

Abstract
A common virulence mechanism among bacterial pathogens is the use of specialized secretion systems that deliver virulence proteins through a translocation channel inserted in the host cell membrane. During Yersinia infection, the host recognizes the type III secretion system mounting a pro-inflammatory response. However, soon after they are translocated, the effectors efficiently counteract that response. In this study we sought to identify YopD residues responsible for type III secretion system function. Through random mutagenesis, we identified eight Y. pseudotuberculosis yopD mutants with single amino acid changes affecting various type III secretion functions. Three severely defective mutants had substitutions in residues encompassing a 35 amino acid region (residues 168–203) located between the transmembrane domain and the C-terminal putative coiled-coil region of YopD. These mutations did not affect regulation of the low calcium response or YopB-YopD interaction but markedly inhibited MAPK and NFκB activation. When some of these mutations were introduced into the native yopD gene, defects in effector translocation and pore formation were also observed. We conclude that this newly identified region is important for YopD translocon function. The role of this domain in vivo remains elusive, as amino acid substitutions in that region did not significantly affect virulence of Y. pseudotuberculosis in orogastrically-infected mice.
Introduction
Pathogenic Yersinia species require the function of a type III secretion system (T3SS) to successfully establish infection. This specialized secretion machinery delivers a series of effector proteins (YopE, H, J, K, M, O, T) into the host cell that target different cell signaling molecules to interfere with the host immune system [1, 2]. In the prevailing model, effectors are thought to travel through a needle-like conduit and to traverse the host cell membrane through a translocation channel formed by translocator proteins YopB and YopD [3–6]. Located at the tip of the needle is LcrV, a protein that interacts with YopB and YopD allegedly assisting in the assembly of the translocation channel prior to their insertion into the host cell membrane [7, 8].
Activation of the T3SS requires contact of the bacteria with the host cell. However, in vitro, Yop secretion can be stimulated by culturing bacteria at 37°C at low calcium concentration. This low calcium condition also results in growth arrest. Thus, wild type Yersinia is said to exhibit a calcium-dependent growth (CD) at 37°C. Under non-permissive high calcium conditions, YscM1/LcrQ in cooperation with YopD and its cytosolic chaperone SycD/LcrH) block yop mRNA translation [9–15]. Consequently, a yopD mutant produces Yops constitutively and has a temperature-sensitive (TS) growth phenotype irrespective of the calcium concentration. Two chaperon-binding sites have been reported for YopD, one spanning residues 53 to 149 and the other across residues 278 to 292 [16]. Deletion of any of those YopD regions results in a TS growth and deregulation of Yop synthesis [17].
Infection of cultured cells with Yersinia mutants lacking multiple effector Yops results in the formation of open pores in the plasma membrane and subsequent osmotic lysis [18]. This process is referred as “pore formation” and requires the function of YopB and YopD [18–20]. On the other hand, during infection with wild-type Yersinia, effectors YopE and YopT prevent pore formation by inhibiting Rho GTPases [20, 21]. These signaling molecules control a variety of cellular functions including regulation of the actin cytoskeleton. The first evidence that translocon insertion induces a host response was provided by data demonstrating YopB/D-dependent activation of Rho GTPases during Yersinia infection [20]. Interestingly, inhibition of Rho GTPase activity or actin polymerization not only prevents pore formation but also reduces effector translocation [22, 23]. These results indicate that the YopB/D-dependent activation of Rho in the host cell is required for effective translocation.
There is increasing data supporting the concept that in addition to recognizing pathogen-associated molecular patterns, the innate immune system can detect host cell processes induced by pathogens during infection [24, 25]. We have previously shown that during infection with a multi Yop mutant Yersinia, epithelial cells respond to the YopB/D translocon by mounting a proinflammatory response involving activation of mitogen-activated protein kinases (MAPKs), nuclear factor kappa B (NFκB) [25, 26]. This provided further evidence that the host cell recognizes the translocon. A YopB/D-dependent pro-inflammatory response was also demonstrated in infected macrophages lacking TLR receptors [21, 26–28].
YopD belongs to a family of structurally similar “minor translocators” [29]. The 306 amino acid YopD protein contains one central predicted transmembrane alpha helix region, a predicted C-terminal coiled-coil domain and a confirmed C-terminal amphipathic alpha helix [17, 30, 31].
It has been recently proposed that the YopB/D-mediated innate immune response elicited during Yersinia infection of macrophages is due to the recognition of an unidentified translocated PAMP and not by direct sensing of the translocon [32]. The study shows that a muti-Yop mutant induces TNF-alpha expression during infection of TLR4-deficient macrophages, while a translocation-deficient (YopDΔTM) mutant does not. However, the possibility that host cells specifically sense the YopB/D translocon could not be completely ruled out because the YopDΔTM mutant was also partly defective in pore formation, introducing smaller pores with delayed kinetics.
The aim of this work was to shed light into the multiple YopD functions and to study their individual role in virulence. Thus, we created a yopD mutant library to identify single amino acids required for translocation, pore formation or host cell signaling. We identified eight single amino acid substitutions that affected various YopD functions. Interestingly, three substitutions in a discrete C-terminal region significantly impaired pore formation, Yop translocation and host cell signaling without affecting YopD secretion, yop regulation, or YopD-YopB interaction. The role of these mutations in vivo was assessed in an well-established mouse model for Yersinia pseudotuberculosis [33]. We conclude that a previously unidentified domain located between the transmembrane and the putative coiled-coil domains at the C-terminal region of YopD is central for its function.
Materials and Methods
Bacterial strains
Wild type Y. pseudotuberculosis serogroup III strain and its derivatives carry a naturally occurring deletion in the virulence plasmid that inactivates the yopT gene (Table 1) [20]. Deletion of yopD in YP52 (yopEHJD), YP62 (yopED), YP63 (yopD) and IP63 (yopD) was performed by allelic exchange using suicide plasmid pSB890ΔyopD as follows. Recombinant PCR was used to construct ΔyopD, a 394bp DNA fragment containing the sequence corresponding to the 177 nucleotides upstream yopD fused in frame to the 217 bp immediately downstream yopD. The construct was created using the following primers: YopD1A (5′-TGAAGGATCCAGACATGGCAGCGTTAC-3′), YopD1B (5′-GGTTATTCCTCCTTAAACTTAAACAG-3′), YopD1C (5′-GGAGGAATAACC CCATTGATGACCTTGGT-3′), and YopD1D (5′-CGGTGGATCCCAGCTCAGGTCTTGA-3′). Underlined sequences correspond to BamHI restriction sites sequences. In bold is a tail sequence in primer YopD1C that overlaps with the first 12 nucleotides at the 5’ end of primer YopD1B. The resulting DNA fragment (ΔyopD) was cloned into BamHI-digested suicide plasmid pSB890. The resulting plasmid (pSB890ΔyopD) was introduced by electroporation into S17–1λpir, and transformants were selected on LB plates containing tetracycline. pSB890ΔyopD was transferred into YP27(KmR), YP6 (KmR), YP126 and IP2666, by conjugation. Transconjugants were selected on LB kanamycin tetracycline for YP27 and YP6, or Yersinia selective agar plates with tetracycline for YP126 and IP2666, TetR transconjugants were grown for several generations in the absence of tetracycline and then plated on LB agar plates lacking NaCl and supplemented with 5% sucrose to select against the sacB gene carried on pSB890. Sucrose-resistant colonies were tested for a TetS phenotype. The resulting mutants were verified by sequencing.
Table 1. Bacterial strains and plasmids.
| Strain | Relevant features | Reference |
|---|---|---|
| YP126 | Wild type Y. pseudotuberculosis YPIII, naturally YopT-deficient | [46] |
| IP2666 | Wild type Y. pseudotuberculosis YPIII, naturally YopT-deficient | [33] |
| YP18 | YP126ΔyopB | [46] |
| YP27 | yopH::cam,yopE::kan,ΔyopJ | [47] |
| YP29 | yopH::cam,yopE::kan,ΔyopJ,ΔyopB | [47] |
| YP52 | yopH::cam,yopE::kan,ΔyopJ ΔyopD | This study |
| YP52/pYopD | YP52::pYopD | This study |
| YP6 | yopE::kan | [48] |
| YP62 | yopE::kan,ΔyopD | This study |
| YP62/pYopD | YP62::pYopD | This study |
| YP63 | YP126ΔyopD | This study |
| YP63/pYopD | YP63::pYopD | This study |
| IP63 | IP2666ΔyopD | This study |
| pMMB67HE | IPTG-inducible expression | [49] |
| pMMB67HEYopD | pMMB67HE::yopD | [26] |
| pSB890 | Suicide plasmid TetR, sacB | [46] |
| pSB890ΔyopD | pSB890::ΔyopD (177bp upstr yopD fused to 217bp downstr yopD) | This study |
| pSB890yopD AD | pSB890::yopD (−177bp yopD + 217bp) | This study |
Construction of a YopD mutant library
Error-prone PCR was used to create a library of single amino acid substitution in YopD. We used the GeneMorph II EZClone random mutagenesis kit from Stratagene that provides a mixture of two error prone DNA polymerases. yopD cloned into plasmid pMMB67HE (AmpR) was used as a template, and the following primers were used in the mutagenic PCR reaction: (pMMB67HE-Fw [5’ CGACATCATAACGGTTCTGGC 3’] and pMMB67HE-Rv [3’ GCGTTTCACTTCTGAGTTCGGC 3’]). These primers served to amplify a DNA fragment encompassing yopD and flanking pMMB67HE sequences (150bp upstream and 175bp downstream). As recommended to obtain 0–4.5 mutations/kb, the PCR reaction was performed using 100 ng of template DNA and 25 cycles. The amplified DNA was purified, digested with SacI and SphI, and cloned back into pMMB67HE to generate pYopD*. The pYopD* library was electroporated into competent YP62 (yopE::kan,ΔyopD) mutant and plated on LB supplemented with kanamycin (Kan) and carbenicillin (Car) to select for colonies containing the virulence plasmid and pYopD*, respectively. A total of 1144 CarR, KanR colonies were patched into 96-deep-well plate containing 0.4% agar LB supplemented with carbenicillin (Car) for maintenance at 4°C, and into LB glycerol to store at −20°C. Individual colonies of yopED and yopED/pYopD were included in each plate to serve as negative and positive control, respectively, in the screening tests.
