

Abstract
Obstructive biological barriers limit the transport and efficacy of cancer nanotherapeutics. Creative manipulation of tumor microenvironment provides promising avenues towards improving chemotherapeutic response. Such strategies include the use of mechanical stimuli to overcome barriers, and increase drug delivery and therapeutic efficacy. The rational use of gold nanorod-mediated mild hyperthermia treatment (MHT) alters tumor transport properties, increases liposomal gemcitabine (Gem Lip) delivery and anti-tumor efficacy in pancreatic cancer CAPAN-1 tumor model. MHT treatment led to a 3-fold increase in accumulation of 80-nm liposomes and enhanced spatial interstitial distribution. I.v. injection of Gem Lip and MHT treatment led to a 3-fold increase in intratumor gemcitabine concentration compared to chemotherapeutic infusion alone. Furthermore, combination of MHT treatment with infusion of 12 mg/kg Gem Lip led to a 2-fold increase in therapeutic efficacy and inhibition of CAPAN-1 tumor growth when compared to equimolar chemotherapeutic treatment alone. Enhanced therapeutic effect was confirmed by reduction in tumor size and increase in apoptotic index where MHT treatment combined with 12 mg/kg Gem Lip achieved similar therapeutic efficacy as the use of 60 mg/kg free gemcitabine. In conclusion, we demonstrated improvements in vivo efficacy resulting from MHT treatment that overcome transport barriers, promote delivery, improve efficacy of nanomedicines.
Keywords: Biological barriers, cancer nanotherapeutics, cancer therapy, mild hyperthermia, liposomal gemcitabine
1. Introduction
Efficacy of cancer nanotherapeutics is often limited by tumor microenvironment that acts as barriers to effective therapeutic transport [1, 2]. These barriers hinder effective delivery of nanomedicines through vessel walls and penetration across tissue matrix, limiting drug reach to cancer cells [3, 4]. Creative strategies to manipulate the tumor microenvironment and overcome barriers are studied as means to improve chemotherapeutic efficacy [1]. Such efforts include the application of external mechanical stimulus to improve tumor mass transport properties [5]. As an example, photodynamic therapy (PDT) aided by laser irradiation created vascular fenestrations and increased permeability which led to improvements in therapeutic delivery and efficacy in patients with age-related macular disorder (AMD) [6]. Hyperthermia treatment is also another example of an external mechanic stimulus that has been used to kill obstructing cells and thereby overcome biological barriers [7]. Majority of studies in this area have largely been confined to the use of thermal ablation to synergize therapeutic efficacy through cell killing [8], sensitizing cells to chemotherapy [9], radiotherapy [10], enhancing drug accumulation [11], and by increasing tumor re-oxygenation [12]. Thermally ablative conditions are attained when temperature reaches 46–50°C [9] and causes cell death through DNA denaturation [13]. However, the treatment also causes irreversible vascular occlusive damage that hinder delivery of subsequent intravenous therapeutics. This is a major clinical drawback as patients are often given multiple cycles of chemotherapy infusions for which intact vasculature is necessary [14].
The potential benefits of using sub-ablative thermal treatment as tumor transport modulator that improves delivery and efficacy is not well explored. In this work, we evaluated the use of non-damaging mild hyperthermia treatment (MHT) to modulate tumor transport and enhance therapeutic efficacy of liposomal gemcitabine (Gem Lip, 80 nm) in pancreatic ductal adenocarcinoma (PDAC). PDAC is an aggressive disease with a measly 6% 5-year survival rate [15]. Gemcitabine (Gem, 2’,2’-difluoro-2’-deoxycytidine, dFdC) alone or in combination with radiation is the primary PDAC treatment but is negatively affected by poor delivery and rapid Gem metabolism that limits efficacy and reduces overall survival rates [16]. Gem is rapidly metabolized by deoxycytidine deaminase (present in blood) into 2',2'-difluoro-2'-deoxyuridine hydrochloride (dFdU) [17]. Motivated by our recent demonstration showing that MHT treatment increases the permeability of tumor vascular endothelium and enhances trans-vascular transport of macromolecules [18], we studied the use of MHT treatment to improve Gem Lip transport and increase efficacy in pancreatic CAPAN-1 tumor model. We designed PEGylated liposomal carriers with transition temperature higher than 42°C to ensure stability under MHT treatment conditions in order to gain insights into the transport alteration and separate out the therapeutic gains from MHT treatment alone. This is in contrast to the previous use of MHT treatment to trigger drug release from heat-sensitive Low Temperature Sensitive Liposomes (LTSLs) to improve therapeutic efficacy [19].
To test this hypothesis, we generated temporary MHT treatment conditions (without damaging vessels) and evaluated preferential accumulation and spatial distribution of liposomal gemcitabine carriers. We then assessed effects of MHT treatment on intratumor Gem accumulation and gain in therapeutic efficacy using pancreatic CAPAN-1 tumor models. Localized MHT treatment was generated by near infrared [20] irradiation of tumor pre-injected with gold nanorods (GNRs, 44 × 11 nm). PEGylated GNRs was localized in tumor microenvironment via enhanced permeation and retention (EPR) strategy and then used to generate rapid MHT treatment upon NIR irradiation [21]. GNRs were chosen due to their high conversion efficiencies [22] which means that a small amount of particles and low laser power was required to achieve rapid selective heating. While previous efforts have demonstrated the use of GNR-mediated photothermal therapy to improve accumulation of thermo-sensitive polymers [23], these strategies have caused irreversible vascular damage and do not account for the transport contributions associated with MHT treatment. Other previous studies have used water-bath heating to produce MHT conditions, the rate of heating is very slow [24] and uncontrolled [25]. We applied prolonged MHT treatment (20 min) to overcome thermo-tolerance known to occur in cancer cells [26] while staying below threshold settings reported to cause permanent vessel occlusive damage that arise once temperature reaches 43°C and maintained for 240 min [27]. Our study demonstrated that well-timed MHT treatment effectively ‘prime’ tumor microenvironment, increase delivery and intratumor concentration of Gem Lip, and thereby enhanced therapeutic efficacy. MHT treatment provides attractive strategy to overcome barriers, increase intratumor drug concentration to achieve increased therapeutic efficacy while reducing systemic dosing.
2. Materials and Methods
2.1 Ethics Statements
All animal experiments were approved by The Houston Methodist Institutional Animal Care and Use Committee guidelines (Houston, TX) and were performed in accordance and under IACUC-approved protocols AUPs 1010-0029 & 1210-0043 and IVM imaging was covered under protocol AUP 0611-0032.
