Fold induction data represent the mean ± s.d., n = 3 with each containing 8 seedlings. Expression values were normalized to that of the eukaryotic translation initiation factor 4A1 (EIF4A1). The primers used were: EIF4A1 (At3g13920), 5’-GCAGTCTCTTCGTGCTGACA-3’ and 5’-TGTCATAGATCTGGTCCTTGAA-3’; CYP71A12 (At2g30750), 5’-GATTATCACCTCGGTTCCT-3’ and 5’-CCACTAATACTTCCCAGATTA-3’; WRKY30 (At5g24110), 5’-GCAGCTTGAGAGCAAGAATG-3’ and 5’-AGCCAAATTTCCAAGAGGAT-3’; GST6 (At2g47730), 5’-CCATCTTCAAAGGCTGGAAC-3’ and 5’-TCGAGCTCAAAGATGGTGAA-3’; WRKY29 (At4g23550), 5’-ATCCAACGGATCAAGAGCTG-3’ and 5’-GCGTCCGACAACAGATTCTC-3’; WRKY33 (At2g38470), 5’-GGGAAACCCAAATCCAAGA-3’ and 5’-GTTTCCCTTCGTAGGTTGTGA-3’; ERF1 (At3g23240), 5’-TCGGCGATTCTCAATTTTTC-3’ and 5’-ACAACCGGAGAACAACCATC-3’; rack1a (At1g18080), 5’-GCTGAAAAGGCTGACAACAGT-3’ and 5’-GCTCCAGTTAAGGCTTGTGC-3’; rack1b (At1g48630), 5’-TTGTTGAGGATTTGAAGGTTGA-3’ and 5’-CCAGTTCAAGCTTGTGCAGTA-3’; rack1c (At3g18130), 5’-GAGGCAGAGAAGAATGAAGGTG-3’ and 5’-CCAGTTCAAGCTTGTGCAGTA-3’. WRKY gene induction was measured 1 hour after elicitation, whereas CYP71A12, ERF1, and GST6, were measured 6 hours after elicitor treatment.
For microarray analysis, RNA quality was assessed by checking the integrity of RNA on an Agilent 2100 Bioanalyzer (Agilent Technologies). Target labeling was performed according to the protocol given in the Affymetrix GeneChip 3’ IVT Express Kit Technical Manual. Microarray hybridizations and scanning were finished at the Genomics Core, Joslin Diabetes Center, Boston, MA. Microarray CEL files were read into the R statistical analysis software, version 2.15.2. Arrays were analyzed together using the standard RMA procedure as implemented in Bioconductor’s “affy” package, version 1.36.132,33. Fold changes were calculated using log-base-2-transformed expression values by subtracting the mean of control samples from the mean of treated samples. Microarray CEL files were also obtained from previous studies exploring the effects of flg22 and OGs on gene expression4. These two experiments were subjected to the RMA procedure together, but downstream analyses (e.g. fold change computations) were performed separately on the two treatments. The microarray data has been deposited in the GEO database under the submission number: GSE58518.
Callose deposition assay
Elicitor-induced callose deposition in cotyledons of 10-day old Arabidopsis seedlings was detected using aniline blue as described34. Eighteen hours after elicitation, seedlings were fixed under a vacuum in 3:1 ethanol:acetic acid. The clearing solution was changed until the leaves were colorless. Tissues were washed in 70% ethanol and then 50% ethanol for at least 2 hours each time and rehydrated in several brief H2O washes followed by an overnight H2O wash. Samples were then made transparent by several minutes in a vacuum with 10% NaOH followed by a 2-hour incubation at 37°C on a shaking platform. After several more H2O washes, tissues were incubated in the dark at room temperature for at least 4 hours with 0.01% aniline blue in 150 mM K2HPO4 (pH 9.5). After mounting on slides in 50% glycerol, samples were examined using a Zeiss Axioplan microscope (Oberkochen, Germany) utilizing UV illumination and a broadband DAPI filter set (excitation filter 390 nm; dichroic mirror 420 nm; emission filter 460 nm).
Pathogenicity assays
Arabidopsis pathogenicity assays, including infection by P. syringae strain DC3000 with or without pre-infiltration of protease IV or flg22, were performed according to previously described protocols21. Data represent the mean of bacterial titers ± s.d. of ten leaf disks excised from 10 leaves of 5 plants. The infection protection assay was repeated three times with similar results.