Native expression of I168T, G196R and A273T
yopD genes coding for I168T, G196R and A273T were expressed into YP52 and IP2666 by allelic replacement using pSB890-based plasmids, as described above. First, we created pSB890yopD 1AD by megaprimer PCR using pSB890ΔyopD as a template, and a 1.3kb PCR product comprising yopD plus its adjacent 177bp upstream and 217bp downstream regions (yopD 1AD) as megaprimers. PCR product yopD 1AD was amplified from pYV using primers YopD1A and YopD1D (described above). Genes coding for YopDI168T, G196R and A273T were amplified from their pMMB67HE expression plasmid using primers YopDstart (ATGACAATAAATATCAAGAC) and YopDend (TCAGACAACACCAAAAGC). The resulting 921bp PCR products were purified and used as megaprimers to substitute yopD in pSB890yopD 1AD by PCR. The resulting plasmids were used to transform S17-λpir. Allelic replacement in YP63 and IP63 was performed as described above.
Cell culture
HeLa cells (ATCC-CCL-2) were cultured in DMEM (Gibco BRL) supplemented with 10% heat-inactivated fetal calf serum (FCS; Gibco BRL) and 1 mM sodium pyruvate in a 5% CO2 humidified incubator at 37°C. For infection experiments performed to determine IL-8 production and LDH release, 1 × 105 HeLa cells were seeded into wells of a 24-well tissue culture plate 24 h before assay. J774A.1 macrophage cell line origin (ATCC-TIB-67)
Screening of the YopD* library
The ability of the YopD mutants to deliver Yops and to cause pore formation was tested simultaneously by assessing YopJ-mediated cell death in macrophages and osmotic lysis of HeLa cells, respectively. Overnight cultures were made by transferring yopED, yopED/pYopD and yopED/pYopD* colonies from a 0.4% agar LB 96-well plate to a 96-well plate containing 200 μl of LB broth supplemented with kanamycin and carbenicillin, except for the well containing yopED, in which only kanamycin was added. Incubation was carried out at 26°C with shaking in a moist chamber to prevent evaporation. The next day, bacteria were subcultured 1/20 in 96-well plate containing 200μl LB with 20 mM magnesium chloride and 20 mM sodium oxalate (low Ca2+ conditions) for 1 h at 26°C and 2 h at 37°C, with shaking. IPTG was added at the 37°C temperature shift to a final concentration of 50μM. To test for translocation, J774A.1 cells were cultured in 96 well plates at a density of 5x104 cells/well and infected at an estimated MOI of 10 in the presence of IPTG. Gentamicin (100 μg/ml) was added 30 min after infection, and incubation proceeded overnight. LDH release from supernatants of infected cells was analyzed using CytoTox96 cytotoxicity assay kit (Promega). To test for pore formation, HeLa cells were cultured in 96 well plates at a density of 2x104 cells/well and infected at a MOI of 100 in the presence of IPTG. Culture supernatants were collected 3 hours post infection and tested for LDH as described above.
Yop secretion assay
Bacteria were cultured in 25 ml LB at low calcium conditions for 1 h at 26°C and 3 h at 37°C in the presence of 50μM IPTG. Proteins secreted in 1 ml of the culture supernatants were precipitated with 10% trichloroacetic acid (TCA), washed with ice-cold acetone, air-dried and suspended in Laemmli sample buffer. Bacterial pellet and secreted proteins were analyzed by Western blot using YopD Mab clone 248:19 [34] YopB mab clone 190.11 [35], and polyclonal antibodies against LcrV (provided by Matt Nilles, University of North Dakota) and YopH [36]. Anti-rabbit and anti-mouse IR680 or IR800 were used as secondary antibodies. Infrared signal was detected using the Odyssey imaging system (LI-COR Biosience).
YopE-mediated cytotoxicity of HeLa cell
HeLa cells were infected at MOI of 100 for 1h with bacteria cultured under high calcium conditions, which repress the T3SS. Infected cells were examined by phase contrast using an inverted microscopy with a 40× objective. Images were recorded using a digital camera and were mounted using Adobe Photoshop CS5. Images are representative of three different experiments.
LDH assay
HeLa cells in 24 well plates were infected with bacteria grown in LB broth under low Ca2+ condition at a multiplicity of infection of 100. The plates containing the infected cells were centrifuged for 5 min at 700 rpm and incubated at 37˚C with 5% CO2. Samples of culture media from wells containing infected cells were collected 3 hours post infection. Levels of LDH were assayed using the CytoTox 96 assay kit (Promega) as previously described. Results were normalized to yopEHJD/pYopD (100%). The mean and standard deviation values were obtained from four duplicate experiments.
YopD co-immunoprecipitation
Bacterial supernatants were collected after 3 hours incubation at low calcium conditions with 50 μM IPTG, and incubated with 10 μg of YopD mab clone 248:19 with rocking for 1 hour at room temperature. The pellet of a 20μl Protein G-beads slurry (Dynabeads Invitrogen) was added to the immunocomplexes an incubated for 10 min while rocking. The beads were washed three times with PBS/0.01% Tween and resuspended in 2X Laemmli sample buffer, boiled for 5 min and resolved in SDS-PAGE and immunoblotting using anti-YopD 248:19 antibody and anti-YopB monoclonal antibody. Anti-rabbit and anti-mouse IR680 or IR800 were used as secondary antibodies. Infrared signal was detected and quantified using the Odyssey imaging system (LI-COR Biosience).
Uptake of impermeant dyes
Hela cells cultured in 24-well plates with coverslips were infected for 3 h with bacteria grown under low calcium conditions. Uptake of ethidium homodimer-2 (EthD-2) by cells with compromised membranes was tested using the DEAD-LIVE kit (Invitrogen) following the protocol provided by the manufacturers. Cover slips were examined by using a fluorescence inverted microscopy with a 10× objective. Images were recorded using a digital camera and were mounted using Adobe Photoshop CS5.
YopD-dependent activation of proinflammatory signaling
HeLa cells cultured in 6 well plates were infected at MOI 100 for 60 min, washed and lysed as previously described [26]. Immunoblotting was performed using polyclonal antibodies against phospho-ERK, p90RSK, IκBα (Cell Signaling) according to the procedures of the manufacturer. Monoclonal antibody 12G10 against α-tubulin from Developmental Studies Hybridoma Bank, was used as a loading control.
Mouse infection
Eight-week-old female C57BL/6 mice (Jackson labs) were infected with Y. pseudotuberculosis wild type IP2666, IP2666YopDI168T, IP2666YopDG196R and IP2666YopDA273T. Mice were fasted for 18 h prior to infection. Bacterial inocula were prepared from overnight cultures grown in LB medium at 26°C, adjusted to 1010 cfu/ml and washed twice in PBS. In two independent experiments, 3 mice per group were infected orogastrically with 2x109cfu (10 LD50) using a feeding needle and an inoculated volume of 0.2 ml. To record survival, infected mice were monitored three times a day, seven days a week over a 2-week period. Animals displaying serve signs of disease, characterized by hunched posture and immobility upon physical stimulation, were euthanized by CO2 inhalation to minimize suffering. These experiments were carried out in compliance with animal care and use regulations. The survival curve of each mutant was compared to that of the wild type infected mice and the statistical difference and P values were calculated using the Log-rank (Mantel Cox) test.
Ethic Statement
The study was carried out in accordance with a protocol that adhered to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH) and was reviewed and approved (approval #206152) by the Institutional Animal Care and Use Committee at Stony Brook University, which operates under Assurance #A3011–01, approved by the NIH Office of Laboratory Animal Welfare.