2.2. Materials
Ammonium sulfate and formamide were purchased from Sigma-Aldrich (St. Louis, MO); fluorescein isothiocyanate (FITC-) and tetramethyl rhodamine isothiocyanate (TRITC-) labeled dextran dyes (70-kDa); phosphate buffer saline solution (PBS buffer, pH 7.4) were purchased from Invitrogen (Carlsbad, CA); TUNEL assay apoptosis detection kit from Roche (Indianapolis, IN); 5kDa Methyl-PEG5K-thiol was purchased from Laysan Bio (Arab, AL). 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC, Tm = 41°C), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC, Tm = 55°C) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPEmPEG2000) were purchased from Avanti Polar (Avanti Lipids, Inc., Alabaster, AL); polycarbonate membrane filters were purchased from Corning Inc. (Corning, NY); cellulose membranes with a molecular weight cut-off of 10 kDa were purchased from Spectra/Por-Membranes (Spectrum Laboratories, Inc., CA, USA); lissamine™ Rhodamine B 1,2-Dihexadecanoyl-sn-Glycero-3-Phosphoethanolamine and Triethylammonium Salt (rhodamine-DHPE) were obtained from Molecular Probes® (Life Technologies, Grand Island, NY). Tetrahydrouridine (THU) was purchased from Calbiochem (Billerica, MA). Gemcitabine hydrochloride salt [28] was purchased from LC laboratories (Woburn, MA); dFdU (difluorodeoxyuridine), 13C15N2-dFdU (heavy-dFdU) and 13C15N2-gemcitabine (heavy-Gem) were synthesized and purchased from Organomed Corporation (Coventry, RI). Heavy Gem and heavy-dFdU were used as stable-isotope labeled internal standards for isotope-dilution used for LC-MS mass spectrometry. Acetonitrile, methanol, and ammonium acetate were Optima-grade and purchased from Fisher Scientific (Fisher Science Education, Hanover Park, IL).
2.3. Preparation and characterization of fluorescent and Gemcitabine-loaded liposomes
Thermally stable DSPC liposomes were prepared and used to evaluate effect of MHT treatment on tumor transport, delivery and tumor distribution, and size boundaries for MHT-assisted tumor priming. To allow facile analyses, rhodamine-DHPE-labeled liposomal nanoparticles were prepared and used to assess intratumor accumulation and spatial distribution. Rhodamine-DHPE was added to phospholipids during the preparation of lipid film [29] which was designed to possess high thermal stability to ensure that carriers remained stable under MHT tumor treatment. Three different sizes of DSPC liposomes (100, 80, and 49 nm) were formulated and analyzed to determine optimal size for tumoritropic accumulation.
DSPC liposomes were formulated following modification of previous protocols [30]. Briefly, DSPC/Chol/DSPEmPEG2000 (6:3:1 molar ratio) were dissolved in chloroform/methanol mixture (3:1 v/v) to given 40 mg/mL of phospholipids in a round-bottom flask. The organic solvent was removed by rotary evaporation (Heidolph Instruments, Germany) under vacuum to obtain a thin lipid film. Residual organic solvents were further removed by overnight evaporation at room temperature. The lipid film was then hydrated and resuspended in PBS buffer (2 mL) by using a vortex-mixer (700 rpm for 3 min) and water bath (60°C for 3 min). Multilamellar DSPC liposomes were then extruded through a stainless steel extrusion device equilibrated at 60°C (Lipex Biomembranes, Northern Lipids Inc., Vancouver, BC, Canada). The mixture was consecutively extruded through paired polycarbonate membrane filters (800, 400, 200, 100, 80, 50 and 30 nm) for 10 passages. DSPC liposomes of three different sizes (100, 80 and 49 nm) were collected from selected polycarbonate membrane filters (100, 50 and 30 nm, respectively) and used for this study.
Gemcitabine was then encapsulated in DSPC liposomes (Gem Lip) where selected size configuration was based on observed optimal tumor accumulation. Briefly, PEGylated DSPC liposomes were extruded (using similar to previous protocol) and gemcitabine was entrapped within liposomal by pH gradient remote loading method [31]. After extrusion, DSPC liposomes was mixed with 1 mL gemcitabine solution (6 mg/mL) and incubated overnight at 60°C and dialyzed against 0.9% NaCl to remove un-entrapped gemcitabine and ammonium sulfate using 8,000-kDa cellulose ester membrane (Spectrapor, Rancho Dominguez, CA). Purified Gem Lip was then concentrated in a vacufuge (Eppendorf, San Diego, CA) and then reconstituted to 2 mg/mL of gemcitabine in sterile PBS. Gemcitabine concentration was determined by HPLC [32]. The physical-chemical properties of rhodamine-labeled liposomal nanoparticles and Gem Lip were characterized by evaluating average size, polydispersity index distribution (PDI), and zeta potential (ZP) as previously described [33]. The particle size was further characterized by transmission electron microscopy (TEM) and Gem release kinetics from liposomal carriers were evaluated using PBS buffer used as receptor medium according to experimental protocol previously reported [33].
2.4. Initiation of localized tumor mild hyperthermia
To produce localized MHT treatment, GNRs (44 × 11 nm) with strong optical absorption coefficients [34] were fabricated and PEGylated to improve systemic circulation and tumor accumulation as recently described [18]. PEGylated GNRs coated with this strategy showed longer in vivo circulation half-life (~18 h) compared to surfactant stabilized GNRs (≤ 1 h) [18]. Tumor-bearing mice were injected with PEG-coated GNRs at a concentration of 10 mg/kg (200 µL, 1.4 × 1011 particles/mL) by i.v. injection. The particles were allowed to passively accumulate at the tumor site. Tumor specimens were irradiated with a pulsed 810-nm NIR laser (~1W/cm2, 1Hz, Angiodynamics, UK) to attain MHT conditions at 42°C which was capably sustained for 20 min. Temperature increase was monitored using IR thermographic camera (FLIR, Thermacam S60) used to determine tumor surface temperature distribution while microprobes (Oxford Optronics, Oxford, UK) placed at different regions of tumor (core, surface, and base) were used to measure intratumor temperature distribution and homogeneity. For combinational therapy, i.v. injections of GNRs were given on a weekly basis to ensure sufficient GNRs were localized in tumor microenvironment.
2.5. Assessment of liposomal particle tumor accumulation by IVM
The effect of mild hyperthermia on liposomal extravasation and accumulation in CAPAN-1 xenografts was assessed by intravital microscopy (IVM). Tumors were exposed using a skin-flap procedure as previously described [35]. Live animals were imaged on an upright Nikon A1R MP-ready (Nikon, Mellville, NY) laser scanning confocal microscope platform equipped with a resonance scanner, isoflurane anesthesia system, heated stage, and custom coverslip mounts. Anesthetized mice were administered with fluorescent dextran via retro-orbital injection (10 mg/kg, 50 µL PBS) and then injected with 50 µL of rhodamine-labeled DSPC liposomal nanoparticles of three different sizes (100, 80 and 49 nm). The extent of accumulation and distribution was assessed by IVM imaging where FITC dextran dye was used to delineate the vascular region. The retro-orbital route of injection was chosen over tail-vein injection due to its accessibility and reproducibility for quantitative imaging experiments. Fluorescent dyes were acquired with the following settings: FITC-labeled dextran was excited at 488 nm while rhodamine-labeled liposomes were excited at 561 nm and collected at band-pass filters widths of 30–50 nm centered at 525 nm for fluorescent dextran and 579 nm for liposomes. 512 × 256 bit two-channel images of tumors were acquired at 30–60 frames per second from the time of dye injection using 4× objective lens with a pinhole of 1.0 Airy units. Randomly selected non-overlapping regions of interest (ROIs) were selected to minimize local phototoxicity and compensate for tissue heterogeneity and imaged at 10 sec interval. The camera and acquisition settings were kept constant between different treatment groups.
For whole tumor qualitative analyses, we evaluated the amount of dye after MHT treatment via spectrofluorometric analyses. At 5 h after MHT treatment, animals were injected with rhodamine-labeled liposomes (50 µL, 1 mg/mL in PBS buffer) which were allowed to circulate and extravasate into tumor for 1 h (n = 8). The duration of dye circulation between treatment groups was kept constant (i.e. dye was injected at 5 h time-point and allowed 1 h to circulate). This was done to correct for circulating PEGylated liposomes in vessels not cleared prior to analyses. Animals were sacrificed and tumors excised, weighed, and homogenized in formamide (200 µL/mg of tumor) [18]. The amount of dextran dye in tumor homogenate was quantified spectrofluorimetrically in a microplate reader (Synergy H4 hybrid reader, BioTek Instruments Inc., Winooski, VT) using 550/570 nm emission/excitation settings. Rhodamine fluorescence was correlated to amount of liposomes using a standard curve.