Xanthomonas pathogenicity assays in Brassica oleracea were carried out according to previously described protocols35 with modifications. Seeds of broccoli cultivar B. oleracea var. Marathon were sown in Fafard #2 soil mix and grown in a 12-hour light (70 μE·m−2·s−1) cycle at 19°C and 60% relative humidity. Individual seedlings were transferred to 5 × 5 cm pots after one week and kept at a cycle of 16-hour light (150 μE·m−2·s−1) at 23°C followed by 8-hour dark at 20°C and 70% relative humidity. After a further two weeks of growth, the three-week old plants were used for Xanthomonas infiltration. Fresh X. campestris overnight cultures were washed and adjusted to 106 cells/ml in 10 mM MgSO4. A standard infiltration protocol was used to infect 3-week old leaves. After infection, the plants were transferred to a growth chamber with the following conditions: 12-hour light (60 μE·m−2·s−1) at 28°C at 90% relative humidity for two days before being harvested for CFU counting. Data represent the mean of bacterial titers ± s.d. of ten leaf disks excised from 10 leaves of 5 plants. The infection protection assay was repeated three times with similar results.
Co-immunoprecipitation (co-IP)
For co-IP carried out in protoplasts, mesophyll protoplast isolation from leaves of 4-week old Arabidopsis plants and polyethylene glycol (PEG)-mediated DNA transfection were carried out as previously described36. Co-IP was performed as described previously37 with modifications. Briefly, 100 μg (50 μL) of PREY plasmids were used to co-transfect 1 mL Arabidopsis mesophyll protoplasts (5×105 cells) with 100 μg (50 μL) of BAIT plasmids or empty vectors. After 6 hours to allow protein expression, the cells were pelleted and lysed in 200 μL of IP buffer (10 mM HEPES, pH7.5, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 1× Roche EDTA-free protease inhibitor cocktail) by vigorous vortexing for 1 min. Twenty μL of lysate was saved as the input fraction to ensure that the Prey proteins were expressed equally in all samples. The rest of the lysate (180 μL) was mixed with 320 μL IP buffer and vigorously vortexed for 1 min. The resultant clear lysate was centrifuged at 21,000 × g for 10 min at 4°C, and the supernatant was incubated with a 10 μL slurry of anti-FLAG M2 agarose beads (Sigma) or anti-HA magnetic beads (Pierce) for 3 hr at 4°C. The beads were washed 3 times with the IP buffer and once with 50 mM Tris-HCl, pH7.5. The eluate was obtained by boiling the beads in 40 μL of SDS-PAGE loading buffer and the presence of Co-IPed PREY proteins was detected by immunoblotting analysis using HRP-conjugated anti-HA antibody or anti-FLAG (Roche) at 1:10,000 dilution, and the immunoblot signal was visualized using the SuperSignal West Femto kit (Thermo Scientific). The same membrane was stripped and re-used to detect the comparable amounts of IPed BAIT proteins by immunoblot.
Bimolecular fluorescence complementation (BiFC)
For plasmids used in the split-mCherry assay, the coding sequence of the N-terminal fragment (mCherryN, aa1- aa159) or the C-terminal fragment (mCherryC, aa160-aa235) of mCherry was PCR amplified, digested by BamHI/NotI, and inserted into the same digested pAN vector, which contains a double 35S promoter and a NOS terminator, to obtain pcCherryN and pcCherryC plasmids. Genes for protein-protein interaction tests were inserted into the XbaI/BamHI digested pcCherryN or pcCherryC vectors after digestion of their PCR products with XbaI (or SpeI, NheI if the XbaI site was present in the gene) at the 5’ end and with BamHI (or BglII if the BamHI site was present in the gene) at the 3’ end, allowing the expression of a chimeric gene of interest with the coding sequence of mCherryN or mCherryC at the 3’ end.
For binary plasmids used in the BiFC assay in Agroinfiltrated Nicotiana benthamiana leaves, pFGC-RCS (kanamycin resistant) and pPZP-RCS (spectinomycin resistant) binary vectors were constructed by replacing the original sequences between EcoRI and HindIII of pFGC19 and pPZP222 with the multiple cloning site sequence from pUC119-RCS flanked by EcoRI and HindIII38. Subsequently, the entire expression cassette of “gene”-mCherryN was PCR amplified from protoplast expression plasmids, digested by AscI and inserted into the AscI site of pFGC-RCS, while the entire expression cassette containing the “gene”-mCherryC fusion DNA was PCR amplified from protoplast expression plasmids, digested by AscI and inserted into the AscI site of pPZP-RCS. A pair of pFGC-RCS and pPZP-RCS plasmids expressing a pair of genes for protein-protein interaction tests were co-transformed into Agrobacterium GV3101 cells by electroporation, and cells transformed with both binary plasmids were selected by the addition of both kanamycin and spectinomycin to the growth medium. Leaves of 4- to 5-week old N. benthamiana plants were infiltrated with agrobacteria (final OD600 = 0.01) containing constructs expressing the mCherryN fragment fused to GPA1, AGB1 or MAP kinases and the mCherryC fragment fused to RACK1A/B/C. The agroinfiltration experiment was carried out as described previously39.