Results
Construction and screening of a yopD mutant library
To better understand YopD function, we created a yopD library and screened for mutants that were secretion-competent but defective in translocation and/or pore formation. The library was generated using error prone PCR under conditions that created an average of one mutation per clone. The mutated PCR product was cloned into the expression vector pMMB67HE under the control of an IPTG-inducible promoter, resulting in pYopD*. The plasmid library was introduced into E. coli and subsequently moved to Y. pseudotuberculosis YPIII by conjugation. Because translocation of YopE or YopT during infection with wild type Y.pseudotuberculosis inhibits pore formation, and because YPIII naturally lacks YopT, we used a yopED mutant as a recipient of pYopD* (Table 1). This approach allowed for the simultaneous screening of YopJ translocation and pore formation. In addition, the kanamycin cassette inserted to disrupt the yopE gene in yopED mutant permitted the selection of colonies that maintained the virulence plasmid. The amount of YopD and other Yops secreted by yopED/pYopD at 500, 100 and 50 μM IPTG was compared to WT, yopE and yopED mutants (S1 Fig.). A high throughput genetic screening was implemented and 1144 colonies were tested. A concentration of 50 μM IPTG was used to induce YopD expression in cultures and during infections. Defects in Yop translocation and pore formation were assessed simultaneously in 96-well plates. Translocation of YopJ was indirectly measured by its ability to induce TLR4-mediated cell death in J774A.1 macrophages [37]. Percent of LDH release from infected cells was used to score cell death. Pore formation of infected HeLa cells leads to osmotic lysis, a phenotype that can also be measured by LDH release [20]. Because the readout for both assays is LDH release, we confirmed that under the infection conditions used in the macrophage cell death assay (low moi and short infection period before Gentamicin killing of extracellular bacteria, see Material and Methods) pore formation did not occur, i.e, infection with a pore forming strain that lacked YopJ (yopEHJ) did not cause LDH release from macrophages (data not shown).
Colonies with a defect in translocation, pore formation, or both, were subsequently tested for the ability of the mutants to secrete YopD. Those mutants with detectable amounts of secreted YopD by Western blot were subjected to DNA sequencing. We identified over 100 mutants that produced and secreted YopD but were defective in translocation, pore formation or both. Out of 100 mutants sequenced, 16 had a single missense mutation. The translocation, pore formation and YopD secretion phenotypes could be confirmed for 8 of these mutants. The location of the single amino acid substitution in each of those mutants is shown in Fig. 1A.
Fig 1. Secretion of the yopD mutants.
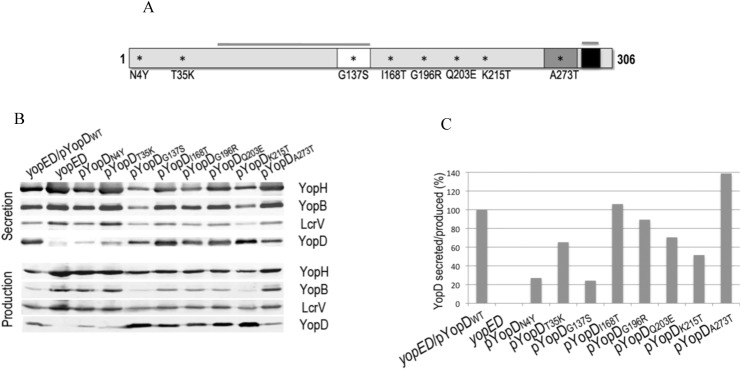
A. Schematic representation of YopD protein indicating a predicted transmembrane domain (white), a putative coiled-coil region (dark grey) and an amphipathic α-helix domain (black). Overlined are the N-terminal and C-terminal chaperon-binding regions (53–149 and 278–292). Amino acid substitutions that interfere with pore formation, translocation or both are indicated by asterisks. B. Immunoblot showing Yop secretion (bacterial supernatant) and production (bacterial lysate) for each of the YopD mutants expressed in a yopED background and grown at 37°C at low calcium conditions. Monoclonal antibodies for the detection of YopD and YopB and polyclonal antibodies for YopH and LcrV are described in Material and Methods. Anti-rabbit and anti-mouse IR680 or IR800 were used as secondary antibodies. Infrared signal was detected using the Odyssey imaging system (LI-COR Biosience). Quantification of the YopD signal intensities was performed using Odyssey imaging system software. The ratio between the amount of YopD secreted and YopD produced was calculated for each mutant and normalized to wild type YopD.
Amino acid substitutions G137S and K215T interfere with calcium–dependent growth
Yersinia with yopD null mutations are defective in yop regulation and have a temperature-sensitive phenotype, growing poorly at 37°C irrespective of the calcium concentration. To determine whether any of the YopD amino acid substitutions affected CD growth, we cultured bacteria at low or high calcium concentration and measured OD600 every 30 minutes for 5 hours. Mutants with amino acid substitutions G137S and K215T exhibit a temperature sensitive phenotype similar to the yopD deletion mutant. The rest of the mutants displayed normal calcium-dependent growth (S2 Fig.). Henceforth we will refer to mutants G137S and K215T as Class I (Table 1).
Substitutions N4Y, T35K and A273T affect YopD synthesis
To test the ability of each mutant to synthesize and secrete YopD, the protein profiles of whole bacteria cells and TCA-precipitated culture supernatants were determined after growth at low calcium conditions. Immunoblot assays using a monoclonal antibody against YopD showed that variable levels of YopD were present in bacterial pellets of the different mutants. Notably, mutants with YopD substitutions N4Y, T35K and A273T produced considerably less YopD than the WT, whereas substitutions G137S and K215T led to yopD overexpression (Fig. 1B).
YopD secretion is decreased by N4Y and G137S residue exchange
To calculate the amount of YopD secreted by the different mutants, we measured the intensities of the YopD bands in the supernatant fractions of the immunoblot. Because of the variability in the amount of YopD produced, we calculated the ratio between secreted and produced YopD (Fig. 1B), and normalized it to that of the WT (Fig. 1C). While secretion of YopD N4Y and G137 were severely affected, the rest of the YopD variants were secreted at levels greater than or equal to 50% of the YopDWT.
YopD mutants with normal calcium-dependent growth overproduced other Yops
We determined whether synthesis and secretion of other Yops were affected by the different YopD amino acid substitutions. As expected, yopED, which is temperature sensitive and defective in down-regulation of yop expression, produced increased amount of YopH, YopB, and LcrV, as determined by immunoblotting (Fig. 1B). The same pattern of Yop overproduction occurred in mutants with substitutions N4Y, T35K and A273T. As shown above, these mutants produced low amounts of YopD. Interestingly, these mutants display normal calcium-dependent growth. On the other hand, amino acid substitutions that affected normal calcium-dependent growth (G137S and K215T) resulted in a considerable downregulation of YopH, YopB and LcrVs synthesis (Fig. 1B). At high calcium conditions, G137S and K215T exhibited a wild type phenotype for Yop synthesis and secretion. This suggests that, at least under the conditions used in this assay, Yop regulation may be uncoupled from calcium-dependent growth in these mutants.
T35K completely abrogates YopE-mediated rounding in Hela cells
Because the Rho GAP activity of YopE negatively controls translocation [22, 23], the lack of yopE causes the bacteria to translocate Yops at higher levels. To better assess the effect of the YopD substitutions in translocation, we transferred the pYopD* plasmids to a yopD background, and we assessed translocation by YopE-mediated rounding of infected HeLa cell. Because very low amounts of translocated YopE can cause disruption of the host cell cytoskeleton, we cultured bacteria under conditions that inhibit T3SS activation before bacterial-host cell contact. As expected, yopD/pYopD caused complete cell rounding one hour after infection, whereas no cytotoxicity was detected in cells infected with yopD mutant. YopD substitution T35K had a phenotype similar to yopD, whereas N4Y and A273T displayed some, but reduced levels of cytotoxicity (Fig. 2). The rest of the mutants caused cell rounding comparable to the wild type. A recognized caveat of the cytotoxicity assay is its poor ability to detect subtle defects in translocation. These results should be cautiously interpreted since we later found that the translocation phenotype of mutants I168T and G196R differed when the yopD mutations were introduced into the native yopD gene (see Fig. 6).
Fig 2. Effect of the different yopD mutations on translocation.
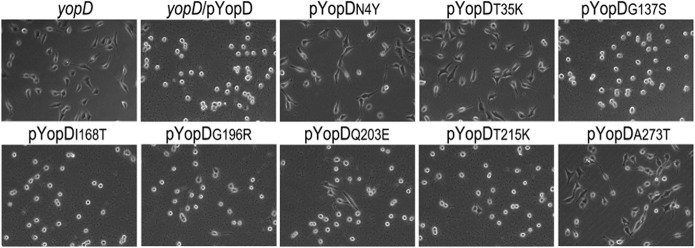
A. yopD variants were expressed ectopically in a yopD single mutant strain and tested for YopE-mediated cell rounding of HeLa cells. Bacteria cultured under conditions that repress the T3SS were used to infect HeLa cells at MOI of 100 for 1h. YopE-mediated cytotoxicity was assessed by inverted microscopy using a 40× objective. Images were recorded using a digital camera. Images are representative of three different experiments.