2.6. Image analyses and particle quantification
The average number of liposomal particles localized in the interstitial space were enumerated from video stills acquired using Nikon NIS element AR software as described previously. In the selected FOVs, a green vessel tracer was used to delineate the vascular region from the interstitial space which was devoid of green (FITC) tracer. The extent of particle extravasation from the vessel walls was determined using an automated intensity profile function (Nikon Elements). For instance, particles located in the interstitial space with minimal vessel tracer were profiled by lowest FITC intensity signal while particles trapped around the vessel walls showed highest intensity of both FITC and rodhamine signals. Cross-sectional intensity profiles of particles were enumerated and plotted as function of distance from nearest vessel wall which was defined by areas with highest FITC signal. Fluorescent settings of FITC and rhodhamine channels were kept same across experimental groups.
2.7. Combinational treatment of mild hyperthermia and chemotherapy
Pancreatic CAPAN-1 tumors were generated in nu/nu mice (Charles River Laboratories, Wilmington, MA) by a one-time injection of 5 × 105 CAPAN-1 cells in hind right flank (ATCC, Manassas, VA). Once tumors reached 5–7 mm in diameter (290–350 mm3) animals were randomly divided into eight different groups (n = 8) and combinational treatment was initiated where MHT treatment was given followed by i.v. injection of Gem Lip or free Gem. Based on our previous findings that showed that MHT treatment transiently increased tumor properties peaking within 24 h after treatment [18], we designed the experiments to test relevance of increasing therapeutic delivery within the suggested time-window as shown in schematic 1. Pancreatic tumor-bearing mice were treated with sustained MHT treatment (20 min) and then infused with Gem 5 h after treatment. Gem Lip was given in doses of 6 and 12 mg dFdC/kg (40 µmol/kg) using carriers containing a total lipid concentration of 2.17 mmol/kg. Therapeutic efficacy was compared to efficacy achieved by free Gem using doses of 6 and 60 mg dFdC/kg of animal. The combination treatment was given twice weekly for three weeks and tumor size monitored for another seven weeks. Tumor growth was measured (caliper) and used to calculate volume by (a2 × b)/2 where, a, represents the length, and, b, represents the width. At the termination of therapy, the animals were sacrificed and tumors extracted for histological examination.
Schematic 1. Illustration of combinational MHT treatment strategy.
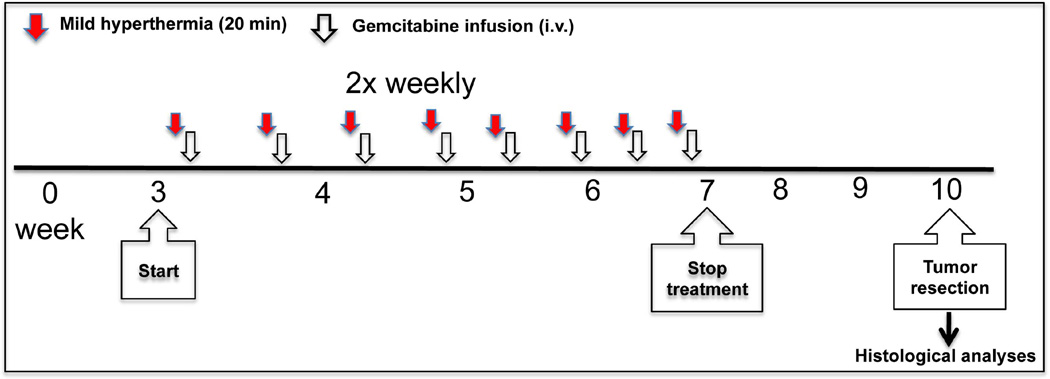
Tumor was given MHT treatment and followed by i.v. injection of gemcitabine (Gem Lip or free Gem) twice weekly for a total of 10 treatments and terminated at 7 week. Tumor growth was monitored until 10 week.
2.8. MHT effect on intratumor gemcitabine accumulation
Mice bearing two bilateral CAPAN-1 tumors (on left and right flanks) were established by one-time injection of 5 × 105 cells (100 µL, HBSS). Once tumors reached 290–350 mm3 in volume, one of the tumors was given MHT treatment and then given i.v. injection of 60 mg/kg of either Gem Lip or free Gem, chosen due to previously demonstrated inhibitory effect on pancreatic cancer cells [17]. The drug was allowed 24 h to circulate and accumulate in tumor before animals were euthanized and tumors excised, snap-frozen, and prepared for LC-MS/MS analyses. Each tumor was weighed, chilled in liquid nitrogen, and pulverized to produce homogenous powdered tumor samples. A small fraction of this sample (~ 30 mg) was re-suspended in water, sonicated, and extracted in acetonitrile. Gem concentration in tissue was determined by LC-MS/MS analyses.
A parallel study evaluated and compared plasma Gem concentration with the use of Gem Lip versus free Gem. At various time-points after i.v. injection (up to 24 h), 25 µL of blood was drawn by retro-orbital bleeding. Gem in plasma was extracted and prepared for LC-MS analyses to determine time-dependence and relationship with tumor accumulation. The Gem concentration in tumor tissue and blood samples was determined by LC-MS/MS analyses using a MRM methodology on a Waters Acquity UPLC system coupled to a Waters Xevo TQ mass spectrometer under control of MassLynx (v4.1; Waters Corp.) software. A detailed protocol used for sample preparation and Gem analyses is provided in the Supplementary information.
2.9. Preparation of samples for LC/MS/MS analyses
2.9.1. Tissue and blood preparation
Approximately ~ 30 mg of pulverized tumor tissue was weighed out and re-suspended in 3 volumes of MilliQ water containing 40 fmol/µL of heavy Gem, 1000 fmol/µL heavy-dFdU, and 1 mg/mL of THU. Samples were sonicated for (6 × 30 sec at 85% power level, 4°C), centrifuged for 10 min (14,000 ×g), and supernatant was recovered and collected in a new tube. Acetonitrile (3:1, v/v) were added and vortexed thrice at a 30-sec interval allowing 15 min of rest. Suspension was centrifuged at 14,000 ×g for 10 min and supernatant was lyophilized to dryness and re-constituted in water containing 0.1% (w/v) of ammonium acetate (3 volumes the original sample weight). Blood samples were processed in a similar protocol where 25 µL of sample was diluted in 3 volumes of MilliQ water and similarly processed for LC-MS/MS analyses using aforementioned method. A portion of processed supernatant was diluted 2-fold in the same buffer and transferred to a 96 well plate and 2.5 µL was injected into LC-MS/MS. Each injection contained 100 fmol heavy Gem and 2500 fmol heavy dFdU (equivalent to 0.42 mg of the original weight).
2.10. TUNEL staining and apoptosis examination
The effect of combinational therapy was further evaluated by deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay following the manufacturer’s protocol [36]. Tumor tissue obtained at the end of therapeutic experiments were fixed in formalin, dried in ethanol, cut into 10-µm-thick slices, and prepared for TUNEL assay. TUNEL-positive cells were identified by brown staining of nuclei which were counted and compared across treatment groups. Ten independent microscopic field-of-views were manually quantified and used for statistical comparisons.
