

Abstract
Genetic causes of focal and segmental glomerulosclerosis (FSGS) typically involve mutations and allele variants of genes expressed in podocytes or, more rarely, in glomerular basement membranes. In this report, we describe a 60-year-old woman with chronic kidney disease whose kidney biopsy showed FSGS. Immunoglobulins and C3 were undetectable in immunofluorescence studies. Electron microscopy showed subendothelial fluffy granular material with occasional double contour formation suggestive of capillary wall injury, and prompting work-up for a prothrombotic state. Evaluation of the alternative pathway of complement revealed a novel polymorphism in short consensus repeat (SCR) 12 of complement factor H (CFH; c.2195C>T, p.Thr732Met) and a previously reported but largely uncharacterized polymorphism in complement factor C3 (c.463A>C, p.Lys155Gln). Dysregulation of the alternative pathway is associated with atypical hemolytic syndrome (aHUS) and Dense Deposit Disease, but heretofore has not been associated with FSGS. This case highlights the expanding spectrum of complement-mediated glomerular disease, and shows that FSGS with features of capillary wall injury should prompt evaluation for abnormalities in the alternative pathway. This case also expands the list of genetic polymorphisms that can be associated with an FSGS phenotype.
Index Words: FSGS, Chronic thrombotic microangiopathy, Alternative pathway of complement, Factor H, C3
INTRODUCTION
Focal and segmental glomerulosclerosis (FSGS) is a clinicopathologic entity characterized clinically by proteinuria, and pathologically by segmental sclerosis of the glomerular capillary tufts on light microscopy, segmental IgM and C3 staining on immunofluorescence studies, and foot process effacement on electron microscopy.(1) It is classified as primary/idiopathic or secondary to reflect an unknown or known etiology, respectively. Secondary FSGS can result from glomerular, interstitial, vascular, or systemic diseases, or a compensatory or adaptive response as in obesity. (1, 2) There are also several genetic causes of FSGS, including mutations in ACTN4 (FSGS1), TRPC6 (FSGS2), CD2AP (FSGS3), APOL1 (FSGS4), INF2 (FSGS5), NPHS2 (podocin), MYO1E (myosin IE), and LAMB2 (laminin S). Of these genes, ACTN4, TRPC6, CD2AP, INF2, NPHS2 and MYO1E encode proteins that are expressed in the podocyte while LAMB2 encodes a glomerular basement membrane protein.(3–8) To correlate pathologic findings with the underlying etiology and to standardize nomenclature, FSGS is also classified by histologic patterns. (9, 10)
Dysregulation of the alternative pathway (AP) of complement results in C3 glomerulopathies (C3G) and atypical hemolytic syndrome (aHUS). (11–14) Both C3 glomerulonephritis (C3GN) and Dense Deposit Disease (DDD) are types of C3G, and result from fluid dysregulation of the AP with deposition of activated and terminal complement components in the mesangium and along capillary walls, leading to an inflammatory response.(13, 15–18) aHUS, in contrast, results from local dysregulation of the AP with activation of the terminal complement cascade (TCC), formation of membrane attack complex (MAC), vascular injury, and thrombotic microangiopathy.
In this report, we describe a woman with chronic kidney disease whose kidney biopsy showed FSGS who was found to have variants in the genes for C3 and complement factor H (CFH). The former is the cornerstone protein of the complement cascade, and the latter is the key regulator of the AP. These findings suggest that mutations in complement genes can result in an FSGS phenotype, and expand the spectrum of the diseases resulting from dysregulation of the AP.