Fig 6. Phenotype of the Y. pseudotuberculosis expressing yopD mutations in its native location.
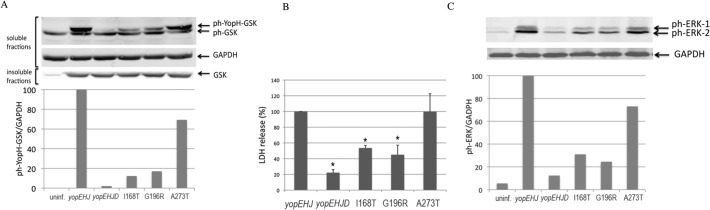
HeLa cells were infected with yopEHJ, yopEHJD, and yopEHJD expressing yopDI168T, G196R and A273T in their native location. A. Effector translocation was determined by infecting HeLa cells at a MOI of 100 for 2h. Cells were washed, lysed with 1% Triton X100, and soluble and insoluble fractions were separated by SDS-PAGE and analyzed by immunoblotting with rabbit anti-phospho-GSK-3β (Ser9). Monoclonal antibody against GAPDH and total GSK-3β (Ser9) antibody were used as a loading control for the soluble and insoluble fractions, respectively. Quantification of the signal intensities was performed using Odyssey imaging system software. Results are expressed as the ratio between total ph-ERK signal and GAPDH. B. Pore formation was determined as described in Fig. 4 by analyzing the amount of LDH released from culture supernatants of infected cells. Results were normalized to yopEHJ (100%). Error bars represent the standard deviation of the mean values obtained from three duplicate experiments. * P<0.005 determined by t-test. C. YopD-dependent activation of proinflammatory signaling was performed as described in Fig. 5. MAPK activation was analyzed in cell lysates by immunoblotting using rabbit anti-phospho ERK. A monoclonal antibody against GAPDH was used as a loading control and quantification of the signal intensities was performed using Odyssey imaging system software. Results are expressed as the ratio between total ph-ERK signal and GAPDH.
Pore formation is compromised in mutants Class II and III
The pore formation phenotype of the mutants selected in the screening was confirmed by infecting HeLa cells seeded on coverslips in 24-well plates at a MOI of 100. Because a yopEHJ mutant causes more pronounced pore formation than a single yopE mutant [20], we transferred pYopD* to a yopEHJD background. Three hours after infection, supernatants of uninfected and infected cells were tested for LDH release (Fig. 3A and S3 Fig.). All but N4Y and G137S were significantly defective in pore formation, with T35K, I168T, G196R and Q203E being the most severely affected. These results were confirmed by the uptake of DNA-binding impermeant dye ethidium homodimer-1 by infected HeLa cells (Fig. 3B). In this assay, only cells with compromised membranes take up the impermeant dye. Thus, infection with T35K, I168T, G196R and Q203E cause very low levels of pore formation as evidenced by only a few cells stained with red nuclei. As shown above, T35K was produced at much lower levels than YopDWT, whereas production and secretion of I168T, G196R and Q203E mutants were normal. We will therefore call pore formation-defective mutants T35K Class II, and I168T, G196R and Q203E Class III (Table 2).
Fig 3. Effect of the different YopD mutations on pore formation.

A. HeLa cells were left uninfected or infected with yopEHJD, yopEHJD/pYopD and yopEHJD expressing the different yopD mutants at MOI of 100. After 3 h infection, culture supernatants were removed and tested for LDH release. Background LDH released from uninfected cells was subtracted from infected wells. Results were normalized to yopEHJD/pYopD (100%). Error bars represent the standard deviation of the mean values obtained from four duplicate experiments. ** P<0.0001, * P<0.0005 determined by t-test. B. HeLa cells on coverslips were infected as described above for the LDH assay, and stained with DEAD-LIVE kit as described in Material and Methods. Cells with disrupted membranes exhibit a red nuclei staining.
Table 2. Phenotypes of the four different classes of YopD mutants.
| Class | Mutation | Growth type | YopD production | YopD secretion | Pore formation | Signal |
|---|---|---|---|---|---|---|
| I | G137S | TS | high | low | interm | interm |
| K215T | TS | high | normal | interm | interm | |
| II | T35K | CD | low | normal | low | low |
| III | I168T | CD | normal | normal | low | low |
| G196R | CD | normal | normal | low | low | |
| Q203E | CD | normal | normal | low | low | |
| IV | N4Y | CD | low | low | interm | interm |
| A273T | CD | low | low | interm | interm |
YopD mutants Class II and III fail to trigger a pro-inflammatory response
Epithelial cells respond to infection with a Yersinia multi-yop mutant by triggering a series of pro-inflammatory signals [21, 26]. This response is dependent on the translocon and includes MAPKs and NFκB activation, and IL-8 production We have previously found that although a yopEHJB mutant does not trigger IL-8 production, infection with a yopEHJD mutant produces detectable levels of IL-8 [21, 26]. Therefore, at that time we concluded that YopD was dispensable for the proinflammatory response caused by the translocon. However, when we investigated the kinetics of MAPKs and NFκB activation we found that yopEHJD could not activate signal at early time points because, as shown in S4A Fig., levels of ph-ERK induced by yopEHJD and yopEHJB infection are comparable to those of uninfected cells. However, we could detect MAPKs and NFκB activation at 4-h infection (S4A Fig.). This delayed activation of proinflammatory signals could account for the modest IL-8 production found in our previous study. To determine what YopD residues are required for signal activation, we tested phosphorylation of the MAP kinase ERK in response to infection with yopEHJD/pYopD*. HeLa cells were left uninfected or infected for 1 h, and cell lysates were analyzed by immunoblotting using antibodies to ph-ERK (Fig. 4A and S5A Fig.). The ph-ERK signal intensities were quantified and the values were normalized to tubulin (Fig. 4B). Robust MAPK activation was observed when HeLa cells were infected with yopEHJD/pYopD, whereas much weaker MAPK phosphorylation was detected in cells infected with the yopEHJD mutant (Fig. 4). Class II (T35K) and Class III (I168T, G196R and Q203E) mutants were defective in MAPK signaling activation (Fig. 4). The results of the MAPK activation were confirmed by assessing the activation of p90RSK, a substrate of the MAPK/ERK (S5B and S5C Fig.). We also tested the effect of the different YopD amino acid substitutions on NFκB activation by measuring degradation of the NFκB inhibitor IκBα. IκBα degradation correlated with ERK activation (S5B Fig. and Fig. 5D). Intermediate levels of MAPK and NFκB activations were detected in cells infected with yopEHJD/pYopD N4Y, A273T and G137S. G137S belongs to TS Class I mutants. Because N4Y and A273T exhibit normal CD growth and have an intermediate pore formation and signaling phenotype, they are grouped as Class IV (Table 2).
Fig 4. YopD-dependent activation of proinflammatory signaling.
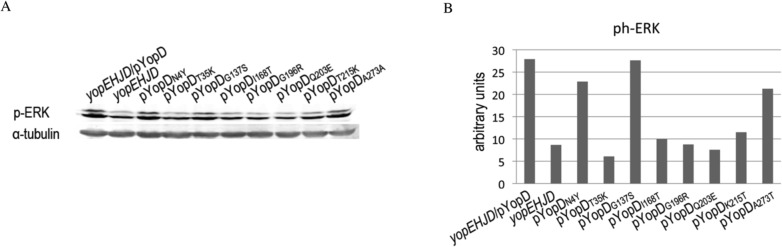
A. HeLa cells infected with yopEHJD, yopEHJD/pYopD and yopEHJD expressing the different YopD mutants at MOI of 100 for 1h. Cells were washed, lysed with sample buffer 1X, separated by SDS-PAGE and analyzed by immunoblotting with rabbit anti-phospho ERK. Monoclonal antibody against tubulin was used as a loading control. Quantification of the signal intensities was performed using Odyssey imaging system software. Values were normalized to tubulin.
Fig 5. Interaction of the different YopD mutants with YopB.