2.11. Statistics
Statistical analysis was performed using a Wilconox (JMP) software for unpaired t-test (between two groups) and one-way analysis of variance (ANOVA) for comparisons of three or more groups. A p < 0.05 value was considered statistically significant.
3. Results
3.1. Generation of sustained tumor mild hyperthermia conditions
The feasibility of using near-infrared [20] irradiation to generate sustained tumor mild hyperthermia was first evaluated. We have recently fabricated GNRs (44 × 11 nm) with strong optical absorption and further modified them by surface PEGylation which increased circulation half-life to 18 h compared to ≤ 1 h for surfactant CTAB- stabilized GNPs [18]. PEGylated GNRs were systemically administered and allowed 72 h to passively accumulate in tumor microenvironment. ICP-MS analyses revealed that 103 µg of GNRs/g of tumor (13% w/w of injected dose) accumulated in tumor while majority of particles were eliminated from circulation by splenic clearance (Figure 1 A). Strong near-infrared absorbance means that small quantities of GNRs are needed to generate MHT conditions under NIR laser irradiation (Supplementary, Fig S1). After GNR localization, CAPAN-1 tumors were exposed to laser irradiation which resulted in a rapid rise in temperature for the group injected with GNRs as compared to the control group, as is illustrated by thermal camera surveillance (Figure 1 B). Further evaluation of intratumor temperature changes using microprobes (at core, surface and tumor base) showed that MHT treatment strategy caused temperature increase that permeated the entire tumor mass and relatively evenly distributed (Figure 1 C). The NIR heating strategy was rapid, elevating tumor temperature to ~ 42°C at the base within 90 sec of initiation (Figure 1 C).
Figure 1. Gold-nanorod mediated Mild hyperthermia (MHT) treatment led to sustained heating but caused minimal cellular damage compared to ablative hyperthermia therapy.

A) in vivo bio-distribution of PEGlylated gold nanorods showing tumor accumulation of 13% of injected dose after 72 h of circulation; B) thermographic images showing selective and homogenous heating of tumor receiving 10 mg/kg GNR injection; C) intratumor temperature profile in various regions of tumor showing permeated MHT treatment; D) MHT caused no significant cell apoptosis at 24 h after treatment as shown by TUNEL assay showing minimal damages (few brown spots) while ablative hyperthermia(50°C) caused extensive cell apoptosis as indicated by brown staining.
We chose to create MHT conditions at 42°C so as to stay above liposomal transition temperature while the treatment was sustained for 20 min to overcome thermo-tolerance associated with cancer treatments. We reasoned that treatment below thermal ablation thresholds (43°C for 240 min) would negate destructive effects of hyperthermia but positively affect tumor transport properties through vessel permeabilization and increased perfusion. TUNEL assay staining confirmed that MHT treatment caused minimal cell apoptosis when evaluated 24 h after treatment; while ablative hyperthermia (~ 50°C) increased apoptotic cell population relative to tumor with no treatment (Figure 1 D). These results confirmed that MHT treatment alone would not cause significant cell damage nor therapeutic effect.
3.2. Physical-chemical characterization demonstrated thermally stable liposomal nanoparticles
Liposomal nanoparticles that can withstand heat treatment were fabricated in order to obtain drug carriers suitable to study and delineate transport-associated effects caused by MHT treatment. DSPC phospholipids that have high transition temperature (Tm = 55°C) were chosen to avoid gel-to-liquid crystal rearrangement (transition) induced by heat treatment. Liposomes of various sizes were fabricated by extrusion through polycarbonate membrane filters of differing sizes. We assessed stability of DSPC liposomal nanoparticles using a temperature-controlled water-bath (50°C for 20 min). Their stability was essential to ensure that any enhancements in combination therapy studies and treatment would primarily be attributable to transport alterations and not merely from heat-induced liposomal degradation. The results showed that heating up to 50°C did not significantly change particle size and polydispersity index (PDI), suggesting thermal stability under MHT conditions (Figure 2). In particular, DSPC liposomes are stable both at 37°C and 50°C with no change in size (Figure 2 A). This is in contrast to DPPC liposomes, fabricated from lipids with lower Tm (~ 41°C), which are stable at 37°C but not after heat treatment (50°C). The average size of DPPC liposomes increased 2-fold after heat treatment (Figure 2 B). Further analyses showed that the PDI of DSPC liposomes remained unchanged after heat treatment whereas the DPPC became unstable upon treatment, resulting in 3-fold increase in PDI (Figure 2 C, D). The average size of 80-nm DPPC liposomes increased from 80 nm to 152 nm while the PDI increased from 0.2 to 0.6 after heat treatment (Figure 2 B, D). Significant changes in physical-chemical properties for DPPC liposomes were apparent but not statistically different for DSPC liposomes (Figure 2). Further TEM characterization confirmed the size and thermal stability of DSPC liposomal carriers where there was no significant difference in size when the temperature was raised from 37°C to 50°C (Supplementary information, Figure S3). Conversely, we observed size increase for 80-nm DPPC liposomes arising from temperature elevation from 37°C to 50°C. Moreover, the shape of liposomes are slightly modified showing a partial fusion of nanoliposomes and consistent with the observed increase in size distribution (Figure 2 D).
Figure 2. Characterization of DSPC liposomes showing un-altered size and PDI after treatment in 50°C water-bath and suggest thermal stability under MHT conditions.

A) DLS size measurement show no significant increase is particle size after heat treatment (50°C) versus liposomes at 37°C; B) DPPC liposomes in contrast shows a 2- to 3-fold increase in size after heat treatment relative to control; C) DSPC liposomes stability was also confirmed by un-altered polydispersity index (PDI) after heat treatment; D) as compared to 3-fold increase in PDI for DPPC liposomes. Error bars represent triple sample readings.
We further evaluated the in vitro release kinetics of Gem from 80-nm DSPC and DPPC nanoliposomes where we demonstrated that ~ 55% of Gem is gradually released into sink buffer within 48 h, for both carriers (Supplementary Information, Figure S4). Our findings were consistent with previous literature studies which demonstrated that the formation of a gel-like structure from ammonium sulfate solution in the liposomal core of nanoliposomes retards the release of Gem [37]. This phenomenon has been shown to be independent of particle size of liposomes as previously reported for Doxil® medicine [38]. The DSPC liposomes were subsequently used to study enhanced tumoritropic delivery when combined with MHT treatment.