CASE REPORT
Clinical History and Laboratory Data
A 60-year-old Caucasian woman was referred with a long history of proteinuria and chronic kidney disease. The patient had a history of mild proteinuria since childhood. She also had hypertension that was relatively well controlled. Although she had a 15-pack year smoking history, she had not smoked for 12 years. Vital signs included a blood pressure of 113/61 mm Hg, pulse of 63 beats/minute, and weight of 76 kilograms (body mass index, 32 kg/m2). Cardiovascular, respiratory, and neurological examinations were unremarkable. There was no history of diabetes or use of non-steroidal analgesic drugs, however there was a strong family history of kidney disease. Her father died at the age of 57 years, and his medical history included proteinuria and end stage kidney disease; both of her daughters have proteinuria; and her first cousin had end stage kidney disease necessitating kidney transplant. Proteinuria and rising serum creatinine from a baseline of 1.1 mg/dL (97.2 micromol/L, corresponding to an estimated glomerular filtration rate [eGFR] of 54 mL/min/1.73 m2 [calculated by the 4-variable MDRD [Modification of Diet in Renal Disease] Study equation) to 1.7 mg/dL (150.3 micromol/L; eGFR, 33 mL/min/1.73 m2) prompted two kidney biopsies over a period of 2 years. Rheumatoid factor and lupus anticoagulant tests were negative. The most recent laboratory findings are shown in Table 1.
Table 1.
Laboratory findings
| Laboratory result | Reference range | |
|---|---|---|
| Hemoglobin | 10.6 | 14–18.0 g/dL |
| Erythrocyte sedimentation rate | 53 | 0–22 mm/h |
| Platelets | 166 | 150–400 103/μl |
| WBC count | 5.8 | 3.5–10.5 ×109/L |
| Total protein | 7.2 | 6.3–7.9 g/dL |
| Albumin | 3.2 | 3.5–4.8 g/dL |
| Glucose | 127 | 70–100 mg/dL |
| Sodium | 140 | 136–144 mEq/L |
| Potassium | 3.8 | 3.6–5.1 mEq/L |
| Serum urea nitrogen | 43 | 4–20 mg/dL |
| Creatinine | 2.0 | 0.64–1.2 mg/dL |
| eGFR | 25 | >60 ml/min/1.73 m2 |
| C3 | 145 | 79–152 mg/dL |
| C4 | 28 | 16–38 mg/dL |
| Hemoglobin A1c | 5.6 | 4–6.0% |
| Cholesterol | 280 | |
| Bilirubin, total | 0.5 | 0.1–1 mg/dL |
| Ferritin | 393 | 11–307 μg/dL |
| Urinalysis | Few hyaline casts | |
| Urinary protein (g/d) | 0.7–1.2 | |
| Peripheral smears | No schistocytes, normal smear | |
| Serum electrophoresis | monoclonal proteins not detected |
Abbreviations: eGFR, estimated glomerular filtration rate; WBC, white blood cell.
Conversion factors for units: glucose in mg/dL to mmol/L, x0.05551; creatinine in mg/dL to micromol/L, x88.4; cholesterol in mg/dL to mmol/L, x0.02586; biliruin in mg/dL to micromol/L, x17.1. No conversion necessary for sodium and potassium in mEq/L and mmol/L.
Kidney Biopsy
In a biopsy performed 2.5 years before time of writing, eleven glomeruli were available for evaluation, five of which were globally sclerosed. Glomeruli with segmental sclerosis were not present. The glomeruli showed mild glomerulomegaly, mild mesangial matrix expansion, and thickened glomerular capillary walls with segmental double contour formation. The glomeruli did not show any evidence of crescents, necrosis, or endocapillary proliferation. There was also no interstitial inflammation present. There was mild tubular atrophy and interstitial fibrosis, which involved approximately 20% of the cortex. Arteries and arterioles were unremarkable. No evidence of immune deposits (including C3 deposits) in the glomeruli was detected by immunofluorescence microscopy. Electron microscopy showed mild segmental foot process effacement, with segmental basement membrane thickening and subendothelial expansion by fluffy granular material. Few capillary loops also showed segments of double contour formation due to entrapment of cellular elements and new basement membrane formation. Electron dense deposits were not present along the capillary loops or in the mesangium. Representative light and electron microscopy are shown in top panel of Figure 1. Of note is the segmental thickening (left half of glomerulus) of the glomerular basement membranes on light microscopy.