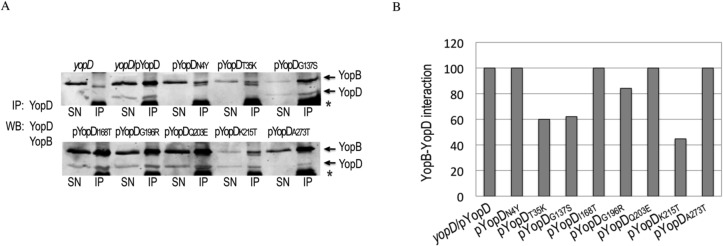
A. YopB-YopD complexes secreted in bacterial supernatants of yopD, yopD/pYopD and yopD expressing the different yopD mutants were precipitated by 1 h incubation with YopD mab (clone 248:19) followed by incubation with Dynabeads Protein G (Invitrogen). After 3 washes, beads were resuspended in 2X Laemmli sample buffer, and boiled. The eluted material (IP) and an aliquot of the bacterial supernatants (SN) were resolved in SDS-PAGE. Western blot was performed using anti-YopD and anti-YopB Mabs, and anti-mouse IR680 or IR800 secondary antibodies. Bands corresponding to YopB and YopD are indicated by arrows. Also present in the IP samples are a band originated from the beads (marked with an asterix), and a weak band of unknown origin that migrates right below YopB. B. Signal intensities of immunoprecipitated YopB and YopD were calculated using Odyssey imaging system software (LI-COR Biosience). YopB-YopD interaction for each mutant was calculated as the ratio between immunoprecipitated YopB and YopD. Results were normalized to yopD/pYopD.
Interaction of YopD mutants with YopB
To determine whether the defect in translocation, pore formation or signaling was due to a defective interaction between the YopD variants and YopB we performed YopD co-immunoprecipitations on secreted Yops in culture supernatants. Fig. 5A shows that YopD monoclonal antibody effectively pulled down YopB from yopED/pYopD bacterial supernatant but not from that of yopED. Because the levels of YopB and YopD secretion vary among the different mutants, the degree of YopD-B interaction was expressed as the amount of pulled-down YopB relative to that of pulled-down YopD (Fig. 5B). With the exception of T35K, G137S and K215T (Class I and II), which exhibited 40–60% reduced YopB-YopD interaction, all the other YopD variants interacted with secreted YopB to high levels. Consistent with previous results in which secreted His-YopBDE complexes were purified from bacterial cultures using nickel affinity chromatography [38], YopD antibody also pulled down YopE, but not LcrV (not shown). Furthermore, a monoclonal antibody against LcrV also failed to pull down YopD from culture supernatants (not shown), suggesting that the YopBDE complex is not composed of aggregated Yops.
Native expression of Class III mutants confirm multiple defects in translocon function
To determine whether the phenotypes of the yopD mutants could be confirmed when expressed from the native yopD gene, we selected three yopD mutants with normal calcium dependent growth; two with strong pore formation and signaling defects representing Class III (I168T, G196R) and one with a more intermediate phenotype from Class IV (A273T). Thus, the three yopD mutations were moved to its native location in yopEHJD (YP52), as described in Material and Methods, and the resulting strains were tested in parallel for translocation, pore formation and MAPK activation.
To quantitatively test for effector translocation in these mutants, we used the glycogen synthase kinase GSK peptide tag-based reporter system [39]. When the tagged effector protein is translocated into the eukaryotic cell, the GSK peptide is phosphorylated by host cell kinases. The phosphorylated fusion protein can then be detected in the cell lysate by immunoblot using an anti ph-GSK antibody. Thus, yopEHJD (YP52) strains expressing the yopD mutations in their native location were transformed with the arabinose-inducible expression plasmid pBADyopH-gsk by electroporation. Arabinose (0.2%) was added to the bacterial cultures and was kept during infection of HeLa cells to induce the expression of YopH-gsk fusion protein. Two hours after infection, cells were washed with PBS, and lysed with 1%Triton X-100. Soluble and insoluble fractions of uninfected and infected cells were analyzed by immunoblotting using antibodies to phospho-GSK-3β (Ser9). Eukaryotic glyceraldehyde 3-phosphate dehydrogenase (GAPDH) signal was used as a loading control of the soluble fraction, and total GSK as a control for the insoluble fraction. A strong band of phosphorylated YopH-GSK was detected in cells infected with yopEHJ and yopEHJ expressing yopD A273T, whereas much less translocation occurred in cells infected with yopEHJ expressing Class III yopD mutations I168T and G196R (Fig. 6A). These results did not correlate with the translocation phenotypes assessed by HeLa cell rounding when the mutations were expressed ectopically (Fig. 2).
In parallel, the same strains were tested for pore formation activity by LDH release in HeLa cells 3 hours post infection. Although the phenotypes were less dramatic than those observed with the ectopic expression of these YopD mutants, the defect in pore formation could be confirmed for Class III mutants I168T and G196R (Fig. 6B).
MAPK activation was also assessed as described above, by infecting HeLa cells for 1 h and analyzing cell lysates by immunoblotting using antibodies to ph-ERK. In accordance to what was observed when the yopD mutations were expressed ectopically, infection with yopEHJYopDA273T elicited a strong ERK phosphorylation, while a poorer MAPK activation, was detected upon infection with mutants Class III I168T and G196R (Fig. 6C).
Based on these results we postulate that YopD amino acid I168 and G196, and most likely also Q203, constitute a new domain in YopD important for translocation, pore formation, and host signaling.
In vivo studies
To determine if substitution I168T or G196R would impact virulence, we infected mice orogastrically with wild type Y. pseudotuberculosis IP2666, IP2666 natively expressing I168T, G196R and, as a control, A273T. Secretion of YopD and other Yops was not affected by the endogenous expression of the yopD variants (S6 Fig.). After infection, survival was followed up for 2 weeks. The survival of mice infected with IP2666 expressing any of the yopD mutations was not significantly different from that of the mice infected with wild type IP2666 (I168T: χ2 2.51, P = 0.1131; G196R: χ2 1.59, P = 0.2077; A273T: χ2 2.3, P = 0.1297, Fig. 7). These results indicate that although substitutions I168T and G196R affect translocation, pore formation and epithelial cell recognition in vitro, these amino acids changes do not decrease virulence under the conditions used in this mouse model.
Fig 7. Residues I168, G196 and A273 are not required for virulence of Y. pseudotuberculosis IP2666 in a mouse model.
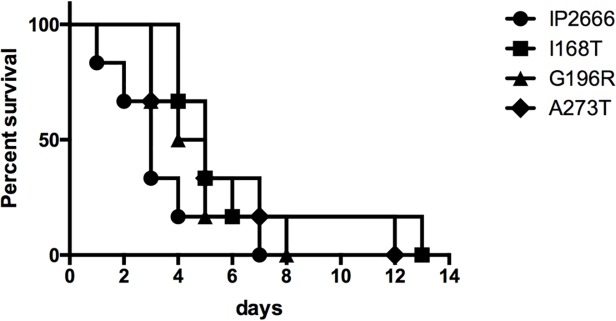
Eight-week old C57BL/6J mice were infected orogastrically with 2×109 CFU of wild type Y. pseudotuberculosis IP2666 and its derivative expressing yopD I168T, G196R and A273T. Mouse survival was monitored for 14 days. Results shown are pooled from two independent experiments using 3 mice per group in each experiment. Statistical analysis using log rank test showed no significant differences between the survival curves of each of the mutants and that of the wild type.
Discussion
YopD plays a multifaceted role in Yersinia virulence. Inside the bacterial cell YopD is in complex with its chaperon and, in cooperation with YscM1/LcrQ, prevents yop transcription during non-permissive condition for Yop secretion. Once secreted, YopD interacts with itself and YopB to form a multimeric ring that can insert in lipid membranes [6]. Insertion of this translocation channel in the membrane of the host cell has several consequences, i.e. it mediates delivery of effectors inside the host cell, it triggers Rho GTPase activation, it leads to cell lysis if the effector Yops do not inhibit Rho GTPases activation [20] [18], and it is directly or indirectly sensed by the innate immune system to mount a pro-inflammatory response [20, 21, 25, 32]. We propose that epithelial cells’ activation of Rho GTPase by the translocon might be detected as a “pattern of pathogenesis”.
Frequently, translocation-deficient yopD mutants also show a defect in Yop synthesis regulation, complicating the study of such mutants. Here we have used a genetic screen to isolate randomly generated YopD variants defective in YopD function. This comprehensive mutational analysis allowed the identification of four different types of mutants (Table 2).
Summary of YopD mutants and their phenotypes compared to wild type YopD. Growth type was determined at 37°C with and without calcium, “TS” refers to temperature sensitive, and “CD” to 37°C calcium-dependent growth; YopD production and secretion were determined in a bacterial culture at low calcium conditions in the pellet and supernatants, respectively; pore formation was tested in HeLa cells by LDH release and uptake of impermeant dyes; signal activation of MAPK and NFκB was assessed in HeLa cells. “interm” refers to intermediate phenotype.