3. MHT treatment improves liposomal tumor delivery
Next, we evaluated and compared the preferential tumor accumulation of the three different liposomal nanoparticles after MHT treatment. This was conducted to determine optimal size carrier with which to study combinational therapy. Whole tumor fluorometric analyses showed that MHT treatment increased accumulation in a size-dependent fashion. At 5 h after MHT treatment, there was over 3-fold increase in accumulation for 49-nm liposomes, ~ 2.5-fold increase for 80-nm drug carriers and no statistically significant increase in accumulation for 100-nm liposomal carriers (Figure 3). These suggested that there is a threshold size of carrier particle that can be enhanced by MHT treatment and would dictate the particle size chosen for drug delivery. Since the 80-nm liposomes showed significant enhancement in accumulation compared to the control group and provided a suitable aqueous volume for drug encapsulation, we evaluated their preferential tumor interstitial accumulation and spatial distribution after MHT treatment using IVM. IVM imaging showed no discernible difference in the levels of liposomal accumulation at 1 h after MHT treatment as majority of fluorescent liposomes remained in circulation and minimal transvascular extravasation was observed (Figure 4). After 5 h of circulation, there was significantly higher liposomal accumulation in tumors that received MHT treatment relative to untreated group (Figure 4 B). To determine effect of mild hyperthermia on particle delivery and whether there was enhancement in liposomal extravasation resulting from treatment, we analyzed the locations of rhodamine-labeled particles relative to neighboring vessels defined by green FITC tracer. Analyses of video stills revealed that MHT treatment enhanced particle extravasation distribution in the interstitial space, where bright red fluorescent particles were dispersed an average of 21 ± 2.5 µm away from the nearest vessels whereas the majority of liposomal particles in the untreated group were dispersed 3.7 ± 1.9 µm from the nearest vessels (Figure 4 C). Furthermore, tumors receiving MHT treatment displayed a higher particle frequency which were mostly spread out throughout the interstitial space; while tumors in control group showed particles with lower particle frequency and mostly located in or around the vessel walls (Figure 4 C). Quantitative fluorescent analyses further revealed that MHT treatment increased the overall accumulation when analyzed 5 h after MHT treatment (Figure 4 D). These findings suggested a MHT treatment as a potent strategy to prime tumor microenvironment and propel therapeutics closer to cancer cell milieu.
Figure 3. MHT treatment increases tumoritropic accumulation of DSPC liposomes in a size-dependent manner.

Extent of liposomal accumulation was examined at 5 h after MHT and 1 h of in vivo circulation before whole tumor excision and digestion. Quantitative fluorescent analyses (521/585 nm Ex/Em) revealed significant accumulation for 49-nm (3-fold) and 81-nm (2-fold) nanoliposomes and showed no significant enhancement for 100-nm nanoliposomes. Statistical significance is denoted by ** p < 0.002, * p < 0.024 for n = 8 per treatment.
Figure 4. MHT treatment enhances accumulation and spatial distribution of nanoliposomes.

A) Intravital microscopy images obtained 1 h after MHT treatment showing nanoliposomes (red); the liposomal accumulation in treatment group was indistinguishable from control group where nanoparticles remained in circulation (green, vessels; B) while analyses at 5 h after treatment revealed significant nanoparticle accumulation (red) in tumor interstitial space compared to untreated group; C) further analyses showed increased nanoparticle spatial distribution with tumor receiving MHT treatment showing liposomal dispersion of 21 ± 2.5 µm away from the nearest tumor vessels versus 3.7 ± 1.9 µm for untreated tumors; D) MHT treatment showed 3-fold increase in accumulation of nanoparticles at 5h and no significant enhancement at 1 h after treatment.
3.4. MHT treatment enhances therapeutic efficacy of liposomal gemcitabine
To evaluate effects of combining MHT treatment with Gem on therapeutic efficacy on CAPAN-1 tumor growth, nu/nu tumor mice bearing were divided into eight treatment groups (n = 8). The use of NIR irradiation to generate sustained MHT has been demonstrated in Figure 1 and its use to enhance delivery and spatial localization is shown in Figure 3. In this experiment, effect of Gem Lip (2 mg/mL, 80 nm) combined with MHT treatment was evaluated based on ability to inhibit tumor growth while the concentration of Gem in nanoliposomal formulations was determined by HPLC analyses using a standard curve (Supplementary information, Figure S5). Once tumor volumes reached ~ 300 mm3, the treatment was initiated by giving MHT treatment followed by i.v. injection of Gem Lip or free Gem after 5 h. The treatment was given twice weekly and continued for 4 weeks. Tumor growth curves of CAPAN-1 tumors are shown in Figure 5. The vehicle control (DSPC liposomes alone) and MHT treated groups showed the fastest tumor growth and little inhibition in growing (Figure 5 A). This confirmed that empty liposomes or MHT treatment alone caused no therapeutic or tumor inhibitory effect. After 4 treatments (day 14), combinational therapy showed notable reduction in tumor growth as compared to the control groups. More significantly, combinational therapy showed increased tumor inhibition after 6 treatments (day 28) where groups treated with MHT alone continued to grow rapidly. This trend continued even after cessation of chemotherapeutic treatment (day 29–49) where group receiving MHT and Gem Lip showed drastic inhibition in tumor growth (Figure 5 A). While treatment with Gem Lip alone also inhibited tumor growth, combinational therapy was more efficacious (Figure 5 A). MHT treatment improved the efficacy of 12 mg/kg Gem Lip, achieving similar efficacy as the use of 60 mg/kg free Gem which is a 5-fold reduction in dosing. In contrast, there was negligible gain in tumor inhibition when MHT treatment was combined with 60 mg/kg free Gem infusion as compared to chemotherapy alone (Figure 5 B). This may due to the fact that free Gem is rapidly deactivated and cleared from circulation as compared to liposomal formulation. Tumor weight analyses on day 49 after treatment confirmed the effect of MHT treatment on enhancement of anti-tumor effect of Gem Lip (Figure 5 C, D). Significant differences in tumor sizes observed at treatment cessation (day 28) continued through until termination of experiment (Figure 5 C, D). No statistically significant difference in tumor size was found between MHT, vehicle alone, and infusion of 6 mg/kg of Gem mono-therapy groups. There was no significant animal weight loss at any point during this therapy, suggesting no cytotoxicity from either chemotherapy or MHT treatment (Supplementary information, Figure S6). Thus, the combination therapy of MHT and Gem increased anti-tumor efficacy in CAPAN-1 tumor model.
Figure 5. MHT treatment improves Gem-dependent tumor growth inhibition and reduction in tumor weight.

A) Combinational tumor therapy using MHT and i.v. infusion of Gem Lip at 6 and 12 mg/kg over 7 weeks. Tumor receiving with Gem Lip + MHT (twice weekly) showed a 2-fold increase in tumor inhibition compared to chemotherapy alone with notable inhibition starting on day 12 and continuing through day 49. B) Growth curves shows combinational therapy using free Gem slightly reduced tumor size compared to Gem treatment alone whereas use of Gem Lip treatment significantly increased tumor inhibition and achieved 5-fold dose reduction. C) Representative images of excised tumors at day 49 showing smallest to tumor size for combination use of Gem Lip + MHT relative to mono-therapy treatment. D) Inhibition is confirmed by reduction in tumor weights on day 49 where significant reduction is observed with Gem Lip plus MHT group while no significant difference was seen between MHT only and untreated group. Seven animals were used per group. Statistical significance is denoted by ** p < 0.039, *** p < 0.0032, ** p < 0.043 for n =7 for group.
3.5. Combinational therapy increased proportion of apoptotic cell population
At the end of therapeutic experiments, we examined the extent of cell kill resulting from combinational therapy and compared it to effect of mono-therapy alone. Figure 6 A shows representative TUNEL assay micrographs from each treatment group. The combination of Gem Lip and MHT treatment led to the highest apoptotic cell population compared to mono-therapies alone (Figure 6 A). The proportion of apoptotic cell population was significantly higher than the use of Gem Lip alone. In contrast, combination of free Gem and MHT treatment showed a smaller increase in apoptotic cell population compared to treatment with free Gem alone. The assay also confirmed that MHT treatment alone caused minimal tumor damage through apoptosis (Figure 6 B). These results corroborate earlier observations regarding differences in tumor inhibition obtained from use of Gem Lip and free Gem. The difference is probably attributable to their differing in vivo pharmacokinetics and disparate sizes.