Figure 1.
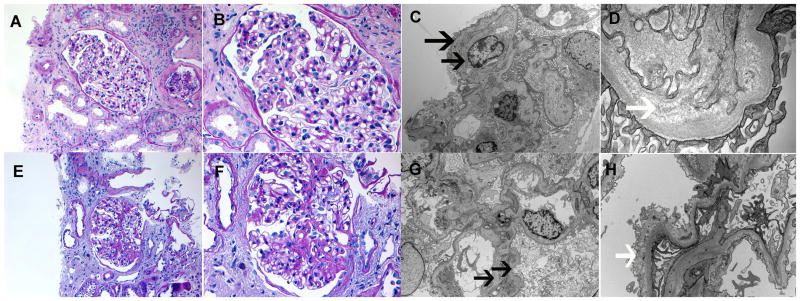
Kidney biopsies. Biopsy 1 (Top panel): A–B. Light microscopy showing segmental thickening of the glomerular basement membranes (Perioidic acid Schiff stain; original magnification, 20× and 40×, respectively). Electron microscopy showing (C) double contour formation along capillary walls (4200x), and (D) subendothelial expansion with fluffy granular material (33000x). Biopsy 2 (bottom panel): E–F Light microscopy showing segmental sclerosis (Perioidic acid Schiff stain; original magnification, 20× and 40×, respectively). Electron microscopy showing (G) double contour formation along capillary walls (5000x), and (H) subendothelial expansion with fluffy granular material (12000x). Black arrows, double contours; white arrows, subendothelial fluffy material.
In a repeat biopsy performed 2 years later, twelve glomeruli were present, eight of which were globally sclerosed. Two glomeruli showed segmental sclerosis of the glomerular tufts and adhesion to Bowman’s capsule. The remaining glomeruli showed mild glomerulomegaly, mild mesangial matrix expansion, and thickened glomerular capillary walls with segmental double contour formation. There was moderate tubular atrophy and interstitial fibrosis involving approximately 40% of the cortex. Arteries and arterioles showed mild sclerosis of the intima. Immunofluorescence microscopy yielded no staining indicative of immune deposits in the glomeruli. Electron microscopy showed mild foot process effacement, and segmental subendothelial expansion with fluffy granular material. Few loops also showed new basement membrane formation resulting in segments of double contours. Electron dense deposits were not present along the capillary loops or in the mesangium. Representative light and electron microscopy images are shown in the bottom panel of Figure 1.
Diagnosis
On the basis of the microscopy findings, we made a diagnosis of FSGS. The presence of subendothelial fluffy granular material and double contours prompted an evaluation for thrombotic microangiopathy, and we performed functional and genetic evaluation of the complement cascade. Using standard protocols, plasma and serum were prepared, and genomic DNA was extracted from whole blood. Testing for autoantibodies to CFH and CFB was performed as previously described and results were negative.(19) Autoantibodies to C3 convertase (C3 nephritic factors) were evaluated by enzyme-linked immunosorbent assay, immunofixation electrophoresis, and C3 convertase stabilizing assay (with and without properdin), as previously described19; in each case, the results were negative. In addition, no hemolysis was detected. For genetic analysis (summarized in Table 2), the coding regions and intron/exon boundaries of the genes encoding CFH, C3, CFI, CFB, CHR5, THBD, and MCP were amplified from genomic DNA and sequenced bi-directionally as described previously.(20) Results were significant for two variants in complement cascade proteins, for which the patient was heterozygous. The first variant, c.2195C>T (p.Thr732Met), was found in short consensus repeat (SCR) 12 of CFH. The second variant, c.463A>C (p.Lys155Gln), was found in C3. In addition, the patient was homozygous for the CFH V62 risk allele and heterozygous for the CFH H402 risk allele in SCR7. (20, 21) The C3 risk alleles G102 and L314 were not detected.
Table 2.