Arabidopsis protoplasts 18 hours after transfection and N. benthamiana leaf pieces 2 days after agroinfiltration were imaged using a Leica DM-6000B upright fluorescence microscope with phase and differential interference contrast equipped with a Leica FW4000 digital image-acquisition and processing system.
Split firefly luciferase complementation (SFLC)
For plasmids used in the SFLC assay, the genes for protein-protein interaction tests were inserted into the XbaI/BamHI digested pcFLucN or pcFLucC vectors37 after digestion of their PCR products with XbaI (or SpeI, NheI if the XbaI site is present in the gene) at the 5’ end and with BamHI (or BglII if the BamHI site is present in the gene) at the 3’ end, allowing the expression of a chimeric gene of interest with the coding sequence of FLucN or FLucC at the 3’ end.
SFLC experiments carried out in protoplasts were performed as described previously37. Briefly, 10 μg (5 μL) of PREY plasmids were used to co-transfect 100 μL of Arabidopsis mesophyll protoplasts (5×105 cells) with 10 μg (5 μL) of BAIT plasmids. One μg UBQ10::GUS plasmid was used in each transfection as an internal normalization control. After 6 hours to allow for protein expression, the luminescence of each sample was recorded by a GloMax-Multi microplate multimode reader (Promega) with the integration time set as 1 sec.
Extended Data
Extended Data Figure 1. Protease IV-triggered GUS staining in CYP71A12pro:GUS transgenic Arabidopsis seedlings.
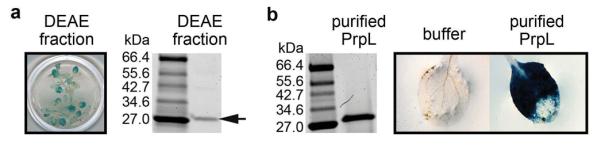
a, Activation of CYP71A12pro:GUS by a DEAE fraction of the PA14 secretome (left) and purification of the eliciting activity by DEAE chromatography (right). b, Activation of CYP71A12pro:GUS in 10-day old seedlings by 100 nM purified PrpL. The experiments in panels a and b were repeated three times with similar results.
Extended Data Figure 2. Trypsin does not activate MAPK cascade or elicit an oxidative burst in Arabidopsis.
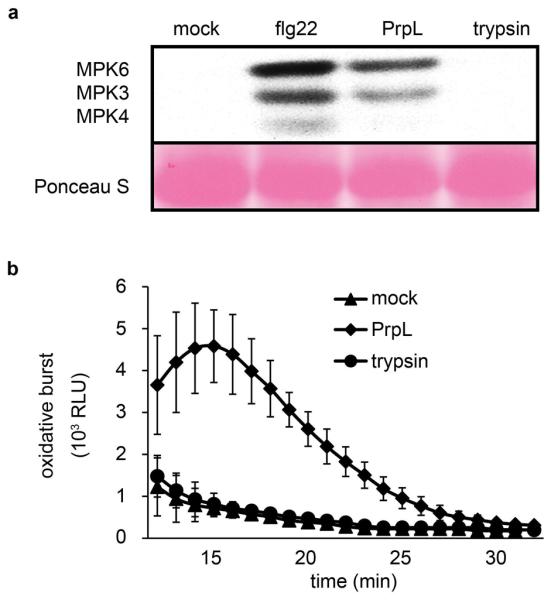
a, Western blot depicting activation of MAPKs by 40 nM flg22, or 40 nM purified PrpL, or trypsin in 10-day old seedlings. The same molecular weight region of the Western blot is shown as in Fig. 1b. b, Chemiluminescence assay showing elicitation of an oxidative burst in 10-day old seedlings by 20 nM purified PrpL or trypsin. Error bars represent standard deviation; n = 16 individual seedlings.
Extended Data Figure 3. Transcriptional analysis of purified protease IV.

a, Genome-wide transcriptomic profiles obtained with Affymetrix Arabidopsis ATH1 GeneChips® of 10-day old seedlings treated with 20 nM purified PrpL and comparison to published flg22- and oligogalacturonide (OG)-responses. A Venn diagram shows the similarity of expression behavior (|fold change| > 2) in response to the three treatments. b, Defense gene induction levels measured by RT-qPCR in 10-day old Col-0 seedlings treated with 20 nM purified PrpL or 20 nM flg22 for 1 hour (WRKY29, 30, and 33) or 6 hours (GST6, ERF1, and CYP71A12). The data represent the mean ± s.d., n = 3 biological replicates, each containing 8 seedlings.