Six yopD mutants (Class II-IV) have normal low calcium response but are defective in translocon function. Class II and III mutants have the strongest phenotype. Importantly, mutants in Class III have amino acid substitutions in a discrete region located between the transmembrane and the putative C-terminal coiled-coil domain (residues 168 to 203). This region has been found to be partly unfolded in several YopD-homologues. Besides allowing YopD’s secretion through the needle, this disordered structure is thought to be required for its interaction with host cell partners [29]. Significantly, residues I168, G196 and Q203 are conserved among YopD homologues in Pseudomonas, Aeromonas and Photorhabdus species (S7 Fig.). The amino acid substitutions in this region range from radical in Q203E (uncharged to acidic), and G196R (uncharged to basic) to semi-conservative in I168T (hydrophobic to polar) Independently, the library screening identified other mutants within the 168–203 region that were excluded from the study because they exhibited more than one missense mutation. Among them was a mutant that resulted in a hydrophobic to polar amino acid substitution at position 168 (I168N), suggesting that the hydrophobic residue at position 168 may play an important role in translocon function. Three others such mutants included YopD variants with substitutions in amino acids 181, 183 and 185 (not shown). Although the consequences of these residues changes would have to be tested independent from the other mutations, it is possible that those amino acids are also part of this previously unrecognized distinct region within the C-terminus of YopD that is required for translocation, pore formation, and activation of the MAPK and NFκB pathways in HeLa cells.
We have previously shown that Yop effectors prevent membrane damage by counteracting a signal triggered by the insertion of the translocon [20]. This signal involves Rho GTPase activation and leads to localized actin polymerization. We postulate that counteracting this signal is essential for the host cell membrane resealing process to prevent cell lysis. We envision that when translocation pores are not promptly sealed, impermeant dyes can enter the cell. At this point, the membrane permeabilization process is reversible. If, for some reason, the membrane cannot be repaired, osmotic lysis occurs and the cytoplasmic content is released. Consistent with this model is our unpublished observation that infection of HeLa cells with a single yopK mutant, which presumably inserts larger pores [40], results in strong membrane blebbing and high uptake of impermeant dyes. However, because yopK mutant can translocate Yops the process can be reversed, cell lysis does not occur, and LDH is not released. The transient inhibition of membrane resealing might be necessary to facilitate delivery of effectors. Supporting this model is our previous data showing that inhibition of Rho GTPases or actin polymerization decreases effector translocation, and conversely loss of YopE-mediated GAP activity leads to hypertranslocation [22, 23]. We reason that substitutions I168T, G196S and Q203E lead to defects in Rho GTPase signaling that impact translocation and pore formation. Because these mutants also failed to trigger MAPK, our model predicts that it is this very signal that is recognized as a pattern of pathogenesis by host cell to trigger an inflammatory response. Different scenarios can explain the defect in signaling. One possibility is that this C-terminal domain is by itself involved in Rho-GTPase activation. Interestingly, that region has been shown to interact with the cytoplasm of the host cell in the enteropathogenic E. coli YopD homologue EspB, suggesting that these residues could directly or indirectly interact with host cell signal proteins [41]. Another possibility is that Rho activation requires correct membrane insertion of a properly assembled YopB/D channel. Although results from the immuno-precipitation studies suggest that I168T, G196S and Q203E do not affect interaction between secreted YopB and YopD, it is possible that different interactions are required to form a signal-competent structure. It should be noted that the role of this domain in Yersinia infection is uncertain, as results from orogastrically-infected mice indicate that substitutions I168T and G196R, do not significantly affect virulence. It was surprising to find that mutants with a marked defect in translocation were not attenuated. On the other hand, it still possible that subtle differences in virulence could not be demonstrated with the conditions used in our in vivo studies.
Substitution T35K has dramatic effects on the translocation and pore formation functions of YopD. The lower level of T35K secretion is not likely to account for those defects as even lower levels of secreted YopD were identified in N4Y and yet this mutant is fully capable of causing pore formation. A serious defect in the interaction between and T35K and YopB can also be ruled out by the results obtained from the immuno-precipitation assay. Thus, although at lower levels than wild type YopD, T35K was able to pull down YopB from culture supernatants. Alternatively, T35K substitution could potentially be disrupting an N-terminal coiled-coil (CC) structure predicted with by Costa et al using the COILS server [42]. In that low probability CC region, threonine 35 would correspond to the d residue from a heptad repeat hxxhcxc, of hydrophobic (h) and charged (c) amino-acid residues. When we used the same program to test whether a threonine to lysine substitution at position 35 would disrupt such a structure, the probability of forming a CC domain did not change. This suggests that the YopD defects caused by the T35K substitution might not be attributed to the disruption of a putative N-terminal CC domain. More studies would need to be done to determine the cause of the T35K substitution’s dramatic phenotype.
Class I mutants exhibit a temperature sensitive growth. Mutants defective in the low calcium response produce Yops constitutively and have a temperature sensitive growth at 37°C irrespective of the calcium concentrations [9, 17]. Previous comprehensive sequential in-frame deletion mutagenesis studies of YopD have identified two main regions required for regulatory control: one spanning residues 53 to 149 and the other 278 to 292 [17]. This regions overlaps with domains required for interaction of YopD with its chaperone LcrH/SycD. Glycine 137 is located within the amino-terminal LcrH/SycD binding site. Lysine 215, however, is an area of the protein not previously associated with regulation. As the LcrH/SycD is required for stabilization of its substrates and both mutants are produced at high levels, it is unlikely that these mutants have a defect in their interaction with their chaperone. It is possible that G137S and K215T substitutions interfere with normal calcium dependent growth by preventing YopD interaction with another component of the T3SS required for normal calcium-dependent growth. These YopD partners might include YscV, which has been identified as a suppressor of the temperature-sensitive mutation in YopD [12].
Substitutions G137S and K215T also lead to hyperproduction of YopD and, interestingly, these mutants produced markedly reduced amounts of YopB. Because YopB and YopD share the LcrH/SycD chaperone, this phenomenon might be the result of a competition for a common chaperon. Although a single chaperone molecule could conceivably bind to both translocators simultaneously, data from the crystal structure of a YopD peptide bound to LcrH/SycD suggests that this is not likely the case [43]. Interestingly, inconsistent to what it would be expected if mutants with substitutions G137S and K215T had a defect in yop regulation, expression of LcrV and YopH was not increased in those mutants. These results suggest that in these mutants calcium dependent growth can be uncoupled from yop regulation.
A predicted transmembrane domain has been identified between residues 128 to 149 of YopD [17, 44]. Although G137 is located within the transmembrane domain, the Glycine to Serine substitution did not significantly affect translocation or pore formation. Accordingly, results obtained using the prediction of transmembrane regions and orientation from the EMBnet server, indicate that the G137S substitution is not likely to affect the transmembrane domain (not shown). The moderate defect in pore formation and translocation caused by G137S and K215T YopD substitutions might simply be attributed to the lower amount of YopB and LcrV produced and secreted by those mutants.
When expressed ectopically, N4Y and A273T showed a translocation defect, whereas their pore formation was not considerably reduced. We were not expecting to identify mutants defective in either translocation or pore formation, as we envision the two processes to be connected. Previous studies have identified mutants defective exclusively in translocation [17]. These studies analyzed pore formation by infection of red blood cells with bacteria that lacked YopK, which supposedly insert much larger pores [40]. Here, we have tested pore formation in HeLa cells using uptake of Ethidium homodimer and LDH release. YopD mutants previously reported to cause lysis in red blood cells, such as YopDΔ53–68 and Δ128–149 [17], test negative in our two pore formation assays (data not shown). So, clearly, these two assays measure different cellular events. Notably, when A273T was expressed in it native location translocation levels were restored to close to 70% of that of the WT. This suggests that slight differences in the amount of protein produced when yopD is expressed ectopically versus in its native location may affect the proper pore stoichiometry.
Asparagine at position 4 lies within a previously identified N-terminal YopD secretion signal [45]. We have found that this residue exchange not only impacts secretion, as it would be expected, but most noticeably, it reduces YopD production. A possible role of the 5′ end of yopD in translation control has been suggested previously [45]. Although decreased levels of YopD production were also detected in the A273T substitution, secretion was not as greatly affected as N4Y, which is expected, as the C-terminus does not seem to be involved in secretion.
Although more work needs to be done to fully understand their role in T3S, the mutants identified in this genetic screening provide a useful tool to further explore the different functions of YopD.
Supporting Information
Wild type (YP126), yopE (YP6), yopED (YP62) and yopED/pYopD bacterial cultures were grown at 37°C at low calcium conditions with different concentrations of IPTG added. Equivalent amounts of cultures supernatants containing secreted Yops were resolved by SDS-PAGE and the gel was stained with Coomassie Blue. Positions of molecular weight standards (kDa) are indicated on the left and bands corresponding to the different Yops are indicated on the right.
(TIF)
Yersinia yopED, yopED/pYopD and yopED expressing the different yopD variants were cultured in LB broth at low calcium (LC) and high calcium (HC) at 37°C for 5 hours. Absorbance at OD600 was measured every 30 minutes and growth curves were constructed for each mutant.
(TIF)
Pore formation was determined as described in Fig. 3 by analyzing the amount of LDH released from culture supernatants of uninfected and infected cells. Results from a typical experiment before normalization and subtraction of the LDH content present in uninfected wells are shown. LDH release is expressed in arbitrary units.