Genetic Analysis of the Alternative Pathway
| Gene | Sequence or CNV Analysisa | Risk Allele Genotyping | ||
|---|---|---|---|---|
| Allele 1 | Allele 2 | Description | No. of Copies | |
| CFH | c.2195C>T, p.Thr732Metb | ref | c.184G (p.Val62)d | 2 |
| -- | -- | c.1204C (p.His402)e | 1 | |
| C3 | c.463A>C, p.Lys155Glnc | ref | c.304G (p.Gly102)f | 0 |
| c.941T (p.Leu314)g | 0 | |||
| CFB, CFI, CFHR5, CD46 (MCP), THBD | ref | ref | NA | NA |
| CFHR3-CFHR1 | ref | ref | NA | NA |
Note: For sequence analysis, sequences were amplified by the polymerase chain reaction from 20 ng of genomic DNA and sequenced bidirectionally using an automated capillary sequencer. Abbreviations and definitions: cDNA, complementary DNA; CFHR5, CFH-related protein 5; MCP, membrane cofactor protein; NA, not applicable; ref, sequence obtained matches reference sequence; rs, reference single-nucleotide polymorphism [number]; THBD, thrombomodulin
Analysis by sequencing except for the CFHR3-CFHR1 region, which was CNV (copy-number variation) analysis performed by multiplex ligation-dependent probe amplification.
Cytosine to thymine substitution at nucleotide 2,195 of the cDNA, corresponding to a threonine to methionine change at amino acid 732.
Adenine to cytosine substitution at nucleotide 463 of the cDNA, corresponding to a lysine to glutamine change at amino acid 155.
Guanine at nucleotide 184 of the cDNA (rs800292), corresponding to a valine (not isoleucine) at amino acid 62.
Cytosine at nucleotide 1,204 of the cDNA (rs1061170), corresponding to a histidine (not tyrosine) at amino acid 402.
Guanine at nucleotide 304 of the cDNA, corresponding to a glycine (not arginine) at amino acid 102.
Thymine at nucleotide 941 of the cDNA, corresponding to a leucine (not proline) at amino acid 314.
As a result of these additional investigations, we refined the diagnosis to FSGS with chronic thrombotic microangiopathy associated with dysregulation of the alternative pathway of complement resulting from mutations in CFH and C3.
DISCUSSION
This case of FSGS results from mild chronic glomerular vascular injury as evidenced by subendothelial expansion by fluffy granular material and segments of double contour formation. No active lesions of thrombotic microangiopathy were present, but the vascular injury prompted an evaluation of the complement pathway which was remarkable for variants in two complement cascade genes, CFH and C3.
There are 20 SCR domains in CFH, each ~60 amino acids in length. The 4 SCRs at the amino terminus bind C3b, and are important in regulating C3 convertase. There are reports of mutations in these domains being associated with DDD and C3GN. (13, 15) SCRs 19-20 are located at the carboxy terminus, and are required for CFH binding to host surfaces. These domains also interact with the C3b degradation products iC3b and C3d. (22) Mutations in SCRs 19-20 have a causal association with atypical hemolytic uremic syndrome (aHUS). (23, 24) Our case featured a polymorphism in SCR12 of CFH on a permissive background of the V62 and H402 risk alleles of CFH. To our knowledge, p.Thr732Met has not previously been reported. We present no direct evidence that the variant is disease related. However, it was not seen in a screen of over 7000 persons (representing 14,000 chromosomes) performed as part of the NHLBI (National Heart, Lung, and Blood Institute) Exome Sequencing Project, a program for high-throughput sequencing of the protein-coding regions of well-phenotyped populations from studies such as the Women’s Health Initiative, the Framingham Heart Study, and others. Moreover, software predictions of the potential consequences of this amino acid change suggest functional relevance. In particular, the ConSeq score, a measure of the structural and functional importance of particular amino acids as calculated using the ConSeq server (conseq.bioinfo.tau.ac.il) is 8 (1, low predicted importance; 9, high) and predicts the residue is solvent exposed. In addition, predictive analysis by the PolyPhen2 tool (genetics.bwh.harvard.edu/pph2), suggests that this amino acid substitution is “possibly damaging”. Together, the population data and functional predictions provide strong presumptive evidence that the variant is disease related. With respect to the permissive CFH background, several studies have shown that the V62 and H402 variants lead to increased baseline AP activity even in apparently healthy controls. (18) Studies have also shown that the H402 variant is associated with poorer AP control secondary to decreased binding to both endothelial cells and lipid peroxidation products like malondialdehyde, which accumulates in many pathophysiologic processes.(25, 26)