Extended Data Figure 4. Protease IV-triggered responses are dependent on proteolytic activity.
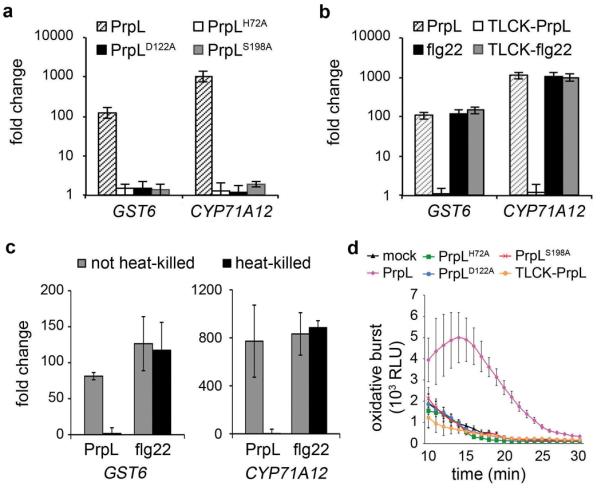
a, Induction of defense-related genes by 20 nM purified PrpL or inactive variants of PrpL measured by RT-qPCR. b, Induction of defense-related genes by 20 nM purified PrpL or 20 nM flg22, or 20 nM TLCK-treated PrpL or 20 nM TLCK-treated flg22 measured by RT-qPCR. c, Induction of defense-related genes by 20 nM PrpL or 20 nM heat-treated PrpL or 20 nM flg22 or 20 nM heat-treated flg22 measured by RT-qPCR. d, Chemiluminescence assay showing elicitation of an oxidative burst by 20 nM purified PrpL, 20 nM inactive variants of PrpL, or 20 nM TLCK-treated PrpL. The data represent the mean ± s.d., n = 3 biological replicates with each experiment contains eight seedlings (a, b, c), and n = 16 individual seedlings (d).
Extended Data Figure 5. Sequence analyses of Xanthomonas argC genes.

a, The protein sequence alignment between P. aeruginosa PA14 PrpL and X. campestris pv. raphani strain 1946 ArgC (Xcr ArgC). b to d, Three independent presumptive null mutations in the Xanthomonas argC gene: an insertion of G, a single nucleotide mutation, and a deletion. The extra G is highlighted in black in b; the single nucleotide substitution is indicated by an arrow in c; and the single base deletion is highlighted in black in d. The resulting premature stop codons are highlighted in red. Sequences were aligned to the argC allele in X. campestris pv. raphani strain 1946 (Xcr-1946), from which the argC gene was cloned. X. campestris pv. campestris strains 8004 (Xcc-8004); X. campestris pv. campestris strains BP109 (Xcc-BP109); X. fuscans subsp. fuscans strain 4834-R (Xf-4834-R); X. campestris pv. vesicatoria (Xcv). e. Activation of CYP71A12pro:GUS in 10-day old seedlings by culture filtrate from X. campestris strain Xcr-1946, Xcc-8004, or Xcc-BP109, and X. campestris strain 8004 complemented with a functional argC gene (8004/argC) or transformed with empty vector (8004/vector). Detection of HA-ArgC with an anti-HA antibody. The GUS staining was repeated three times with similar results and the representative images shown were selected from at least three images.
Extended Data Figure 6. G proteins are required for protease IV response.
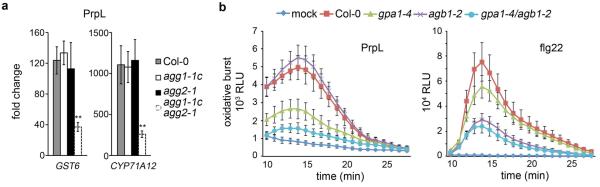
a, Induction of CYP71A12 and GST6 gene expression by 20 nM purified PrpL in 10-day old wild-type Col-0, gγ single mutants (agg1-1c and agg2-1), or a gγ1γ2 double mutant measured by RT-qPCR. b, Chemiluminescence assay showing elicitation of an oxidative burst by 20 nM purified PrpL or 20 nM flg22 in wild-type Col-0 or G protein T-DNA mutants. The data represent the mean ± s.d., n = 3 biological replicates with each containing 8 seedlings (a), and n = 16 individual seedlings (b); **P < 0.01, Student’s t-test.
Extended Data Figure 7. Interactions between RACK1 and Gβ or MAP kinases.