Figure 6. TUNEL assay illustrates increased apoptotic cell population from combinational use of MHT treatment with i.v. injection of Gem Lip.

A) Representative TUNEL assay images illustrating increased apoptotic cell population from combination of MHT and Gem Lip (12 mg/kg); B) Quantitative analyses show significant increased in cell apoptosis for Gem Lip + MHT compared to mono-therapies alone. Statistical significance represents apoptotic cell population over 10 representative ROIs.
3.6. MHT improved therapy through increased intratumor gemcitabine
Tumor challenge experiments revealed improvement in tumor inhibition resulting from the use of MHT treatment. To further associate these improvements to enhancement in drug delivery, we evaluated amounts of Gem present in tumor after a single of MHT treatment. Two bilateral tumors (right and left flank) were established in nu/nu mice and treated once they reached ~ 300 mm3. LC-MS/MS Gem analyses revealed that MHT treatment led to a 3-fold increase in intratumor dFdC concentration (active form of Gem) and a 4-fold increase in dFdU (Gem metabolite) when 60 mg/kg Gem Lip was i.v. injected 5 h after MHT treatment (Figure 7 A). Similar enhancements in delivery were observed for tumors receiving MHT treatment before free Gem injection. In these tumors, dFdU concentrations were significantly higher than in untreated tumors (Figure 7 B). The ratio of dFdC/dFdU present in tumor receiving MHT treatment was higher than untreated group, suggesting that treatment led to a higher concentration in the active form of Gem (Figure 7 C). While therapeutic enhancement was observed with combination treatment using free Gem, majority of drug found in tumor was dFdU, drug metabolite, which is inactivated and ineffective in inhibiting tumor growth (Figure 7 D, E). However, intratumor dFdC concentration was not statistically higher probably because free Gem is rapidly metabolized to dFdU upon injection (Figure 7 D, E). Additional pharmacokinetic studies confirmed that dFdC is rapidly converted into dFdU when administered as a free drug (free Gem) while there was a prolonged presence of dFdC when given as liposomal formulation (Gem Lip) (Supplementary information, Figure S7). In this set of experiments, MHT treatment led to significantly higher amounts of dFdU as reflected by a lower ratio dFdC/dFdU (Figure 7 F). While MHT treatment enhanced the delivery of free Gem, the drug was already converted to inactive form. This may explain why MHT treatment combination with free Gem only led to marginal improvements in therapeutic efficacy whereas MHT treatment with Gem Lip resulted in a drastic tumor inhibition.
Figure 7. MHT treatment enhances intratumor Gem concentration.

A) Increased dFdC concentration in tumors receiving MHT relative to untreated after one-time i.v. injection of 60 mg/kg of Gem Lip; B) corresponding increases was also observed for drug metabolite (dFdU) after Gem Lip injection; C) MHT treatment led to a measurable increase in the ratio of active form: inactive form (dFdC/dFdU); D) In contrast there was no significant elevations in dFdC with administration of 60 mg/kg free Gem; E) whereas majority of drug enhancement was in the form of dFdU; F) Lower ratio dFdC/dFdU resulting from significant increases in dFdU but no corresponding increases in dFdC. Statistical significance derived from (n = 13).
4.0. Discussion
Overcoming tumor microenvironment barriers that impede delivery of nanomedicines is one of the challenges toward improving therapeutic efficacy in cancer treatment [3]. To this end, various strategies are explored as means to manipulate the tumor microenvironment by pre-treating “priming” tumor microenvironment prior to therapy. These have included tumor pre-treatment with small molecules to normalize vascular perfusion and interstitial pressure [39], improvement of drug delivery through paclitaxel-induced apoptosis of vascular endothelial cells [5], and application of mechanical stimuli such as High Intensity Focused Ultrasound [40] [41] to drive nanomedicines through vascular and interstitial barriers.
In this study, we demonstrated the use of gold nanorod-mediated MHT treatment to overcome vascular and interstitial barriers, enhance delivery of thermally stable Gem Lip which ultimately increased intratumor Gem concentration and enhanced growth inhibition of pancreatic CAPAN-1 tumors. GNRs were chosen to produce MHT because they possess high-energy conversion coefficients [21] and were used in combination with low-laser irradiation to generate sustained tumor MHT conditions. NIR light is preferred because it can penetrate through several centimeters of cancer tissue allowing whole tumor priming [42]. This strategy produced a rapid site-specific MHT treatment attained within 90 sec and sustained for ~ 20 min by pulsed irradiation (Figure 1). MHT treatment was more rapid than water-bath heating which takes more than 2 min to reach desired temperatures [43]. It also allowed a more accurate and deeper actuation of tumor microenvironment properties (Figure 1 A). NIR heating caused homogeneous heat distribution that permeated the entire tumor and also minimized cellular damage was showed by TUNEL assay (Figure 1 B-D). The ability to controllably administer MHT treatment negated thermal ablative drawbacks which have been associated with the use of other heating modalities such as HIFU [41].
We then fabricated three different liposomal formulations in sizes by extrusion and tested their thermal stability and preferential tumor accumulation upon MHT treatment. DSPC liposomes showed robust thermal stability when incubated in a 50°C water-bath for 20 min (Figure 2). After thermal treatment, there were no significant changes in particle size and PDI, suggesting nanoparticle stability under MHT conditions used in this study (Figure 2 C-DSupplementary Information, Figure S3). The thermal stability of DSPC liposomes depends on their material compositions. To avoid these drawbacks, the design and use thermally stable DPSC liposomes was an infallible strategy to separate out contributions solely from tumor transport and negate any potential enhancements due to carrier degradation from thermal treatment. Further, the liposomal architectural structure provided by combination DSPC and cholesterol in liposomal composition and pH-driven loading allowed efficient and high entrapment efficiency (~ 90%) of Gem (Supplementary Information, Figure S2). Our findings are consistent with the previous reported use of DSPC lipids to fabricate robust liposomal delivery systems. For example, DPSC used to encapsulate lapatinib (chemotherapeutic) which are confined inside nanochannel delivery systems (nDS) to achieve controlled drug release [44]. Furthermore, the remote-loading procedure allowed the fabrication of nanoliposomes, which entrapped high percentage of Gem whereby ~ 55% of Gem was shown to be released within 48 h of injection (Supplementary Information, Figure S4).
In this study, we demonstrated that MHT treatment can enhance the delivery, localization, and interstitial dispersion of liposomal carriers that are less than 100 nm in size. Through whole tumor quantitative analyses, we showed enhancement in accumulation of 49-nm and 80-nm liposomal formulations. In particular, there was a 3-fold increase tumoritropic in accumulation for 80-nm DPSC liposomes compared to control (Figure 3). Further intratumor analyses revealed that MHT treatment enhanced interstitial dispersion of drug carriers, propelling delivery of DSPC liposomal 21 ± 2.5 µm away from the nearest tumor vessels. This is compared to the interstitial dispersion of 3.7 ± 1.9 µm for untreated tumors (Figure 4 C). The overall tumor localization of liposomal carriers was also enhanced relative to the control group when analyses at 5 h after treatment (Figure 4 D). These findings suggested that MHT treatment enhances drug availability by improving penetration depth of drug carriers and thereby propelling drugs [28] closer to tumor cell milieu.