In addition, we found a heterozygous polymorphism, c.463A>C (p.Lys155Gln), in C3. This variant has been previously reported, with a reported minor allele frequency (MAF) of 0.006, and was identified in 28 of 10,730 chromosomes (MAF of 0.0026) as part of the NHLBI Exome Sequencing Project. It is noteworthy that a two amino-acid deletion in the MG7 domain of C3 (the loss of the aspartate 923 and glycine 924) has been reported to result in fluid phase dysregulation of AP with the consequent development of DDD.(27) The C3 variant in our case could therefore contribute to AP dysregulation. Certainly, a genetic cause is consistent with the strong family history of kidney disease.
This case demonstrates a new pattern of glomerular injury, i.e., chronic glomerular vascular injury leading to chronic kidney disease and FSGS, that is associated with single-nucleotide polymorphisms in CFH and C3 in the context of a permissive genetic background comprising the V62 and H402 variants of CFH. Given that the mutations are found in AP genes, one might expect to find a proliferative glomerulonephritis (C3GN or DDD) or aHUS instead. As mentioned previously, mutations in CFH, particularly at the amino terminus, result in a proliferative glomerulonephritis such as C3GN or DDD from fluid phase dysregulation of the AP and deposition of complement factors and products along the glomerular capillary walls and mesangium. This case did not show any evidence of complement deposition in the glomeruli or along the capillary walls or in the mesangium. Mutations in C3 have been reported in DDD. However, there was no evidence of DDD. Other CFH mutations, particularly at the carboxy terminus, lead to aHUS. However, arteries showed only mild sclerosis of the vessel walls with no evidence of an active thrombotic microangiopathy. Instead, there was evidence of chronic endothelial injury (which prompted evaluation of the AP) with ensuing changes including FSGS and tubulointerstitial scarring.
The variants in CFH and C3 that we found in this case likely resulted in AP dysregulation at the microvascular level, and capillary wall injury. This was evidenced by subendothelial expansion by fluffy granular material and double contour formation. There was only very mild medium or large vessel sclerosis noted. Genetic causes of mild chronic vascular injury and capillary wall injury with double contour formation have been noted in prothrombotic states such as prothrombin gene mutation and Factor V Leiden. (28) To our knowledge, this is the first case to document dysregulation of AP via polymorphisms in CFH and C3 alongside biopsy findings of this kind of capillary wall injury.
The two biopsies done over a period of 2 years allow for follow-up of the progression of the disease. The first biopsy showed capillary wall injury, but with no evidence of FSGS (we cannot rule out FSGS since the lesion may have not been present on biopsy material). The second biopsy 2 years later clearly shows the FSGS and increasing tubular atrophy and interstitial fibrosis.
This case broadens the spectrum of lesions resulting from dysregulation of the AP of complement (Figure 2). In addition to this list of recognized lesions (29, 30), we would like to add secondary FSGS due to chronic vascular injury. Our case also expands the clinical presentation of patients with dysregulation of AP. Patients with C3GN or DDD often present with hematuria and proteinuria, which can be sometimes rapidly progressive, indicating active glomerulonephritis. aHUS also presents with acute kidney injury with features of active thrombotic microangiopathy as evidenced by schistocytes in the peripheral smear, hemolytic anemia, and thrombocytopenia. Kidney biopsy in aHUS shows an acute lesion with swelling of the endothelial cells, and thrombi, debris, and platelet plugs in the capillary lumen. This case shows that AP dysregulation can also manifest as low grade proteinuria and chronic kidney disease. Kidney biopsy in such cases shows FSGS with evidence of vascular injury.