a, Split-mCherry assay in 4-week old Agrobacterium-infiltrated Nicotiana benthamiana leaves. Images were pseudocolored for visualization. Bar = 100 μm. RACK1A, B, C proteins were fused with the C-terminal half of mCherry and the potential interaction partner proteins were fused with the N-terminal half of mCherry. b, Split-mCherry assay in Arabidopsis protoplasts. RACK1A protein was fused with the C-terminal half of mCherry and the potential interaction partner proteins were fused with the N-terminal half of mCherry. GFP protein was included in each experiment to serve as a transfection control. Images were pseudocolored for visualization. Bar = 10 μm. c, Relative interaction intensity between RACK1A and G proteins or MAP kinases measured by split firefly luciferase complementation. RACK1A protein was fused with the FLucN or FLucC to pair with G proteins or MAP kinases fused with the other half of firefly luciferase. Both constructs were co-expressed in protoplasts for 6 h and the complemented luciferase activity was used to relatively quantify protein-protein interactions. UBQ10::GUS was included in each experiment to serve as a transfection normalization control. The data represent the mean ± s.d., n = 3 technical replicate samples. d, Protoplasts were co-transfected with GPA1-HA or AGB1-HA and RACK1B/C-FLAG or a control vector. Co-IP was carried out with an anti-FLAG antibody. (Top) The expression of GPA1 or AGB1 protein. (Middle) AGB1, but not GPA1 co-immunoprecipitates with RACK1 proteins. (Bottom) Pull-down of RACK1 proteins by anti-FLAG antibody. Protoplasts were treated with 100 nM purified PrpL for 15 min. e, Co-IP between GPA1 or AGB1 and RACK1A was carried out in wild-type Col-0 or gαβ mutant Arabidopsis mesophyll protoplasts. Numbers left of blots represent marker size in kDa. f, Mass spectrophotometric analysis of endogenous proteins pulled down by FLAG-tagged MEKK1(K361M). A peptide conserved in all three RACK1 proteins is shown. The experiments in panels a and b were repeated three times with similar results.
Extended Data Figure 8. Protease IV-triggered defense responses in wild-type Col-0 and MAP kinase mutants.

a, Induction of WRKY30 and WRKY33 gene expression by 20 nM purified PrpL in 7-day old seedlings of wild-type Col-0 and transgenic mpk3,6-es1/2 and mkk4,5-es1/2 plants in the absence or presence of estradiol. b, Western blot depicting activation of MPK3 and MPK6 by 40 nM purified PrpL in 7-day old seedlings of wild-type Col-0 and transgenic mkk4,5-es1 plants in the absence or presence of estradiol. The same molecular weight region of the Western blot is shown as in Fig. 1b. c, Induction of WRKY30 and WRKY33 gene expression by 20 nM purified PrpL in 10-day old wild-type Col-0 and mekk1/pMEKK1::MEKK1(K361M) mutant seedlings. d, Induction of WRKY30 and WRKY33 gene expression by 20 nM purified PrpL in 4-day old wild-type Col-0 and mekk1 null mutant seedlings. e, Western blot depicting activation of MPK3 and MPK6 by 40 nM purified PrpL in 10-day old wild-type Col-0 and mekk1/pMEKK1::MEKK1(K361M) mutant seedlings or 4-day old wild-type Col-0 and mekk1 null mutant seedlings. The same molecular weight region of the Western blot is shown as in Fig. 1b. The data represent the mean ± s.d., n = 3 biological replicates with each containing 8 seedlings (a, c, d); **P < 0.01; *** P < 0.001, Student’s t-test vs. Col-0 controls.
Extended Data Figure 9. RACK1 proteins are required for protease IV response.

a, Western blot depicting activation of MAPKs by 40 nM purified PrpL or 40 nM flg22 in 5-day old seedlings of wild-type Col-0 and individual rack1::T-DNA insertion mutants. The same molecular weight region of the Western blot is shown as in Fig. 1b. b, Induction of CYP71A12 by 20 nM purified PrpL or 20 nM flg22 in 5-day old seedlings of wild-type Col-0 and individual rack1::T-DNA insertion mutants. c, RT-qPCR analysis of rack1a, rack1b, and rack1c transcript levels in the 5-day old Col-0 or amiR-rack1-es1 and amiR-rack1-es2 seedlings. d, RT-qPCR analysis of rack1a, rack1b, and rack1c transcript levels in Arabidopsis protoplasts transfected with amiR-RACK1-4 or amiRNA control. e, Western blot depicting activation of MAPKs by 40 nM purified PrpL or 40 nM flg22 in Arabidopsis protoplasts transfected with amiR-RACK1-4 or amiRNA control. The same molecular weight region of the Western blot is shown as in Fig. 1b. The data represent the mean ± s.d., n = 3 biological replicates (b, c, d); *P < 0.05; **P < 0.01, Student’s t-test.