MHT treatment enhances liposomal penetration by creating vascular fenestrations through which particles can extravasate and simultaneously causing increased vascular flow. In our recent work, we have demonstrated enhancement in vascular permeability after MHT treatment [18]. Through real-time intravital microscopy studies, we observed a 200% increase in extravasation of 70-kDa dextran dye which occurred at 5 h after MHT treatment and persisted for up to 24 h. Further analyses of flow dynamics showed that MHT treatment also increases tumor vascular flow, leading to up to 6-fold increase in particle concentration [45]. These observations and findings are consistent with other studies which have shown that hyperthermia treatment cause delayed inflammatory-associated responses such as enhanced permeability [46], improved perfusion and vascular shutdown [14]. The use of other parallel heating modalities such as MR-guided HIFU have been shown to be effective strategies to improve interstitial and intercellular drug delivery of thermo-sensitive liposomes [47] but are also associated with transport drawbacks such as vascular occlusion [47]. In our study, we seek to harness benefits of hyperthermia treatment while avoiding vascular occlusion that serves as further hindrance to therapeutic delivery.
Therapeutic strategy that combines MHT treatment and chemotherapeutic delivery was designed to exploit the “window-of-opportunity” characterized by enhanced tumor transport observed at 5h after treatment. We demonstrated that combinational therapy using MHT treatment used to ‘prime’ microenvironment enhanced the therapeutic efficacy of liposomal gemcitabine in pancreatic CAPAN-1 cells (Figure 5). MHT treatment combined with 12 mg/kg of Gem Lip led to a 4-fold reduction in tumor growth and reduced tumor weight, achieving efficacy equivalence of using 60 mg/kg free Gem (Figure 5). This result was further confirmed by increased proportion of apoptotic cell population in the combinational therapy group compared to control (Figure 6). The effectiveness of MHT treatment was also corroborated by a 3-fold increase in intratumor Gem accumulation after a single dose of treatment as evaluated by relative amounts of dFdC (active) and dFdU (Gem metabolite) (Figure 7).
MHT treatment promoted the interstitial transport delivery and tumor efficacy of Gem by improving interstitial transport probably due creation of vascular fenestrations and perturbation of vascular endothelium, which allowed extravasation of Gem Lip into the interstitium. This strategy caused minimal irreversible damage, unlike thermal ablation. Previous effort have mostly demonstrated the use of MHT treatment to target and improve efficacy using drug-loaded LTSLs nanomedicines that degrade upon heat treatment [47]. The present study established MHT treatment as a modulator of tumor environment, allowing enhancement in extravasation of thermally stable nanomedicines (liposomes) into tumor.
5. Conclusion
In conclusion, we have demonstrated the rational utility of mild hyperthermia to prime tumor microenvironment, improve tumor transport properties, and enhance delivery, dispersion, and efficacy of chemotherapeutics [28] while minimizing damage to tumor vessels and drug carriers We showed preferential accumulation of thermally stable liposomes at 5 h after MHT treatment and a 2-fold increase in intratumor Gem concentration after a single MHT treatment course. Further tumor challenge experiments revealed that a 3-week combinational MHT therapy increased therapeutic efficacy and tumor inhibition (3-fold) when Gem Lip was used with MHT relative to the control group. The treatment strategy also reduced drug dosing where infusion of 12 mg/kg Gem Lip led to tumor inhibition efficiency similar to the treatment with 60 mg/kg of Gem - 5-fold reduction. MHT treatment promises to open up venues with which to improve therapeutic efficacy of various nanotherapeutics (including thermally stable nanocarriers) and reduce cytotoxic due to dosing.
Supplementary Material
Acknowledgments
The authors acknowledge Matthew G. Landry for schematics and Dr. Yeon Ju Lee for figure and manuscript proofreading. The authors gratefully acknowledge funding support from the following sources: Department of Defense grants W81XWH-09-1-0212 and W81XWH-12-1-0414, National Institute of Health grants U54CA143837 and U54CA151668, the CPRIT grant RP121071 from the State of Texas, and the Ernest Cockrell Jr. Distinguished Endowed Chair.
References
- 1.Chauhan VP, Jain RK. Nat. Mater. 2013;12:958. doi: 10.1038/nmat3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK. Ann. Rev. Chem. Biomol. Eng. 2011;2:281. doi: 10.1146/annurev-chembioeng-061010-114300. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK. J. Clin. Oncol. 2013;31:2205. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sano K, Nakajima T, Choyke PL, Kobayashi H. ACS Nano. 2013;7:717. doi: 10.1021/nn305011p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu D, Wientjes MG, Lu Z, Au JLS. J. Pharmacol. Exp. Ther. 2007;322:80. doi: 10.1124/jpet.107.121632. [DOI] [PubMed] [Google Scholar]
- 6.Fingar V, H J. Clin. Laser Med. Surg. 1996;14:323. doi: 10.1089/clm.1996.14.323. [DOI] [PubMed] [Google Scholar]
- 7.Diagaradjane P, Shetty A, Wang JC, Elliott AM, Schwartz J, Shentu S, Park HC, Deorukhkar A, Stafford RJ, Cho SH, Tunnell JW, Hazle JD, Krishnan S. Nano Lett. 2008;8:1492. doi: 10.1021/nl080496z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Overgaard J, Bentzen SM, Gonzalez D, Hulshof MCM, Arcangeli G, Dahl O, Mella O. The Lancet. 1995;345:540. doi: 10.1016/s0140-6736(95)90463-8. [DOI] [PubMed] [Google Scholar]
- 9.Vertrees RA, Das GC, Popov VL, Coscio AM, Goodwin TJ, Logrono R, Zwischenberger JB, Boor PJ. Cancer Biol. Ther. 2005;4:1144. doi: 10.4161/cbt.4.10.2074. [DOI] [PubMed] [Google Scholar]
- 10.Wilson R, Xiaojing Z, Linghong G, Andrew S, Xiuying H, Yeping X, Sunil G, Samir P, Xuejun S, Jie C, Ronald M, James ZX. Nanotechnology. 2009;375101 [Google Scholar]
- 11.Wang L, Lin X, Wang J, Hu Z, Ji Y, Hou S, Zhao Y, Wu X, Chen C. Adv. Funct. Mater. 2014;24:4197. [Google Scholar]; Yang J, Yao M-H, Du M-S, Jin R-M, Zhao D-H, Ma J, Ma Z-Y, Zhao Y-D, Liu B. Chem. Commun. 2015 [Google Scholar]
- 12.Atkinson RL, Zhang M, Diagaradjane P, Peddibhotla S, Contreras A, Hilsenbeck SG, Woodward WA, Krishnan S, Chang JC, Rosen JM. Sci. Transl. Med. 2010;2:55ra79. doi: 10.1126/scitranslmed.3001447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, Felix R, Riess H. Crit. Rev. Oncol. Hematol. 2002;43:33. doi: 10.1016/s1040-8428(01)00179-2. [DOI] [PubMed] [Google Scholar]