Figure 2.
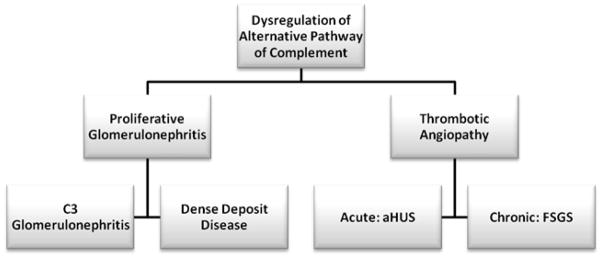
Spectrum of lesions noted with dysregulation of the AP of complement.
To summarize, although genetic causes of FSGS typically involve mutations of the podocyte proteins and occasionally the glomerular basement membranes, we describe a case of FSGS associated with single-nucleotide polymorphisms in CFH and C3, suggesting FSGS can be caused by dysregulation of the AP of complement. Evidence of vascular injury in FSGS warrants an in depth evaluation of the AP of complement.
Acknowledgments
Support: This research was supported in part by NIH grant DK074409 (to Dr sethi and Dr Smith), and a Fulk Family Foundation award (Mayo Clinic) to Dr Sethi.
Footnotes
Financial Disclosure: The authors declare that they have no relevant financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.D’Agati VD, Kaskel FJ, Falk RJ. Focal Segmental Glomerulosclerosis. New England Journal of Medicine. 2011;365:2398–2411. doi: 10.1056/NEJMra1106556. [DOI] [PubMed] [Google Scholar]
- 2.Gbadegesin R, Lavin P, Foreman J, Winn M. Pathogenesis and therapy of focal segmental glomerulosclerosis: an update. Pediatric Nephrology. 2011;26:1001–1015. doi: 10.1007/s00467-010-1692-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kestila M, Lenkkeri U, Mannikko M, et al. Positionally Cloned Gene for a Novel Glomerular Protein Nephrin Is Mutated in Congenital Nephrotic Syndrome. Molecular Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 4.Zenker M, Aigner T, Wendler O, et al. Human laminin beta-2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Human Molecular Genetics. 2004;13:2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 5.Santin S, Garcia-Maset R, Ruiz P, et al. Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int. 2009;76:1268–1276. doi: 10.1038/ki.2009.381. [DOI] [PubMed] [Google Scholar]
- 6.Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nature Genetics. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding -actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 24:251–256. doi: 10.1038/73456. 200. [DOI] [PubMed] [Google Scholar]
- 8.Mele C, Iatropoulos P, Donadelli R, et al. MYO1E Mutations and Childhood Familial Focal Segmental Glomerulosclerosis. New England Journal of Medicine. 2011;365:295–306. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Agati VD. The spectrum of focal segmental glomerulosclerosis: new insights. Current Opinion in Nephrology and Hypertension. 2008;17:271–281. doi: 10.1097/MNH.1090b1013e3282f1094a1096. [DOI] [PubMed] [Google Scholar]
- 10.D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. American Journal of Kidney Diseases. 2004;43:368–382. doi: 10.1053/j.ajkd.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 11.Noris M, Remuzzi G. Atypical Hemolytic-Uremic Syndrome. New England Journal of Medicine. 2009;361:1676–1687. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 12.Le Quintrec M, Roumenina L, Noris M, Fremeaux-Bacchi V. Atypical Hemolytic Uremic Syndrome Associated with Mutations in Complement Regulator Genes. Semin Thromb Hemost. 2010;36:641, 652. doi: 10.1055/s-0030-1262886. [DOI] [PubMed] [Google Scholar]