Extended Data Table 1.
P. aeruginosa PA14 transposon mutants screened for activation of CYP71A12pro:GUS.
| gene names |
gene IDs |
type* | gene names |
gene IDs |
type* | gene names |
gene IDs |
type* |
|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||
| aprA | 865 | 1 | toxA | 399 | 2 | cIpB | 130 | 6 |
| aprD | 7385 | 1 | xcpP | 3450 | 2 | hcp1 | 4311 | 6 |
| aprE | 1317 | 1 | xcpQ | 417 | 2 | hcpA | 4107 | 6 |
| aprF | 922 | 1 | xcpR | 812 | 2 | stnR | 1865 | 6 |
| aprl | 4760 | 1 | xcpT | 4498 | 2 | stp1 | 3334 | 6 |
| aprX | 1421 | 1 | xcpW | 3292 | 2 | vgrG2 | 141 | 6 |
| hasAp | 3774 | 1 | xcpZ | 4249 | 2 | gacA | 631 | R |
| hasF | 1253 | 1 | xphA | 4239 | 2 | lasl | 3828 | R |
| cbpD | 1394 | 2 | xqhA | 246 | 2 | rhll | 3829 | R |
| cupB5 | 75 | 2 | exoT | 7001 | 3 | rhIR | 3229 | R |
| lasA | 1299 | 2 | exoU | 339 | 3 | rpoS | 2108 | R |
| lasB | 759 | 2 | exoY | 7430 | 3 | fimL | 627 | S |
| lipA | 2386 | 2 | pscD | 1303 | 3 | flgB | 4759 | S |
| lipC | 2417 | 2 | aaaA | 375 | 5 | flgK | 352 | S |
| pepB | 629 | 2 | eprS | 69 | 5 | fliC | 1029 | S |
| phoA | 914 | 2 | estA | 354 | 5 | fliN | 4482 | S |
| phoB | 3473 | 2 | lepA | 631 | 5 | motA | 2879 | S |
| phoR | 1112 | 2 | tps1 | 556 | 5 | motB | 1935 | S |
| pIcB | 1956 | 2 | tps2 | 600 | 5 | pilA | 7353 | S |
| pIcH | 210 | 2 | tps3 | 555 | 5 | pilD | 2579 | S |
| pIcN | 267 | 2 | tps5 | 545 | 5 | tadZ | 1689 | S |
| pmpA | 93 | 2 | ||||||
Numbers represent the type of secretion system. For example, 2 means Type II secreted protein or Type II secretion machinery protein. R represents regulatory proteins. S represents surface proteins.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 4. Transiently silencing all three rack1 genes abrogates proteases but not flg22-mediated responses.
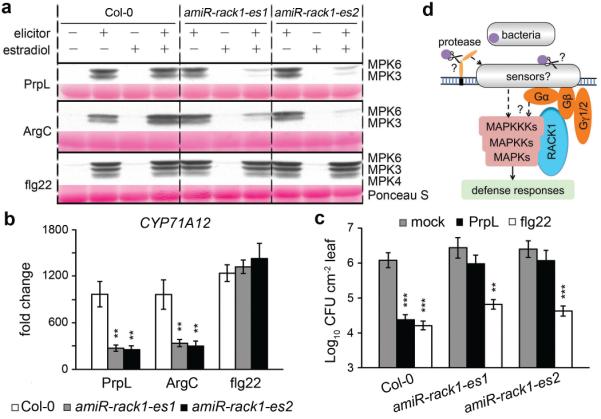
a, Three-day old wild-type Col-0 and transgenic Arabidopsis seedlings from two independent amiR-rack1-es lines were treated with estradiol to activate expression of the artificial microRNA constructs and then two days later were treated with PrpL, ArgC or flg22 and harvested for the MAPK phosphorylation assay. The same molecular weight region of the Western blot is shown as in Fig. 1b.b, Seedlings were treated with estradiol followed by PrpL, ArgC or flg22 as in panel a and then harvested for RT-qPCR analysis of CYP71A12 transcript levels. Water-treated Col-0 was used as a normalization control. c, Protection of 4-week old wild-type Col-0 and transgenic amiR-rack1-es1 or amiR-rack1-es2 plants from P. syringae pv. tomato strain DC3000 infection mediated by PrpL or flg22 24 hours after treatment with estradiol. The data represent the mean ± s.d., n = 3 biological replicates with each experiment containing twelve seedlings (b), and n = 10 leaves from 5 plants (c); **P< 0.01; ***P< 0.001, Student’s t-test vs. Col-0 (b) and vs. mock (c). d. A model of protease-activated novel innate immune signaling pathway in Arabidopsis.