- 14.Melancon MP, Elliott AM, Shetty A, Huang Q, Stafford RJ, Li C. J. Control. Release. 2011;156:265. doi: 10.1016/j.jconrel.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loos M, Kleeff J, Friess H, Büchler MW. Ann. N. Y. Acad. Sci. 2008;1138:169. doi: 10.1196/annals.1414.024. [DOI] [PubMed] [Google Scholar]
- 16.Kim C-E, Lim SK, Kim JS. H. Control. Release. 2012;157:190. doi: 10.1016/j.jconrel.2011.09.066. [DOI] [PubMed] [Google Scholar]; Ciliberto D, Botta C, Correale P, Rossi M, Caraglia M, Tassone P, Tagliaferri P. Eur. J. Cancer. 2013;49:593. doi: 10.1016/j.ejca.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 17.Bornmann C, Graeser R, Esser N, Ziroli V, Jantscheff P, Keck T, Unger C, Hopt U, Adam U, Schaechtele C, Massing U, von Dobschuetz E. Cancer Chemother. Pharmacol. 2008;61:395. doi: 10.1007/s00280-007-0482-z. [DOI] [PubMed] [Google Scholar]
- 18.Kirui D, Koay E, Guo X, Cristini V, Shen H, Ferrari M. Nanomedicine: NBM. 2014;10:1487. doi: 10.1016/j.nano.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landon CD, Park JY, Needham D, Dewhirst MW. Open Nanomed J. 2011;3:38. doi: 10.2174/1875933501103010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maitra A, Iacobuzio-Donahue C, Rahman A, Sohn TA, Argani P, Meyer R, Yeo CJ, Cameron JL, Goggins M, Kern SE, Ashfaq R, Hruban RH, Wilentz RE. Am. J. Clin. Patho. 2002;118:52. doi: 10.1309/3PAM-P5WL-2LV0-R4EG. [DOI] [PubMed] [Google Scholar]
- 21.Huang X, El-Sayed IH, Qian W, El-Sayed MA. J. Am. Chem. Soc. 2006;128:2115. doi: 10.1021/ja057254a. [DOI] [PubMed] [Google Scholar]
- 22.Choi R, Yang J, Choi J, Lim E-K, Kim E, Suh J-S, Huh Y-M, Haam S. Langmuir. 2010;26:17520. doi: 10.1021/la1029728. [DOI] [PubMed] [Google Scholar]
- 23.Buckway B, Frazier N, Gormley AJ, Ray A, Ghandehari H. Nucl. Med Bio. 2014;41:282. doi: 10.1016/j.nucmedbio.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang Z, Wang J, Nie X, Wen T, Ji Y, Wu X, Zhao Y, Chen C. J. Am. Chem. Soc. 2014;136:7317. doi: 10.1021/ja412735p. [DOI] [PubMed] [Google Scholar]
- 24.Kong G, Braun RD, Dewhirst MW. Cancer Res. 2001;61:3027. [PubMed] [Google Scholar]
- 25.Joshi N, Duhan V, Lingwal N, Bhaskar S, Upadhyay P. PLoS ONE. 2012;7:e32067. doi: 10.1371/journal.pone.0032067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapp DS, Hahn GM, Carlson RW. Principles of Hyperthermia. Holland-Frei; Ontario: 2000. [Google Scholar]
- 27.Jansen W, Haveman J. Pathol. Res. Pract. 1990;2:247. doi: 10.1016/S0344-0338(11)80542-X. [DOI] [PubMed] [Google Scholar]
- 28.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. Science. 2010;328:1031. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Celia C, Calvagno MG, Paolino D, Bulotta S, Ventura CA, Russo D, Fresta M. J Nanosci Nanotechnol. 2008;8:2102. doi: 10.1166/jnn.2008.065. [DOI] [PubMed] [Google Scholar]
- 30.Celia C, Trapasso E, Locatelli M, Navarra M, Ventura CA, Wolfram J, Carafa M, Morittu VM, Britti D, Di Marzio L, Paolino D. Colloid Surf. B Biointerf. 2013;112:548. doi: 10.1016/j.colsurfb.2013.09.017. [DOI] [PubMed] [Google Scholar]; Celia C, Malara N, Terracciano R, Cosco D, Paolino D, Fresta M, Savino R. Nanomedicine: NBM. 2008;4:155. doi: 10.1016/j.nano.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Celano M, Calvagno MG, Bulotta S, Paolino D, Arturi F, Rotiroti D, Filetti S, Fresta M, Russo D. BMC Cancer. 2004;4:1. [Google Scholar]
- 32.Bansal SS, Celia C, Ferrati S, Zabre E, Ferrari M, Palapattu G, Grattoni A. Curr Drug Targets. 2013;14:1061. doi: 10.2174/13894501113149990165. [DOI] [PubMed] [Google Scholar]
- 33.Licciardi M, Paolino D, Celia C, Giammona G, Cavallaro G, M F. Biomaterials. 2010;31:7340. doi: 10.1016/j.biomaterials.2010.05.060. [DOI] [PubMed] [Google Scholar]
- 34.Jain PK, Lee KS, El-Sayed IH, El-Sayed MA. J. Phys. Chem. B. 2006;110:7238. doi: 10.1021/jp057170o. [DOI] [PubMed] [Google Scholar]; Hu M, Chen J, Li Z-Y, Au L, Hartland GV, Li X, Marquez M, Xia Y. Chem. Soc. Rev. 2006;35:1084. doi: 10.1039/b517615h. [DOI] [PubMed] [Google Scholar]
- 35.van de Ven AL, Wu M, Lowengrub J, McDougall SR, Chaplain MAJ, Cristini V, Ferrari M, Frieboes HB. AIP Advances. 2012;2:011208. doi: 10.1063/1.3699060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dudeja V, Chugh RK, Sangwan V, Skube SJ, Mujumdar NR, Antonoff MB, Dawra RK, Vickers SM, Saluja AK. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2011;300:G948. doi: 10.1152/ajpgi.00346.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calvagno MG, Celia C, Paolino D, Cosco D, Iannone M, Castelli F, Doldo P, Fresta M. Curr. Drug Deliv. 2007;4 doi: 10.2174/156720107779314749. [DOI] [PubMed] [Google Scholar]
- 38.Barenholz Y. J. Control. Release. 2012;160:117. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 39.Stylianopoulos T, Jain RK. Proc. Natl. Acad. Sci. USA. 2013;110:18632. doi: 10.1073/pnas.1318415110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Itoh Y, Seiki M. J. Cellular Physiol. 2006;206:1. doi: 10.1002/jcp.20431. [DOI] [PubMed] [Google Scholar]
- 41.Dromi S, Frenkel V, Luk A, Traughber B, Angstadt M, Bur M, Poff J, Xie J, Libutti SK, Li KCP, Wood BJ. Clin. Cancer Res. 2008;13:2722. doi: 10.1158/1078-0432.CCR-06-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weissledder R. Nat. Biotechnol. 2001;19:316. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 43.Liu M, Qi L, Zeng Y, Yang Y, Bi Y, Shi X, Zhu H, Zhou Z, Sha J. PLoS ONE. 2012;7:e456954. doi: 10.1371/journal.pone.0045694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Celia C, Ferrati S, Bansal S, van de Ven A, Ruozi B, Zabre E, Hosali S, Paolino D, Sarpietro M, Fine D, Fresta M, Ferrari M, Grattoni A. Adv. Healthc. Mater. 2014;3:230. doi: 10.1002/adhm.201300188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirui DK, Mai J, Palange AL, Guoting Q, van de Ven AL, Liu X, Shen H, Ferrari M. PLoS ONE. 2014;9:e86489. doi: 10.1371/journal.pone.0086489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hancock HA, Smith LH, Cuesta J, Durrani AK, Angstadt M, Palmeri ML, Kimmel E, Frenkel V. sUltrasound Med. Biol. 2009;35:1722. doi: 10.1016/j.ultrasmedbio.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anna Y, Matthieu LC, Mariska DS, Sander L, Holger G, Chrit M. J. Control Release. 2012;161:90. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.