(TIF)
A. HeLa cells were left uninfected or infected with yopEHJ, yopEHJB and yopEHJD for 1hour. Cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with rabbit anti-phospho ERK, and anti-rabbit IR680. Monoclonal antibody against tubulin was used as a loading control (not shown). Quantification of the signal intensities was performed using Odyssey imaging system software. Values were normalized to tubulin. B. Kinetics of MAPK activation was tested at 1, 2 and 4 hours post infection. Cell lysates were analyzed by immunoblotting with rabbit anti-phospho ERK, and anti-rabbit IR680, and anti-IκBα and anti-rabbit IR680.
(TIF)
A. HeLa cells were left uninfected or infected with yopEHJD, and yopEHJD/pYopD at MOI of 100 for 1h. Cells were washed, lysed with sample buffer 1X, separated by SDS-PAGE and analyzed by immunoblotting with rabbit anti-phospho ERK. Monoclonal antibody against tubulin was used as a loading control (not shown). B. HeLa cells infected with yopEHJD, yopEHJD/pYopD and yopEHJD expressing the different yopD mutants at the same conditions as described above. Immunoblotting was performed with rabbit anti-phospho p90RSK, anti-IκBα, and anti-tubulin, independently. C and D. Quantification of the signal intensities was performed using Odyssey imaging system software. Values were normalized to tubulin.
(TIF)
Comassie Blue stained SDS-PAGE gel showing Yop secretion for IP2666 (WT) yopD (IP63) and IP63 endogenously expressing YopDI168T, G196R and A273Y and grown at 37°C at low calcium conditions.
(TIF)
A 55 amino acid region of YopD encompassing residues 168–203, was aligned with the corresponding protein regions of three other YopD homologues: Pseudomonas aeruginosa, Aeromonas spp. and Photorhabdus spp. Identical residues are shown in bold.
(TIF)
Acknowledgments
We are grateful to Uday Tak, and Surabhi Vijayaraghavan, and past and present students of the Clinical Laboratory Sciences program at Stony Brook University: Qi Gao, Joel Attard, Connie Cai, Hanyi Zhao, Wayne Yu, Sze Hung, Kristen Chapman, Eunyoung Cho, Shulin Hang, Katrina Callan and Helen Chao for their assistance with the screening of the YopD library. We thank Emmanuel Adomfeh and Nikolai Cassanova for construction of YP62 and YP62/pYopD. The monoclonal antibody against alpha tubulin, developed by E. M. Nelsen, was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work is supported by a grant from the National Institutes of Health R21-AI073815 awarded to GIV and NIH R01AI099222 to JBB. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Buttner D. Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol Mol Biol Rev. 2012. Jun;76(2):262–310. 10.1128/MMBR.05017-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Viboud GI, Bliska JB. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol. 2005;59:69–89. [DOI] [PubMed] [Google Scholar]
- 3. Sory MP, Cornelis GR. Translocation of a hybrid YopE-adenylate cyclase from Yersinia enterocolitica into HeLa cells. Mol Microbiol. 1994. Nov;14(3):583–94. [DOI] [PubMed] [Google Scholar]
- 4. Rosqvist R, Magnusson KE, Wolf-Watz H. Target cell contact triggers expression and polarized transfer of Yersinia YopE cytotoxin into mammalian cells. EMBO J. 1994. Feb 15;13(4):964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Persson C, Nordfelth R, Holmstrom A, Hakansson S, Rosqvist R, Wolf-Watz H. Cell-surface-bound Yersinia translocate the protein tyrosine phosphatase YopH by a polarized mechanism into the target cell. Mol Microbiol. 1995. Oct;18(1):135–50. [DOI] [PubMed] [Google Scholar]
- 6. Montagner C, Arquint C, Cornelis GR. Translocators YopB and YopD from Yersinia enterocolitica form a multimeric integral membrane complex in eukaryotic cell membranes. J Bacteriol. 2011. Dec;193(24):6923–8. 10.1128/JB.05555-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mueller CA, Broz P, Cornelis GR. The type III secretion system tip complex and translocon. Mol Microbiol. 2008. Jun;68(5):1085–95. 10.1111/j.1365-2958.2008.06237.x [DOI] [PubMed] [Google Scholar]
- 8. Thanassi DG, Bliska JB, Christie PJ. Surface organelles assembled by secretion systems of Gram-negative bacteria: diversity in structure and function. FEMS Microbiol Rev. 2012. Nov;36(6):1046–82. 10.1111/j.1574-6976.2012.00342.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Williams AW, Straley SC. YopD of Yersinia pestis plays a role in negative regulation of the low-calcium response in addition to its role in translocation of Yops. J Bacteriol. 1998. Jan;180(2):350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Francis MS, Lloyd SA, Wolf-Watz H. The type III secretion chaperone LcrH co-operates with YopD to establish a negative, regulatory loop for control of Yop synthesis in Yersinia pseudotuberculosis. Mol Microbiol. 2001. Nov;42(4):1075–93. [DOI] [PubMed] [Google Scholar]
- 11. Wattiau P, Bernier B, Deslee P, Michiels T, Cornelis GR. Individual chaperones required for Yop secretion by Yersinia. Proc Natl Acad Sci U S A. 1994. Oct 25;91(22):10493–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fowler JM, Wulff CR, Straley SC, Brubaker RR. Growth of calcium-blind mutants of Yersinia pestis at 37 degrees C in permissive Ca2+-deficient environments. Microbiology. 2009. Aug;155(Pt 8):2509–21. 10.1099/mic.0.028852-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anderson DM, Ramamurthi KS, Tam C, Schneewind O. YopD and LcrH regulate expression of Yersinia enterocolitica YopQ by a posttranscriptional mechanism and bind to yopQ RNA. J Bacteriol. 2002. Mar;184(5):1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kopaskie KS, Ligtenberg KG, Schneewind O. Translational regulation of Yersinia enterocolitica mRNA encoding a type III secretion substrate. J Biol Chem. 2013. Dec 6;288(49):35478–88. 10.1074/jbc.M113.504811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Y, Anderson DM. Expression hierarchy in the Yersinia type III secretion system established through YopD recognition of RNA. Mol Microbiol. 2011. May;80(4):966–80. 10.1111/j.1365-2958.2011.07623.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Francis MS, Aili M, Wiklund ML, Wolf-Watz H. A study of the YopD-lcrH interaction from Yersinia pseudotuberculosis reveals a role for hydrophobic residues within the amphipathic domain of YopD. Mol Microbiol. 2000. Oct;38(1):85–102. [DOI] [PubMed] [Google Scholar]
- 17. Olsson J, Edqvist PJ, Broms JE, Forsberg A, Wolf-Watz H, Francis MS. The YopD translocator of Yersinia pseudotuberculosis is a multifunctional protein comprised of discrete domains. J Bacteriol. 2004. Jul;186(13):4110–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hakansson S, Schesser K, Persson C, Galyov EE, Rosqvist R, Homble F, et al. The YopB protein of Yersinia pseudotuberculosis is essential for the translocation of Yop effector proteins across the target cell plasma membrane and displays a contact-dependent membrane disrupting activity. EMBO J. 1996. Nov 1;15(21):5812–23. [PMC free article] [PubMed] [Google Scholar]
- 19. Neyt C, Cornelis GR. Insertion of a Yop translocation pore into the macrophage plasma membrane by Yersinia enterocolitica: requirement for translocators YopB and YopD, but not LcrG. Mol Microbiol. 1999. Sep;33(5):971–81. [DOI] [PubMed] [Google Scholar]
- 20. Viboud GI, Bliska JB. A bacterial type III secretion system inhibits actin polymerization to prevent pore formation in host cell membranes. EMBO J. 2001. Oct 1;20(19):5373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Viboud GI, Mejia E, Bliska JB. Comparison of YopE and YopT activities in counteracting host signalling responses to Yersinia pseudotuberculosis infection. Cell Microbiol. 2006. Sep;8(9):1504–15. [DOI] [PubMed] [Google Scholar]
- 22. Mejia E, Bliska JB, Viboud GI. Yersinia controls type III effector delivery into host cells by modulating Rho activity. PLoS Pathog. 2008. Jan;4(1):e3 10.1371/journal.ppat.0040003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aili M, Isaksson EL, Carlsson SE, Wolf-Watz H, Rosqvist R, Francis MS. Regulation of Yersinia Yop-effector delivery by translocated YopE. Int J Med Microbiol. 2008. Apr;298(3–4):183–92. 10.1016/j.ijmm.2008.04.005 [DOI] [PubMed] [Google Scholar]
- 24. Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009. Jul 23;6(1):10–21. 10.1016/j.chom.2009.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bliska JB, Wang X, Viboud GI, Brodsky IE. Modulation of innate immune responses by Yersinia type III secretion system translocators and effectors. Cell Microbiol. 2013. Oct;15(10):1622–31. 10.1111/cmi.12164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Viboud GI, So SS, Ryndak MB, Bliska JB. Proinflammatory signalling stimulated by the type III translocation factor YopB is counteracted by multiple effectors in epithelial cells infected with Yersinia pseudotuberculosis. Mol Microbiol. 2003. Mar;47(5):1305–15. [DOI] [PubMed] [Google Scholar]