Acknowledgments
We thank G. Tena for generating the mekk1/pMEKK1::MEKK1(K361M) transgenic line, Y. Zhang for the summ1-1 mutant, M.C. Suarez-Rodriguez and P.J. Krysan for discussion, the Arabidopsis Biological Resource Center for T-DNA insertion lines, and M. Curtis and U. Grossniklaus for the estradiol-inducible binary vector. We thank S. Lory for P. aeruginosa PAO ADD1976, M.B. Mudgett for pVSP61. We thank N. Clay, X. Dong, S. Somerville, and Ausubel lab members for critical reading of the manuscript. This work was supported by NSERC and Banting Postdoctoral Fellowships awarded to Z.C., NSF grants MCB-0519898 and IOS-0929226 and NIH grants R37-GM48707 and P30 DK040561 awarded to F.M.A., and NSF grant IOS-0618292 and NIH grant R01-GM70567 awarded to J.S.
Footnotes
Author Information:
The authors declare no competing financial interests.
References
- 1.Tena G, Boudsocq M, Sheen J. Protein kinase signaling networks in plant innate immunity. Curr. Opin. Plant Biol. 2011;14:519–529. doi: 10.1016/j.pbi.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 3.Millet YA, et al. Innate immune responses activated in Arabidopsis roots by microbe-associated molecular patterns. Plant Cell. 2010;22:973–990. doi: 10.1105/tpc.109.069658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denoux C, et al. Activation of defense response pathways by OGs and flg22 elicitors in Arabidopsis seedlings. Mol. Plant. 2008;1:423–445. doi: 10.1093/mp/ssn019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traidej M, Marquart ME, Caballero AR, Thibodeaux BA, O’Callaghan RJ. Identification of the active site residues of Pseudomonas aeruginosa protease IV. J. Biol. Chem. 2003;278:2549–2553. doi: 10.1074/jbc.M208973200. [DOI] [PubMed] [Google Scholar]
- 6.Urano D, Chen J-G, Botella JR, Jones AM. Heterotrimeric G protein signalling in the plant kingdom. Open Biol. 2013;3:120186. doi: 10.1098/rsob.120186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ullah H, et al. Structure of a signal transduction regulator, RACK1, from Arabidopsis thaliana. Protein Sci. 2008;17:1771–1780. doi: 10.1110/ps.035121.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dell EJ, et al. The betagamma subunit of heterotrimeric G proteins interacts with RACK1 and two other WD repeat proteins. J. Biol. Chem. 2002;277:49888–49895. doi: 10.1074/jbc.M202755200. [DOI] [PubMed] [Google Scholar]
- 9.Nakashima A, et al. RACK1 functions in rice innate immunity by interacting with the Rac1 immune complex. Plant Cell. 2008;20:2265–2279. doi: 10.1105/tpc.107.054395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J-G, et al. RACK1 mediates multiple hormone responsiveness and developmental processes in Arabidopsis. J. Exp. Bot. 2006;57:2697–2708. doi: 10.1093/jxb/erl035. [DOI] [PubMed] [Google Scholar]
- 11.Asai T, et al. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature. 2002;415:977–983. doi: 10.1038/415977a. [DOI] [PubMed] [Google Scholar]
- 12.Suarez-Rodriguez MC, et al. MEKK1 is required for flg22-induced MPK4 activation in Arabidopsis plants. Plant Physiol. 2007;143:661–669. doi: 10.1104/pp.106.091389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichimura K, et al. (MAPK Group) Mitogen-activated protein kinase cascades in plants: a new nomenclature. Trends Plant Sci. 2002;7:301–308. doi: 10.1016/s1360-1385(02)02302-6. [DOI] [PubMed] [Google Scholar]
- 14.Guo J, Chen J-G. RACK1 genes regulate plant development with unequal genetic redundancy in Arabidopsis. BMC Plant Biol. 2008;8:108. doi: 10.1186/1471-2229-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J-F, et al. Comprehensive protein-based artificial microRNA screens for effective gene silencing in plants. Plant Cell. 2013;25:1507–1522. doi: 10.1105/tpc.113.112235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witzel F, Maddison L, Blüthgen N. How scaffolds shape MAPK signaling: what we know and opportunities for systems approaches. Front. Physiol. 2012;3:475. doi: 10.3389/fphys.2012.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahme LG, et al. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 18.Liberati NT, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motley ST, Lory S. Functional characterization of a serine/threonine protein kinase of Pseudomonas aeruginosa. Infect. Immun. 1999;67:5386–5394. doi: 10.1128/iai.67.10.5386-5394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker JE, Barber CE, Mi-jiao F, Daniels MJ. Interaction of Xanthomonas campestris with Arabidopsis thaliana: Characterization of a gene from X. c. pv. raphani that confers avirulence to most A. thaliana accessions. Mol. Plant-Microbe Interact. 1993;6:216–224. doi: 10.1094/mpmi-6-216. [DOI] [PubMed] [Google Scholar]