- 13.Servais A, Fremeaux-Bacchi V, Lequintrec M, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. Journal of Medical Genetics. 2007;44:193–199. doi: 10.1136/jmg.2006.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 15.Smith RJH, Alexander J, Barlow PN, et al. New Approaches to the Treatment of Dense Deposit Disease. Journal of the American Society of Nephrology. 2007;18:2447–2456. doi: 10.1681/ASN.2007030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sethi S, Fervenza FC, Zhang Y, et al. Proliferative Glomerulonephritis Secondary to Dysfunction of the Alternative Pathway of Complement. Clinical Journal of the American Society of Nephrology. 2011;6:1009–1017. doi: 10.2215/CJN.07110810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gale DP, de Jorge EG, Cook HT, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. The Lancet. 2010;376:794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sethi S, Nester CM, Smith RJH. Membranoproliferative glomerulonephritis and C3 glomerulopathy: resolving the confusion. Kidney Int. 2012 doi: 10.1038/ki.2011.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Meyer NC, Wang K, et al. Causes of Alternative Pathway Dysregulation in Dense Deposit Disease. Clinical Journal of the American Society of Nephrology. 2012;7:265–274. doi: 10.2215/CJN.07900811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abrera-Abeleda MA, Nishimura C, Frees K, et al. Allelic Variants of Complement Genes Associated with Dense Deposit Disease. Journal of the American Society of Nephrology. 2011 doi: 10.1681/ASN.2010080795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abrera-Abeleda MA, Nishimura C, Smith JLH, et al. Variations in the Complement Regulatory Genes Factor H (CFH) and Factor H Related 5 (CFHR5) are Associated with Membranoproliferative Glomerulonephritis Type II (Dense Deposit Disease) J Med Genet. 2006;43:582–589. doi: 10.1136/jmg.2005.038315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jokiranta TS, Hellwage J, Koistinen V, Zipfel PF, Meri S. Each of the Three Binding Sites on Complement Factor H Interacts with a Distinct Site on C3b. Journal of Biological Chemistry. 2000;275:27657–27662. doi: 10.1074/jbc.M002903200. [DOI] [PubMed] [Google Scholar]
- 23.Lehtinen MJ, Rops AL, Isenman DE, van der Vlag J, Jokiranta TS. Mutations of Factor H Impair Regulation of Surface-bound C3b by Three Mechanisms in Atypical Hemolytic Uremic Syndrome. Journal of Biological Chemistry. 2009;284:15650–15658. doi: 10.1074/jbc.M900814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jokiranta TS, Jaakola V-P, Lehtinen MJ, Parepalo M, Meri S, Goldman A. Structure of complement factor H carboxyl-terminus reveals molecular basis of atypical haemolytic uremic syndrome. EMBO J. 2006;25:1784–1794. doi: 10.1038/sj.emboj.7601052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skerka C, Lauer N, Weinberger A, et al. Defective complement control of Factor H (Y402H) and FHL-1 in age-related macular degeneration. Molecular Immunology. 2007;44:3398–3406. doi: 10.1016/j.molimm.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Weismann D, Hartvigsen K, Lauer N, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Barricarte R, Heurich M, Valdes-Canedo F, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. The Journal of Clinical Investigation. 2010;120:3702–3712. doi: 10.1172/JCI43343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goforth RL, Rennke H, Sethi S. Renal vascular sclerosis is associated with inherited thrombophilias. Kid Int. 2006;70:743–750. doi: 10.1038/sj.ki.5001551. [DOI] [PubMed] [Google Scholar]
- 29.Fervenza FC, Smith RJH, Sethi S. Association of a novel complement factor H mutation with severe crescentic and necrotizing glomerulonephritis. Am J Kidney Dis. 2012;60(2) doi: 10.1053/j.ajkd.2012.03.007. •••–•••. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sethi S, Fervenza FC, Zhang Y, et al. C3 Glomerulonephritis: Clinicopathologic findings, complement abnormalities, glomerular proteomic profile, treatment and follow-up. Kidney Int. 2012 doi: 10.1038/ki.2012.212. •••–•••. [DOI] [PMC free article] [PubMed] [Google Scholar]