- 27. Auerbuch V, Golenbock DT, Isberg RR. Innate immune recognition of Yersinia pseudotuberculosis type III secretion. PLoS Pathog. 2009. Dec;5(12):e1000686 10.1371/journal.ppat.1000686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, et al. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe. 2010. May 20;7(5):376–87. 10.1016/j.chom.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mattei PJ, Faudry E, Job V, Izore T, Attree I, Dessen A. Membrane targeting and pore formation by the type III secretion system translocon. FEBS J. 2011. Feb;278(3):414–26. 10.1111/j.1742-4658.2010.07974.x [DOI] [PubMed] [Google Scholar]
- 30. Costa TR, Edqvist PJ, Broms JE, Ahlund MK, Forsberg A, Francis MS. YopD self-assembly and binding to LcrV facilitate type III secretion activity by Yersinia pseudotuberculosis. J Biol Chem. 2010. Aug 13;285(33):25269–84. 10.1074/jbc.M110.144311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang T, Diaz-Rosales P, Costa MM, Campbell S, Snow M, Collet B, et al. Functional characterization of a nonmammalian IL-21: rainbow trout Oncorhynchus mykiss IL-21 upregulates the expression of the Th cell signature cytokines IFN-gamma, IL-10, and IL-22. J Immunol. 2011. Jan 15;186(2):708–21. 10.4049/jimmunol.1001203 [DOI] [PubMed] [Google Scholar]
- 32. Kwuan L, Adams W, Auerbuch V. Impact of host membrane pore formation by the Yersinia pseudotuberculosis type III secretion system on the macrophage innate immune response. Infect Immun. 2013. Mar;81(3):905–14. 10.1128/IAI.01014-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simonet M, Falkow S. Invasin expression in Yersinia pseudotuberculosis. Infect Immun. 1992. Oct;60(10):4414–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Noel BL, Lilo S, Capurso D, Hill J, Bliska JB. Yersinia pestis can bypass protective antibodies to LcrV and activation with gamma interferon to survive and induce apoptosis in murine macrophages. Clin Vaccine Immunol. 2009. Oct;16(10):1457–66. 10.1128/CVI.00172-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chung LK, Philip NH, Schmidt VA, Koller A, Strowig T, Flavell RA, et al. IQGAP1 is important for activation of caspase-1 in macrophages and is targeted by Yersinia pestis type III effector YopM. MBio. 2014;5(4):e01402–14. 10.1128/mBio.01402-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montagna LG, Ivanov MI, Bliska JB. Identification of residues in the N-terminal domain of the Yersinia tyrosine phosphatase that are critical for substrate recognition. J Biol Chem. 2001. Feb 16;276(7):5005–11. [DOI] [PubMed] [Google Scholar]
- 37. Ryndak MB, Chung H, London E, Bliska JB. Role of predicted transmembrane domains for type III translocation, pore formation, and signaling by the Yersinia pseudotuberculosis YopB protein. Infect Immun. 2005. Apr;73(4):2433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ivanov MI, Noel BL, Rampersaud R, Mena P, Benach JL, Bliska JB. Vaccination of mice with a Yop translocon complex elicits antibodies that are protective against infection with F1- Yersinia pestis. Infect Immun. 2008. Nov;76(11):5181–90. 10.1128/IAI.00189-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garcia JT, Ferracci F, Jackson MW, Joseph SS, Pattis I, Plano LR, et al. Measurement of effector protein injection by type III and type IV secretion systems by using a 13-residue phosphorylatable glycogen synthase kinase tag. Infect Immun. 2006. Oct;74(10):5645–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holmstrom A, Petterson J, Rosqvist R, Hakansson S, Tafazoli F, Fallman M, et al. YopK of Yersinia pseudotuberculosis controls translocation of Yop effectors across the eukaryotic cell membrane. Mol Microbiol. 1997. Apr;24(1):73–91. [DOI] [PubMed] [Google Scholar]
- 41. Luo W, Donnenberg MS. Interactions and predicted host membrane topology of the enteropathogenic Escherichia coli translocator protein EspB. J Bacteriol. 2011. Jun;193(12):2972–80. 10.1128/JB.00153-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Costa TR, Amer AA, Fallman M, Fahlgren A, Francis MS. Coiled-coils in the YopD translocator family: A predicted structure unique to the YopD N-terminus contributes to full virulence of Yersinia pseudotuberculosis. Infect Genet Evol. 2012. Dec;12(8):1729–42. 10.1016/j.meegid.2012.07.016 [DOI] [PubMed] [Google Scholar]
- 43. Schreiner M, Niemann HH. Crystal structure of the Yersinia enterocolitica type III secretion chaperone SycD in complex with a peptide of the minor translocator YopD. BMC Struct Biol. 2012;12:13 10.1186/1472-6807-12-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hakansson S, Bergman T, Vanooteghem JC, Cornelis G, Wolf-Watz H. YopB and YopD constitute a novel class of Yersinia Yop proteins. Infect Immun. 1993. Jan;61(1):71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Amer AA, Ahlund MK, Broms JE, Forsberg A, Francis MS. Impact of the N-terminal secretor domain on YopD translocator function in Yersinia pseudotuberculosis type III secretion. J Bacteriol. 2011. Dec;193(23):6683–700. 10.1128/JB.00210-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Palmer LE, Hobbie S, Galan JE, Bliska JB. YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNF-alpha production and downregulation of the MAP kinases p38 and JNK. Mol Microbiol. 1998. Mar;27(5):953–65. [DOI] [PubMed] [Google Scholar]
- 47. Palmer LE, Pancetti AR, Greenberg S, Bliska JB. YopJ of Yersinia spp. is sufficient to cause downregulation of multiple mitogen-activated protein kinases in eukaryotic cells. Infect Immun. 1999. Feb;67(2):708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bliska JB, Galan JE, Falkow S. Signal transduction in the mammalian cell during bacterial attachment and entry. Cell. 1993. Jun 4;73(5):903–20. [DOI] [PubMed] [Google Scholar]
- 49. Furste JP, Pansegrau W, Frank R, Blocker H, Scholz P, Bagdasarian M, et al. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene. 1986;48(1):119–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Wild type (YP126), yopE (YP6), yopED (YP62) and yopED/pYopD bacterial cultures were grown at 37°C at low calcium conditions with different concentrations of IPTG added. Equivalent amounts of cultures supernatants containing secreted Yops were resolved by SDS-PAGE and the gel was stained with Coomassie Blue. Positions of molecular weight standards (kDa) are indicated on the left and bands corresponding to the different Yops are indicated on the right.
(TIF)
Yersinia yopED, yopED/pYopD and yopED expressing the different yopD variants were cultured in LB broth at low calcium (LC) and high calcium (HC) at 37°C for 5 hours. Absorbance at OD600 was measured every 30 minutes and growth curves were constructed for each mutant.
(TIF)
Pore formation was determined as described in Fig. 3 by analyzing the amount of LDH released from culture supernatants of uninfected and infected cells. Results from a typical experiment before normalization and subtraction of the LDH content present in uninfected wells are shown. LDH release is expressed in arbitrary units.
(TIF)
A. HeLa cells were left uninfected or infected with yopEHJ, yopEHJB and yopEHJD for 1hour. Cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with rabbit anti-phospho ERK, and anti-rabbit IR680. Monoclonal antibody against tubulin was used as a loading control (not shown). Quantification of the signal intensities was performed using Odyssey imaging system software. Values were normalized to tubulin. B. Kinetics of MAPK activation was tested at 1, 2 and 4 hours post infection. Cell lysates were analyzed by immunoblotting with rabbit anti-phospho ERK, and anti-rabbit IR680, and anti-IκBα and anti-rabbit IR680.
(TIF)
A. HeLa cells were left uninfected or infected with yopEHJD, and yopEHJD/pYopD at MOI of 100 for 1h. Cells were washed, lysed with sample buffer 1X, separated by SDS-PAGE and analyzed by immunoblotting with rabbit anti-phospho ERK. Monoclonal antibody against tubulin was used as a loading control (not shown). B. HeLa cells infected with yopEHJD, yopEHJD/pYopD and yopEHJD expressing the different yopD mutants at the same conditions as described above. Immunoblotting was performed with rabbit anti-phospho p90RSK, anti-IκBα, and anti-tubulin, independently. C and D. Quantification of the signal intensities was performed using Odyssey imaging system software. Values were normalized to tubulin.
(TIF)
Comassie Blue stained SDS-PAGE gel showing Yop secretion for IP2666 (WT) yopD (IP63) and IP63 endogenously expressing YopDI168T, G196R and A273Y and grown at 37°C at low calcium conditions.
(TIF)
A 55 amino acid region of YopD encompassing residues 168–203, was aligned with the corresponding protein regions of three other YopD homologues: Pseudomonas aeruginosa, Aeromonas spp. and Photorhabdus spp. Identical residues are shown in bold.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.