- 21.Djonović S, et al. Trehalose biosynthesis promotes Pseudomonas aeruginosa pathogenicity in plants. PLoS Pathog. 2013;9:e1003217. doi: 10.1371/journal.ppat.1003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prentki P, Krisch HM. In vitro insertional mutagenesis with a selectable DNA fragment. Gene. 1984;29:303–313. doi: 10.1016/0378-1119(84)90059-3. [DOI] [PubMed] [Google Scholar]
- 23.Hirsch AM, et al. Rhizobium meliloti nodulation genes allow Agrobacterium tumefaciens and Escherichia coli to form pseudonodules on alfalfa. J. Bacteriol. 1984;158:1133–1143. doi: 10.1128/jb.158.3.1133-1143.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng Z, Duan J, Hao Y, McConkey BJ, Glick BR. Identification of bacterial proteins mediating the interactions between Pseudomonas putida UW4 and Brassica napus (canola) Mol. Plant-Microbe Interact. 2009;22:686–694. doi: 10.1094/MPMI-22-6-0686. [DOI] [PubMed] [Google Scholar]
- 25.Smith AW, Iglewski BH. Transformation of Pseudomonas aeruginosa by electroporation. Nucleic Acids Res. 1989;17:10509. doi: 10.1093/nar/17.24.10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J-G, et al. Xanthomonas T3S effector XopN suppresses PAMP-triggered immunity and interacts with a tomato atypical receptor-like kinase and TFT1. Plant Cell. 2009;21:1305–1323. doi: 10.1105/tpc.108.063123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curtis MD, Grossniklaus U. A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol. 2003;133:462–469. doi: 10.1104/pp.103.027979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee L-Y, Fang M-J, Kuang L-Y, Gelvin SB. Vectors for multi-color bimolecular fluorescence complementation to investigate protein-protein interactions in living plant cells. Plant Methods. 2008;4:24. doi: 10.1186/1746-4811-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuo J, Niu QW, Chua NH. Technical advance: an estrogen receptor-based transactivator XVE mediates highly inducible gene expression in transgenic plants. Plant J. 2000;24:265–273. doi: 10.1046/j.1365-313x.2000.00868.x. [DOI] [PubMed] [Google Scholar]
- 30.Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- 31.Engel LS, Hill JM, Caballero AR, Green LC, O’Callaghan RJ. Protease IV, a unique extracellular protease and virulence factor from Pseudomonas aeruginosa. J. Biol. Chem. 1998;273:16792–16797. doi: 10.1074/jbc.273.27.16792. [DOI] [PubMed] [Google Scholar]
- 32.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 33.Gentleman RC, et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clay NK, Adio AM, Denoux C, Jander G, Ausubel FM. Glucosinolate metabolites required for an Arabidopsis innate immune response. Science. 2009;323:95–101. doi: 10.1126/science.1164627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer D, Lauber E, Roby D, Arlat M, Kroj T. Optimization of pathogenicity assays to study the Arabidopsis thaliana - Xanthomonas campestris pv. campestris pathosystem. Mol. Plant Pathol. 2005;6:327–333. doi: 10.1111/j.1364-3703.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 36.Yoo SD, Cho YH, Sheen J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protocols. 2007;2:1565–1572. doi: 10.1038/nprot.2007.199. [DOI] [PubMed] [Google Scholar]
- 37.Li J-F, Bush J, Xiong Y, Li L, McCormack M. Large-scale protein-protein interaction analysis in Arabidopsis mesophyll protoplasts by split firefly luciferase complementation. PLoS One. 2011;6:e27364. doi: 10.1371/journal.pone.0027364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J-F, Park E, von Arnim AG, Nebenfuhr A. The FAST technique: a simplified Agrobacterium-based transformation method for transient gene expression analysis in seedlings of Arabidopsis and other plant species. Plant Methods. 2009;5:6. doi: 10.1186/1746-4811-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King SRF, et al. Phytophthora infestans RXLR effector PexRD2 interacts with host MAPKKKε to suppress plant immune signaling. Plant Cell. 2014;26:1345–1359. doi: 10.1105/tpc.113.120055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.